Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 2: Dynamic Mechanical Analysis



Abstract
1. Introduction
2. Materials and Methods
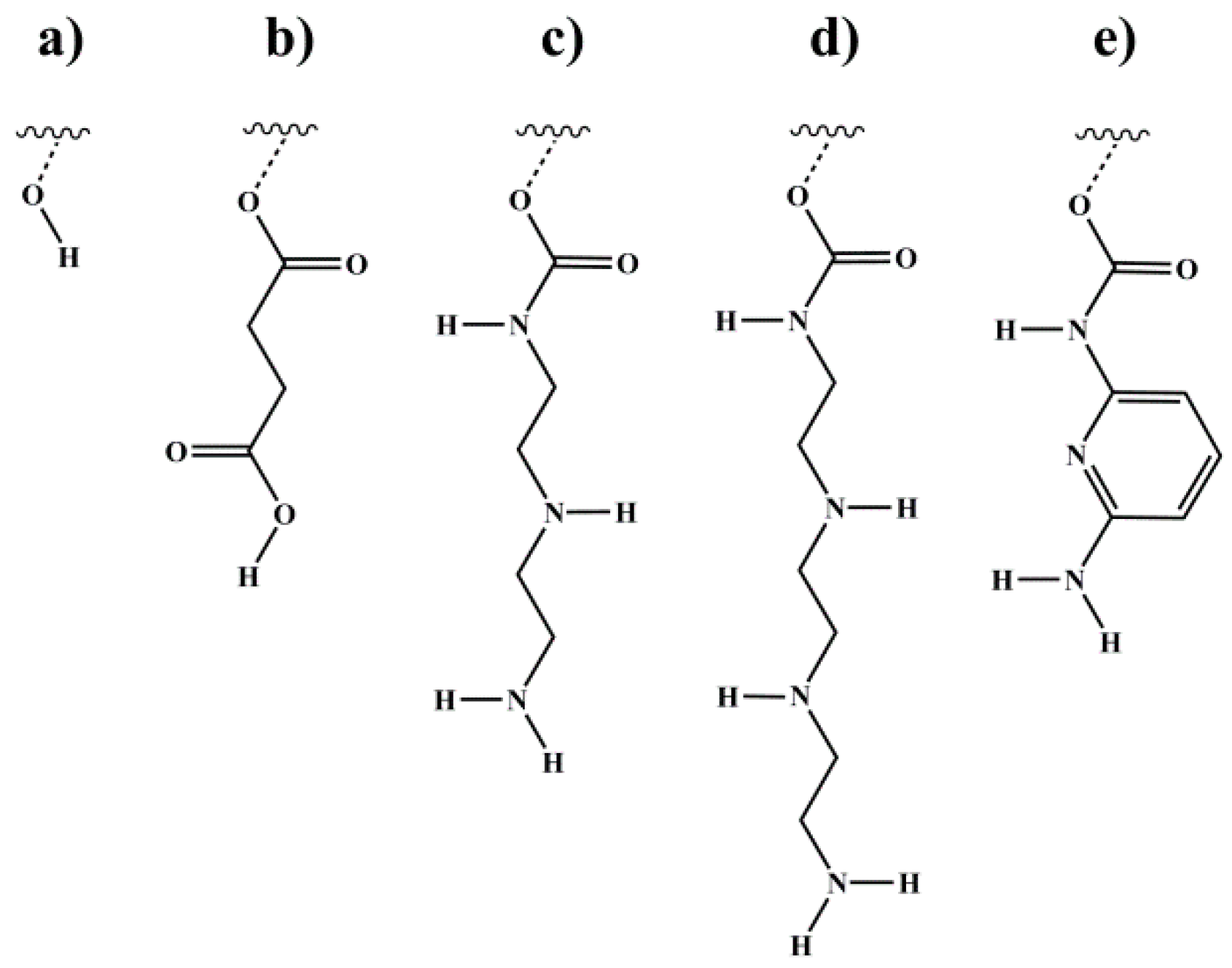
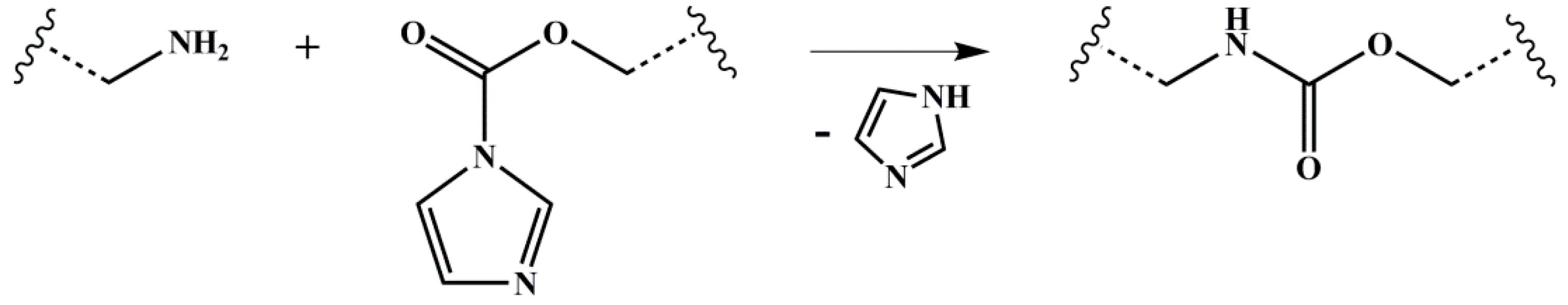
2.1. Synthesis of Functionalized Block Copolymers
2.2. Characterization
2.2.1. 1H NMR Spectroscopy and Calculation of Degree of Functionalization Df
2.2.2. Dynamic Mechanical Analysis
2.2.3. Fourier Transform Infrared Spectroscopy
3. Results and Discussion
3.1. Dynamic Mechanical Analysis
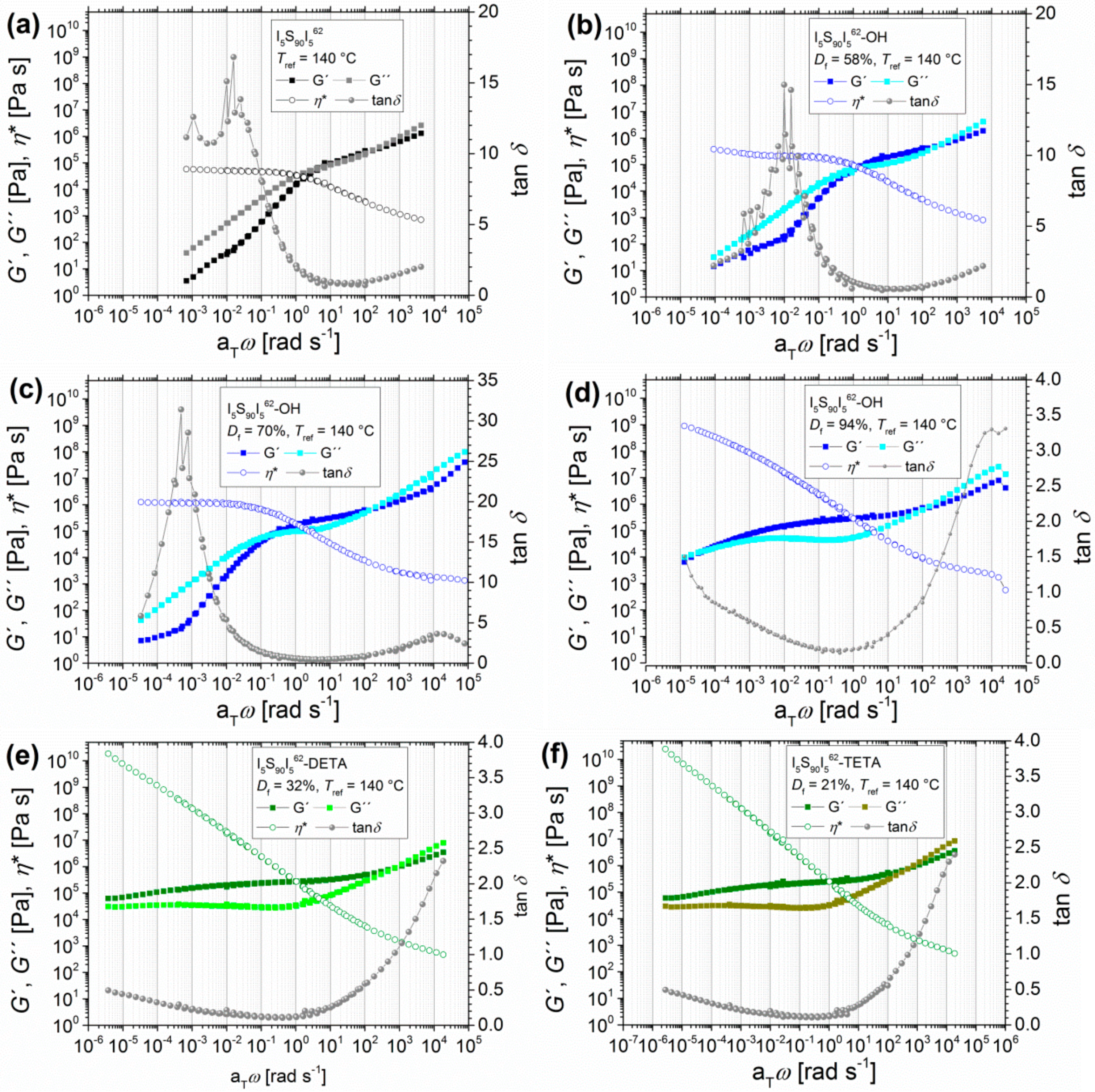
