Visible Light Assisted Organosilane Assembly on Mesoporous Silicon Films and Particles

 ,
, 
Abstract
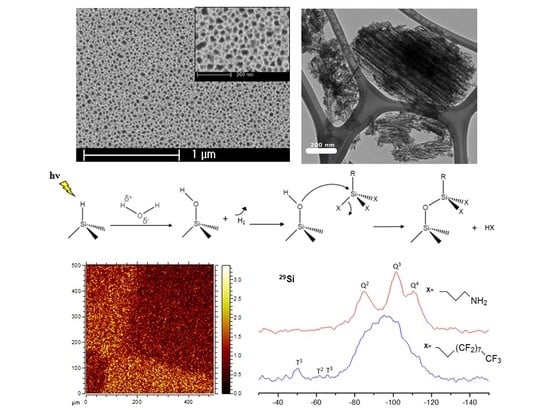
{kind=link}
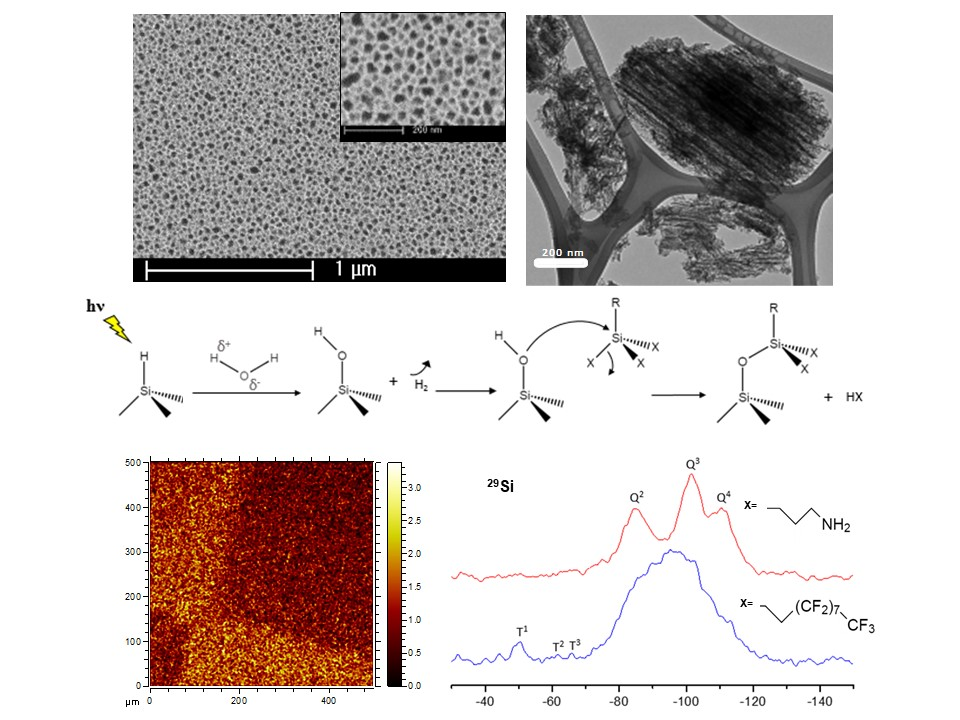
{kind=link}
{kind=link}
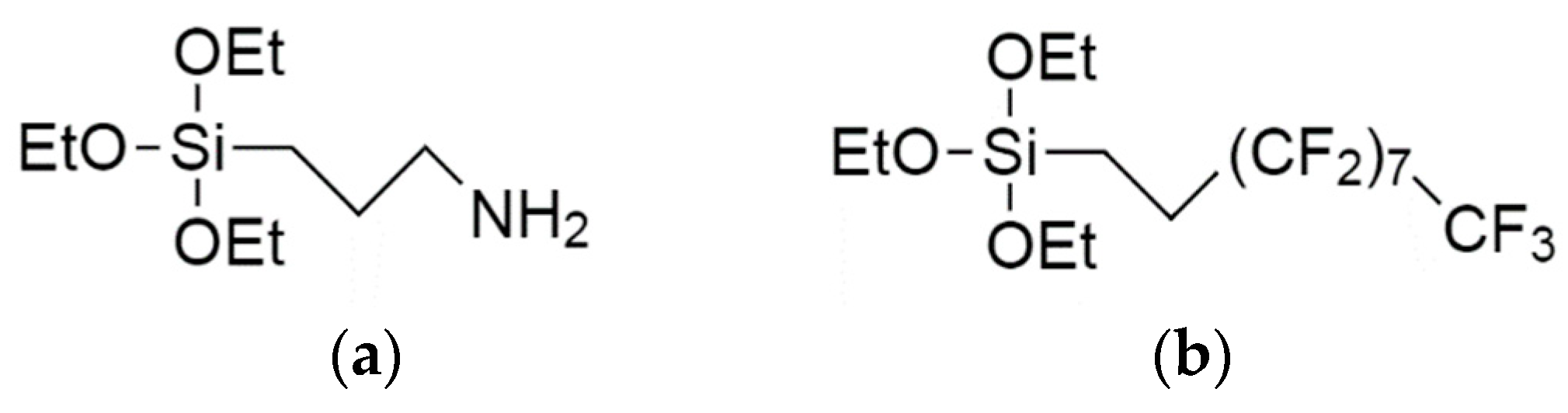
{kind=link}
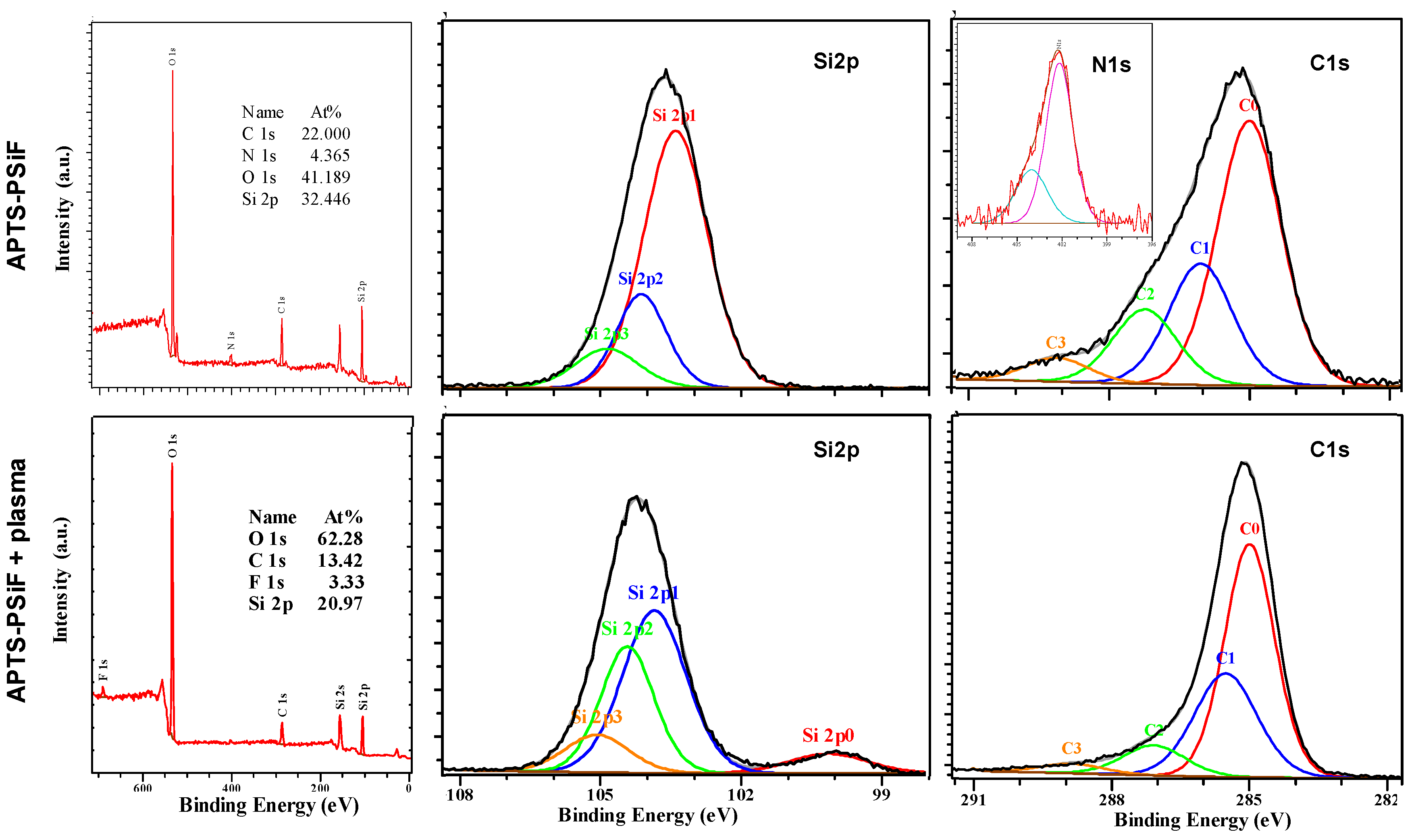
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Preparation of PSiFs and PSiPs
2.2. Organosilane Assembly on PSi
2.3. Characterization
3. Results and Discussion
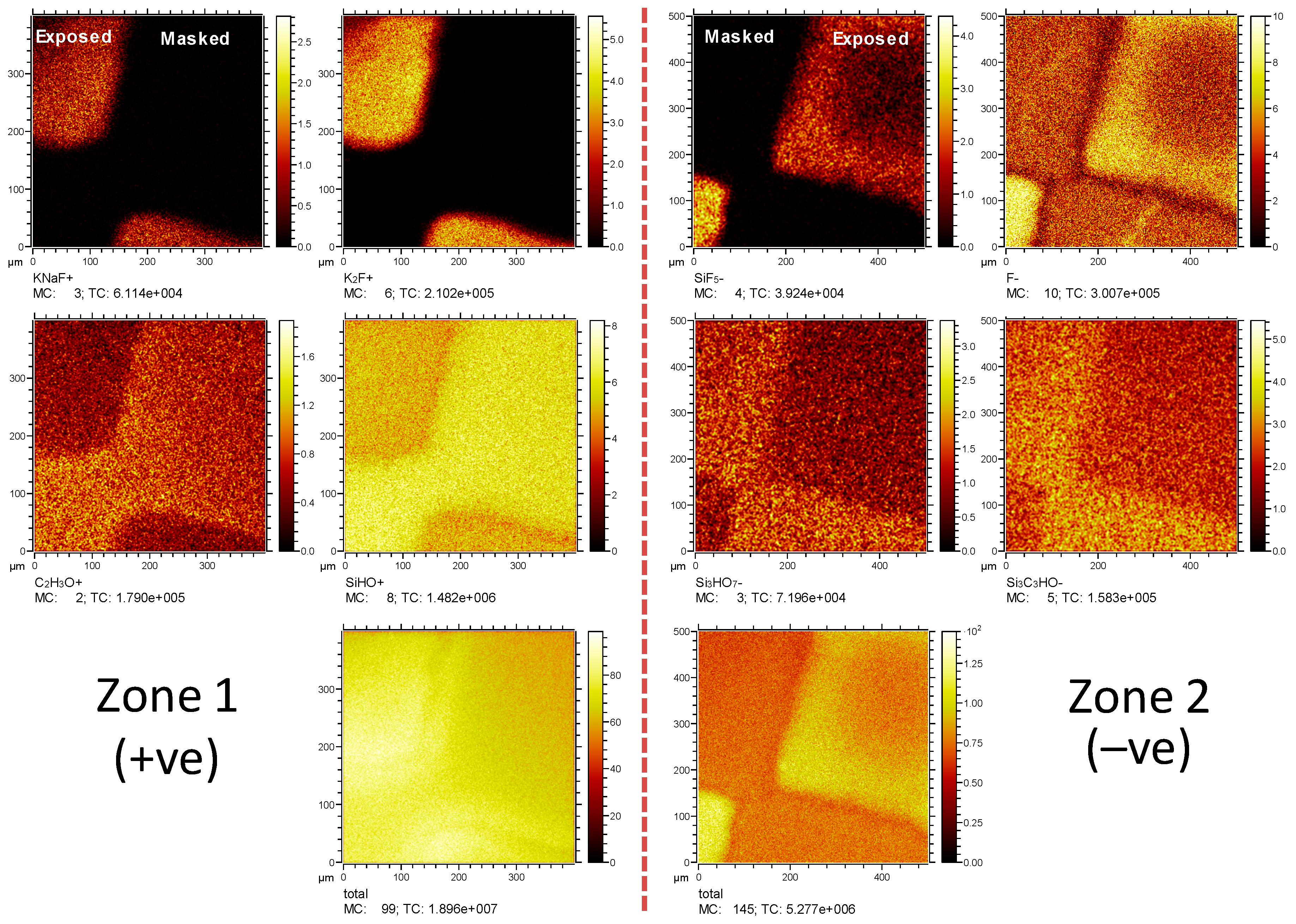
3.1. Characterization of PSiFs
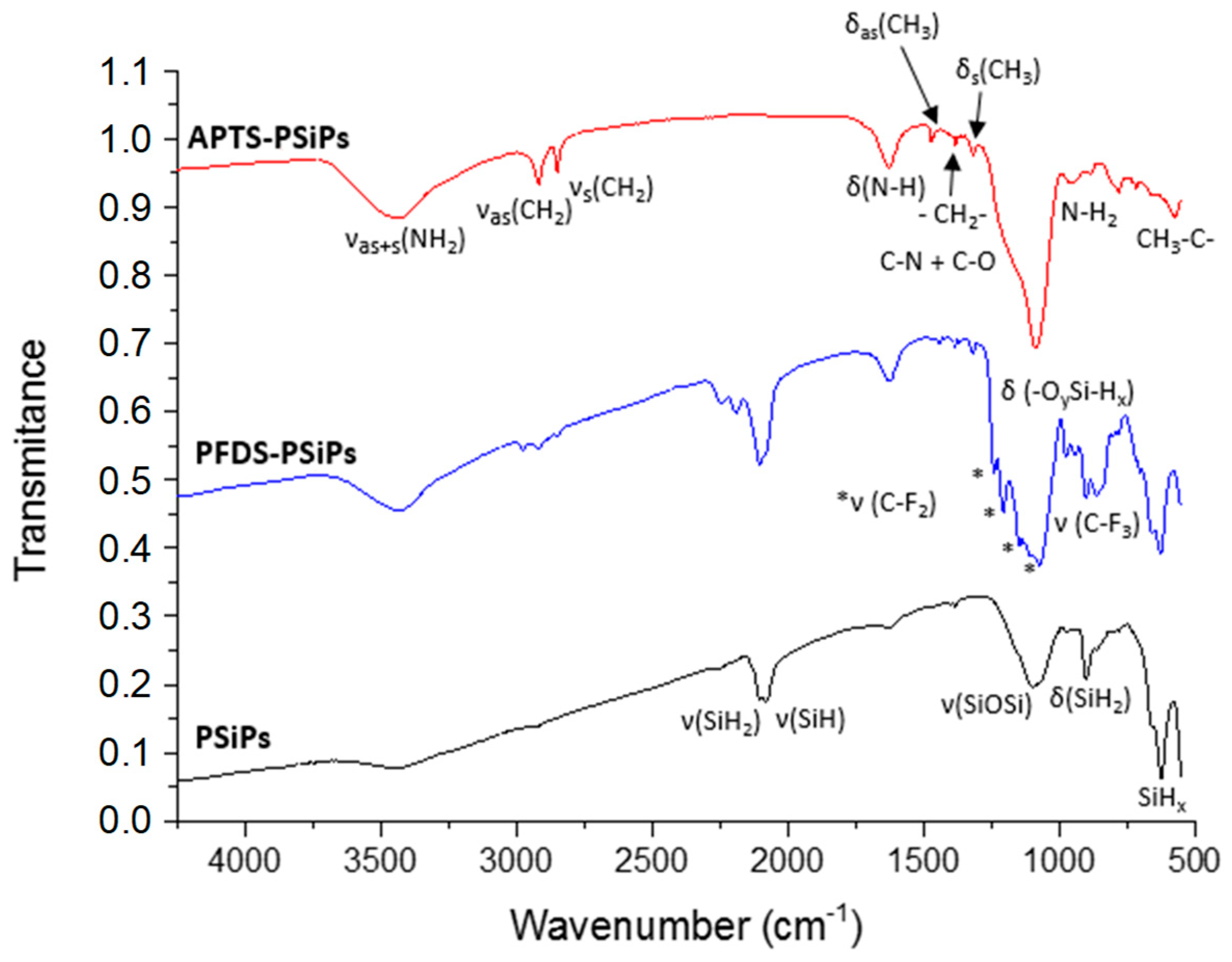
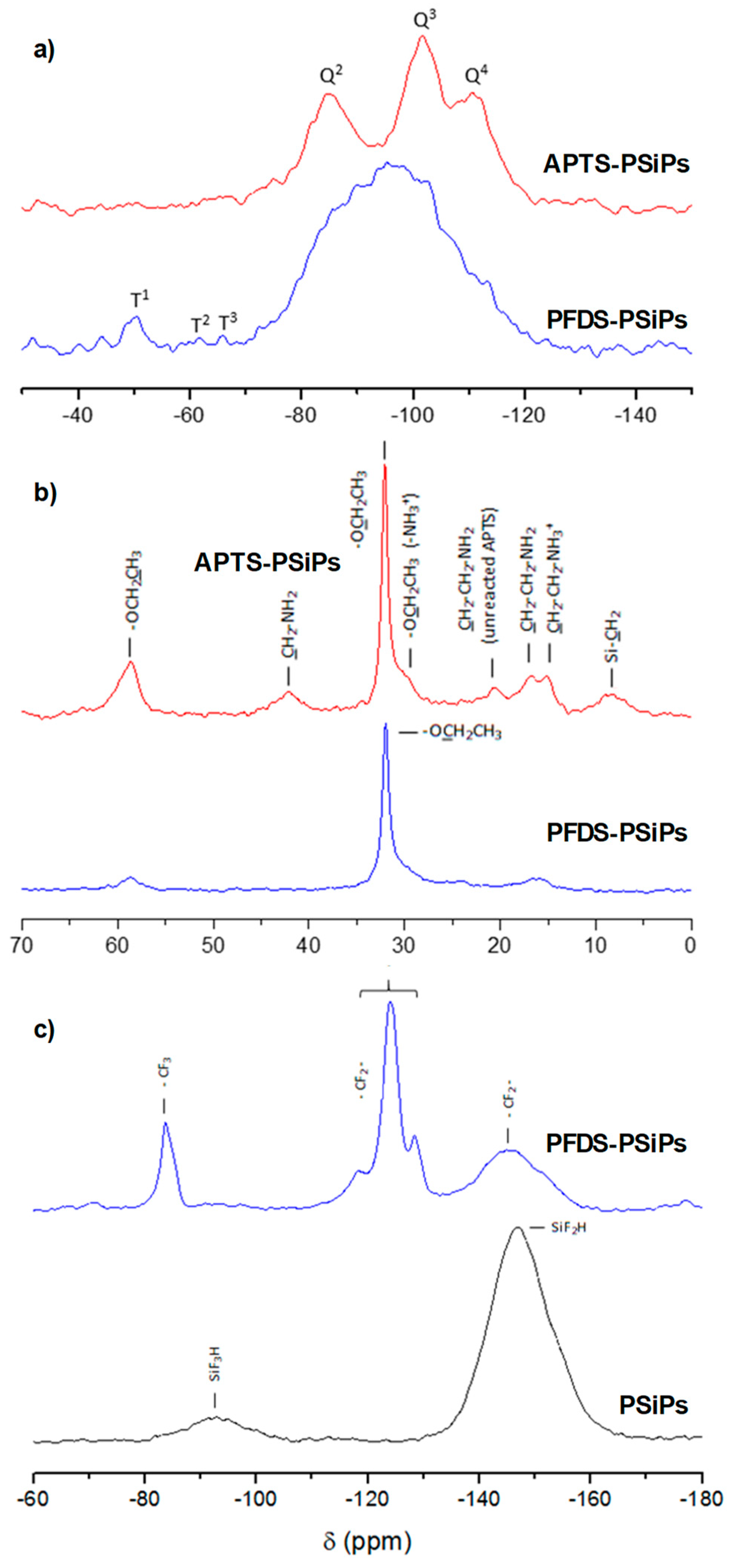
3.2. Characterization of PSiPs
3.3. Reaction Mechanism
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Badr, Y.A.; El-Kader, K.M.A.; Khafagy, R.M. Raman spectroscopic study of CdS, PVA composite films. J. Appl. Polym. Sci. 2004, 92, 1984–1992. [Google Scholar] [CrossRef]
- Sacarescu, L.; Roman, G.; Sacarescu, G.; Simionescu, M. Fluorescence detection system based on silicon quantum dots-polysilane nanocomposites. Express Polym. Lett. 2016, 10, 990–1002. [Google Scholar] [CrossRef]
- Makila, E.; Bimbo, L.M.; Kaasalainen, M.; Herranz, B.; Airaksinen, A.J.; Heinonen, M.; Kukk, E.; Hirvonen, J.; Santos, H.A.; Salonen, J. Amine Modification of Thermally Carbonized Porous Silicon with Silane Coupling Chemistry. Langmuir 2012, 28, 14045–14054. [Google Scholar] [CrossRef]
- Kao, H.M.; Liao, C.H.; Hung, T.T.; Pan, Y.C.; Chiang, A.S.T. Direct synthesis and solid-state NMR characterization of cubic mesoporous silica SBA-1 functionalized with phenyl groups. Chem. Mater. 2008, 20, 2412–2422. [Google Scholar] [CrossRef]