3.1.1. Angular-Frequency-Dependent DMA of Hydroxylated I5S90I562 with Varying Degrees of Functionalization
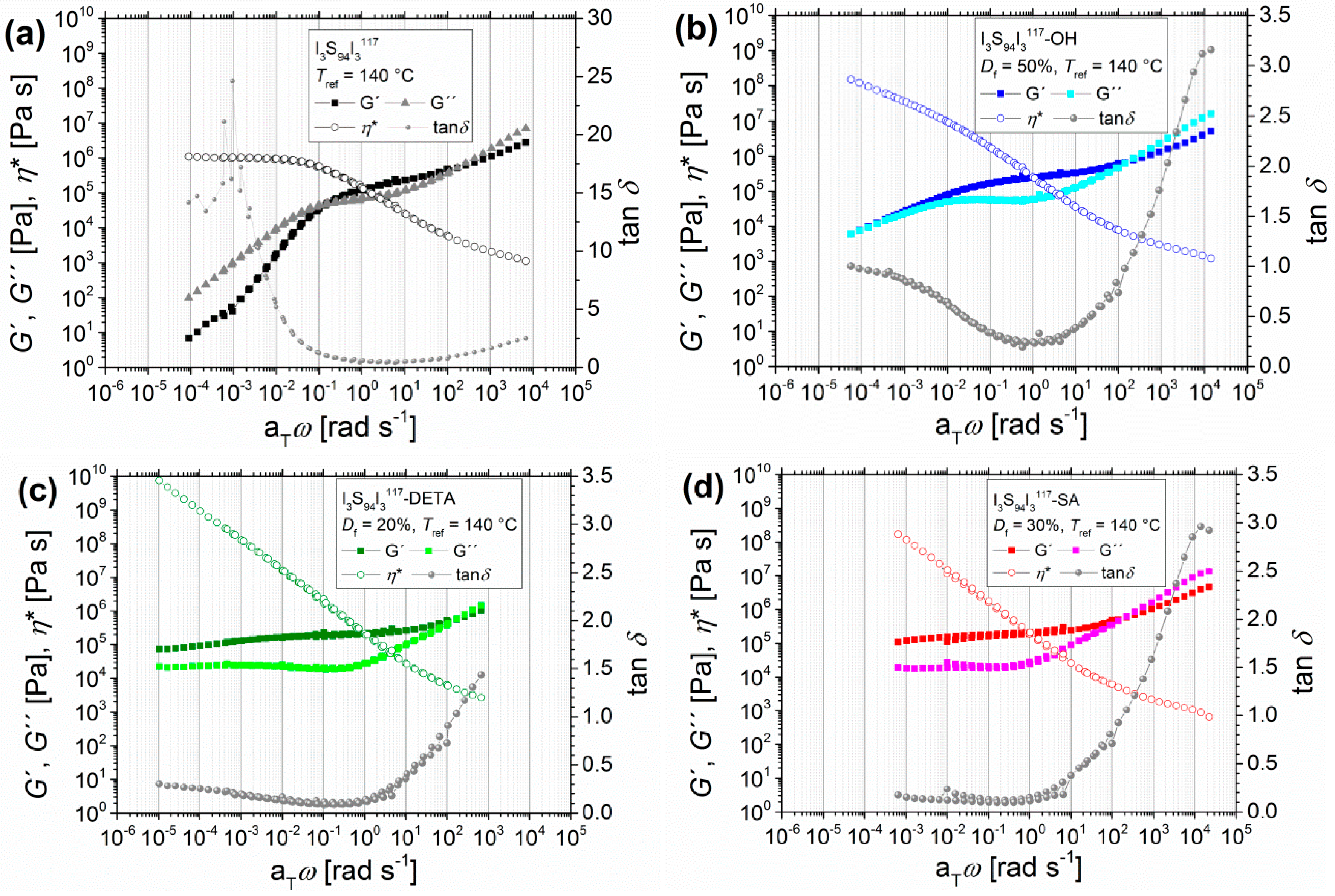
3.1.2. Angular-Frequency-Dependent DMA of Different Modified I5S90I562 and I3S94I3117
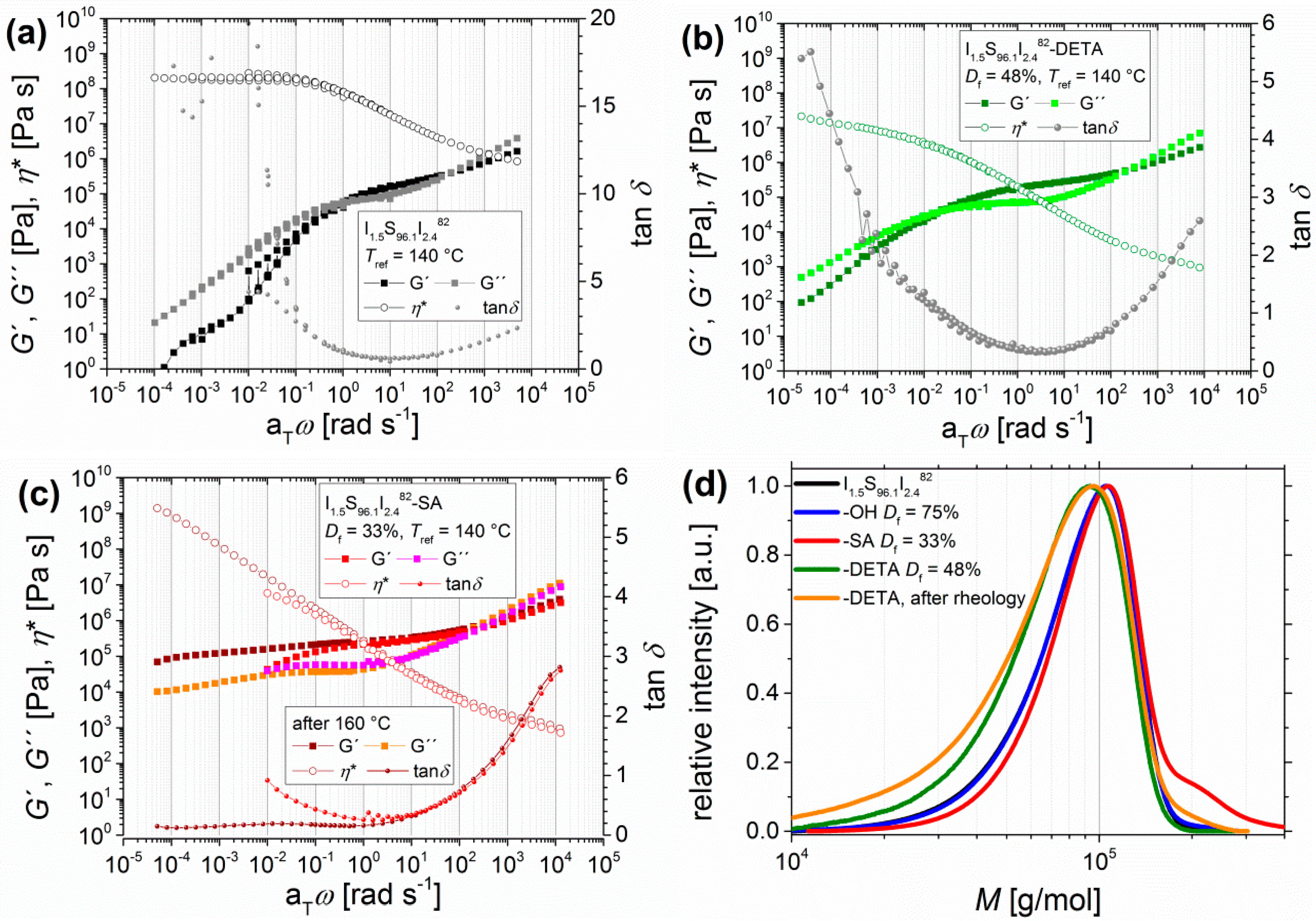
3.1.3. Lowering the PI Contents in ISI and Its Influence on the Results from DMA
3.1.4. Comparison of Functionalized ISI Triblock Copolymers to OH- and SA-functionalized SI Diblock Copolymers
3.1.5. Temperature-Dependent Dynamic Mechanical Analysis