- Tanaka, T.; Godin, B.; Bhavane, R.; Nieves-Alicea, R.; Gu, J.; Liu, X.; Chiappini, C.; Fakhoury, J.R.; Amra, S.; Ewing, A.; et al. In vivo evaluation of safety of nanoporous silicon carriers following single and multiple dose intravenous administrations in mice. Int. J. Pharm. 2010, 402, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Torres-Costa, V.; Ahumada, O.; Cebrian, V.; Gomez-Abad, C.; Diaz, A.; Silvan, M.M. Gold nanoparticle triggered dual optoplasmonic-impedimetric sensing of prostate-specific antigen on interdigitated porous silicon platforms. Sens. Actuators B Chem. 2018, 267, 559–564. [Google Scholar] [CrossRef]
- Dominguez, R.B.; Alonso, G.A.; Munoz, R.; Hayat, A.; Marty, J.L. Design of a novel magnetic particles based electrochemical biosensor for organophosphate insecticide detection in flow injection analysis. Sens. Actuators B Chem. 2015, 208, 491–496. [Google Scholar] [CrossRef]
- Dorvee, J.R.; Sailor, M.J.; Miskelly, G.M. Digital microfluidics and delivery of molecular payloads with magnetic porous silicon chaperones. Dalton Trans. 2008, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Godin, B.; Tasciotti, E.; Liu, X.W.; Serda, R.E.; Ferrari, M. Multistage Nanovectors: From Concept to Novel Imaging Contrast Agents and Therapeutics. Acc. Chem. Res. 2011, 44, 979–989. [Google Scholar] [CrossRef]
- Fan, D.M.; De Rosa, E.; Murphy, M.B.; Peng, Y.; Smid, C.A.; Chiappini, C.; Liu, X.W.; Simmons, P.; Weiner, B.K.; Ferrari, M.; et al. Mesoporous Silicon-PLGA Composite Microspheres for the Double Controlled Release of Biomolecules for Orthopedic Tissue Engineering. Adv. Funct. Mater. 2012, 22, 282–293. [Google Scholar] [CrossRef]
- Chadwick, E.G.; Clarkin, O.M.; Tanner, D.A. Hydroxyapatite formation on metallurgical grade nanoporous silicon particles. J. Mater. Sci. 2010, 45, 6562–6568. [Google Scholar] [CrossRef]
- Zhu, G.X.; Liu, J.T.; Wang, Y.Z.; Zhang, D.C.; Guo, Y.; Tasciotti, E.; Hu, Z.B.; Liu, X.W. In Situ Reductive Synthesis of Structural Supported Gold Nanorods in Porous Silicon Particles for Multifunctional Nanovectors. ACS Appl. Mater. Interfaces 2016, 8, 11881–11891. [Google Scholar] [CrossRef] [PubMed]
- DeLouise, L.A.; Miller, B.L. Quantatitive assessment of enzyme immobilization capacity in porous silicon. Anal. Chem. 2004, 76, 6915–6920. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Kim, S.; Ko, Y.C.; Cho, S.; Sohn, H. DBR-structured smart particles for sensing applications. Colloids Surf. A Physicochem. Eng. Asp. 2008, 313, 398–401. [Google Scholar] [CrossRef]
- Dong, J.P.; Wang, A.F.; Ng, K.Y.S.; Mao, G.Z. Self-assembly of octadecyltrichlorosilane monolayers on silicon-based substrates by chemical vapor deposition. Thin Solid Films 2006, 515, 2116–2122. [Google Scholar] [CrossRef]
- Naveas, N.; Torres Costa, V.; Gallach, D.; Hernandez-Montelongo, J.; Martin Palma, R.J.; Predenstinacion Garcia-Ruiz, J.; Manso-Silvan, M. Chemical stabilization of porous silicon for enhanced biofunctionalization with immunoglobulin. Sci. Technol. Adv. Mater. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Noval, A.; Gallach, D.; Angel Garcia, M.; Ferro-Llanos, V.; Herrero, P.; Fukami, K.; Ogata, Y.H.; Torres-Costa, V.; Martin-Palma, R.J.; Ciment-Font, A.; et al. Characterization of hybrid cobalt-porous silicon systems: Protective effect of the Matrix in the metal oxidation. Nanoscale Res. Lett. 2012, 7. [Google Scholar] [CrossRef]
- Rodriguez, C.; Ahumada, O.; Cebrian, V.; Costa, V.T.; Silvan, M.M. Biofunctional porous silicon micropatterns engineered through visible light activated epoxy capping and selective plasma etching. Vacuum 2018, 150, 232–238. [Google Scholar] [CrossRef]
- Blasco, T.; PerezPariente, J.; Kolodziejski, W. A solid-state NMR study of the molecular sieve VPI-5 synthesized in the presence of a CTABr surfactant. Solid State Nucl. Magn. Reson. 1997, 8, 185–194. [Google Scholar] [CrossRef]
- Petit, D.; Chazalviel, J.N.; Ozanam, F.; Devreux, F. Porous silicon structure studied by nuclear magnetic resonance. Appl. Phys. Lett. 1997, 70, 191–193. [Google Scholar] [CrossRef]
- Brandt, M.S.; Ready, S.E.; Boyce, J.B. Si-29 nuclear magnetic resonance of luminescent silicon. Appl. Phys. Lett. 1997, 70, 188–190. [Google Scholar] [CrossRef]
- Faulkner, R.A.; DiVerdi, J.A.; Yang, Y.; Kobayashi, T.; Maciel, G.E. The Surface of Nanoparticle Silicon as Studied by Solid-State NMR. Materials 2013, 6, 18–46. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, J.R.; Harley, S.J.; Carter, R.S.; Power, P.P.; Augustine, M.P. Using liquid and solid state NMR and photoluminescence to study the synthesis and solubility properties of amine capped silicon nanoparticles. Solid State Nucl. Magn. Reson. 2007, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shanmugharaj, A.M.; Rhee, K.Y.; Ryu, S.H. Influence of dispersing medium on grafting of aminopropyltriethoxysilane in swelling clay materials. J. Colloid Interface Sci. 2006, 298, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.T.; Joo, J.; Gu, L.; Sailor, M.J. Size Control of Porous Silicon Nanoparticles by Electrochemical Perforation Etching. Part. Part. Syst. Charact. 2014, 31, 252–256. [Google Scholar] [CrossRef]
- Munoz-Noval, A.; Torres-Costa, V.; Martin-Palma, R.J.; Herrero-Fernandez, P.; Angel Garcia, M.; Fukami, K.; Ogata, Y.H.; Manso Silvan, M. Electroless nanoworm Au films on columnar porous silicon layers. Mater. Chem. Phys. 2012, 134, 664–669. [Google Scholar] [CrossRef]
- Tacke, R.; Becht, J.; Lopezmras, A.; Sheldrick, W.S.; Sebald, A. Syntheses, x-ray crystal-structure analyses, and solid-state NMS-studies of some zwitterionic organofluorosilicates. Inorg. Chem. 1993, 32, 2761–2766. [Google Scholar] [CrossRef]
- Leichle, T.; Silvan, M.M.; Belaubre, P.; Valsesia, A.; Ceccone, G.; Rossi, F.; Saya, D.; Pourciel, J.B.; Nicu, L.; Bergaud, C. Nanostructuring surfaces with conjugated silica colloids deposited using silicon-based microcantilevers. Nanotechnology 2005, 16, 525–531. [Google Scholar] [CrossRef]
- Munoz Noval, A.; Sanchez Vaquero, V.; Punzon Quijorna, E.; Torres Costa, V.; Gallach Perez, D.; Gonzalez Mendez, L.; Montero, I.; Martin Palma, R.J.; Climent Font, A.; Garcia Ruiz, J.P.; et al. Aging of porous silicon in physiological conditions: Cell adhesion modes on scaled 1D micropatterns. J. Biomed. Mater. Res. Part A 2012, 100A, 1615–1622. [Google Scholar] [CrossRef]
- Gallach, D.; Sanchez, G.R.; Noval, A.M.; Silvan, M.M.; Ceccone, G.; Palma, R.J.M.; Costa, V.T.; Duart, J.M.M. Functionality of porous silicon particles: Surface modification for biomedical applications. Mater. Sci. Eng. B-Adv. Funct. Solid-State Mater. 2010, 169, 123–127. [Google Scholar] [CrossRef]
- Beamson, G.; Briggs, D. High Resolution XPS of Organic Polymers—The Scienta ESCA300 Database; Wiley Interscience: New York, NY, USA, 1992. [Google Scholar]
- Manso-Silvan, M.; Valsesia, A.; Hasiwa, M.; Rodriguez-Navas, C.; Gilliland, D.; Ceccone, G.; Garcia Ruiz, J.P.; Rossi, F. Micro-spot, UV and wetting patterning pathways for applications of biofunctional aminosilane-titanate coatings. Biomed. Microdevices 2007, 9, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Vanea, E.; Simon, V. XPS study of protein adsorption onto nanocrystalline aluminosilicate microparticles. Appl. Surf. Sci. 2011, 257, 2346–2352. [Google Scholar] [CrossRef]
- Naveas, N.; Hernandez-Montelongo, J.; Pulido, R.; Torres-Costa, V.; Villanueva-Guerrero, R.; Ruiz, J.P.G.; Manso-Silvan, M. Fabrication and characterization of a chemically oxidized-nanostructured porous silicon based biosensor implementing orienting protein A. Colloids Surf. B Biointerfaces 2014, 115, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Ohlhausen, J.A.; Zavadil, K.R. Time-of-flight secondary ion mass spectrometry measurements of a fluorocarbon-based self-assembled monoloyer on Si. J. Vac. Sci. Technol. A 2006, 24, 1172–1178. [Google Scholar] [CrossRef]
- Kempson, I.M.; Barnes, T.J.; Prestidge, C.A. Use of TOF-SIMS to Study Adsorption and Loading Behavior of Methylene Blue and Papain in a Nano-Porous Silicon Layer. J. Am. Soc. Mass Spectrom. 2010, 21, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Pietrass, T.; Bifone, A.; Roth, R.D.; Koch, V.P.; Alivisatos, A.P.; Pines, A. Si-29 high resolution solid state nuclear magnetic resonance spectroscopy of porous silicon. J. Non-Cryst. Solids 1996, 202, 68–76. [Google Scholar] [CrossRef]
- Tsuboi, T.; Sakka, T.; Ogata, Y.H. Structural study of porous silicon and its oxidized states by solid-state high-resolution Si-29 NMR spectroscopy. Phys. Rev. B 1998, 58, 13863–13869. [Google Scholar] [CrossRef]
- Innocenzi, P.; Brusatin, G.; Guglielmi, M.; Bertani, R. New synthetic route to (3-glycidoxypropyl)trimethoxysilane-base hybrid organic-inorganic materials. Chem. Mater. 1999, 11, 1672–1679. [Google Scholar] [CrossRef]
- Chang, W.K.; Liao, M.Y.; Gleason, K.K. Characterization of porous silicon by solid-state nuclear magnetic resonance. J. Phys. Chem. 1996, 100, 19653–19658. [Google Scholar] [CrossRef]
- Silvan, M.M.; Messina, G.M.L.; Montero, I.; Satriano, C.; Ruiz, J.P.G.; Marletta, G. Aminofunctionalization and sub-micrometer patterning on silicon through silane doped agarose hydrogels. J. Mater. Chem. 2009, 19, 5226–5233. [Google Scholar] [CrossRef]
- Vasil’ev, S.G.; Volkov, V.I.; Tatarinova, E.A.; Muzafarov, A.M. A Solid-State NMR Investigation of MQ Silicone Copolymers. Appl. Magn. Reson. 2013, 44, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Al-Sabagh, A.M.; Harding, D.R.K.; Kandile, N.G.; Badawi, A.M.; El-Tabey, A.E. Synthesis of Some Novel Nonionic Ethoxylated Surfactants Based on alpha-Amino Acids and Investigation of Their Surface Active Properties. J. Dispers. Sci. Technol. 2009, 30, 427–438. [Google Scholar] [CrossRef]
- Lu, H.T. Synthesis and characterization of amino-functionalized silica nanoparticles. Colloid J. 2013, 75, 311–318. [Google Scholar] [CrossRef]
- Sano, H.; Maeda, H.; Matsuoka, S.; Lee, K.H.; Murase, K.; Sugimura, H. Self-assembled monolayers directly attached to silicon substrates formed from 1-hexadecene by thermal, ultraviolet, and visible light activation methods. Jpn. J. Appl. Phys. 2008, 47, 5659–5664. [Google Scholar] [CrossRef]
- Meskini, O.; Abdelghani, A.; Tlili, A.; Mgaieth, R.; Jaffrezic-Renault, N.; Martelet, C. Porous silicon as functionalized material for immunosensor application. Talanta 2007, 71, 1430–1433. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, C.; Muñoz Noval, A.; Torres-Costa, V.; Ceccone, G.; Manso Silván, M. Visible Light Assisted Organosilane Assembly on Mesoporous Silicon Films and Particles. Materials 2019, 12, 131. https://doi.org/10.3390/ma12010131
Rodriguez C, Muñoz Noval A, Torres-Costa V, Ceccone G, Manso Silván M. Visible Light Assisted Organosilane Assembly on Mesoporous Silicon Films and Particles. Materials. 2019; 12(1):131. https://doi.org/10.3390/ma12010131
Chicago/Turabian StyleRodriguez, Chloé, Alvaro Muñoz Noval, Vicente Torres-Costa, Giacomo Ceccone, and Miguel Manso Silván. 2019. "Visible Light Assisted Organosilane Assembly on Mesoporous Silicon Films and Particles" Materials 12, no. 1: 131. https://doi.org/10.3390/ma12010131
APA StyleRodriguez, C., Muñoz Noval, A., Torres-Costa, V., Ceccone, G., & Manso Silván, M. (2019). Visible Light Assisted Organosilane Assembly on Mesoporous Silicon Films and Particles. Materials, 12(1), 131. https://doi.org/10.3390/ma12010131