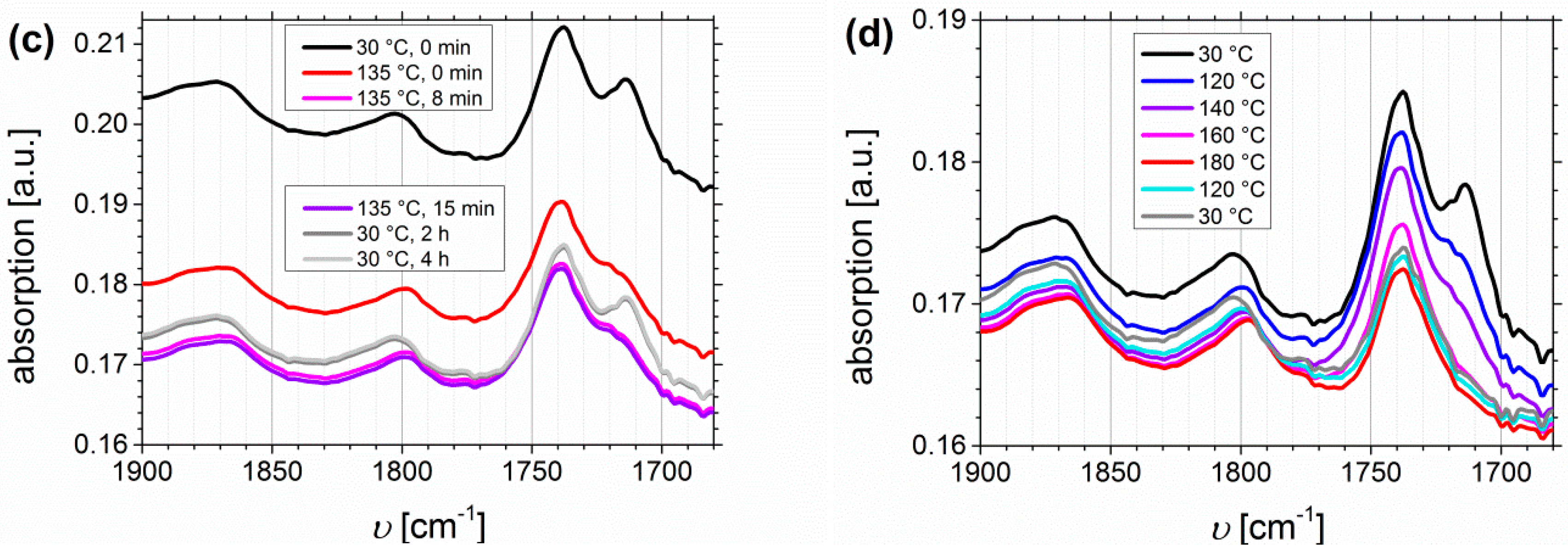
3.2. Fourier Transform Infrared Spectroscopy (FTIR)
DMA-Related Temperature Profile of I1.5S96.1I2.482-DETA and of I1.5S96.1I2.482-SA
3.3. Discussion on Cross-Linking Behavior during Heat Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Burattini, S.; Greenland, B.W.; Merino, D.H.; Weng, W.; Seppala, J.; Colquhoun, H.M.; Hayes, W.; Mackay, M.E.; Hamley, I.W.; Rowan, S.J. A healable supramolecular polymer blend based on aromatic π−π stacking and hydrogen-bonding interactions. J. Am. Chem. Soc. 2010, 132, 12051–12058. [Google Scholar] [CrossRef] [PubMed]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zou, W.; Luo, Y.; Xie, T. Shape memory polymer network with thermally distinct elasticity and plasticity. Sci. Adv. 2016, 2, e1501297. [Google Scholar] [CrossRef] [PubMed]
- Wojtecki, R.J.; Meador, M.A.; Rowan, S.J. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 2011, 10, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Billiet, S.; De Bruycker, K.; Driessen, F. Triazolinediones enable ultrafast and reversible click chemistry for the design of dynamic polymer systems. Nat. Chem. 2014, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Binder, W.H.; Bouteiller, L.; ten Brinke, G.; Ikkala, O.; Rotello, V.M.; Ruokolainen, J.; Srivastava, S.; Xu, H.; Zirbs, R. Hydrogen Bonded Polymers; Binder, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 207, ISBN 9783540685876. [Google Scholar]
- Brunsveld, L.; Folmer, B.J.B.; Meijer, E.W.; Sijbesma, R.P. Supramolecular polymers. Chem. Rev. 2001, 101, 4071–4098. [Google Scholar] [CrossRef] [PubMed]
- Gloe, K.; Heßke, H.; Lindoy, L. Supramolekulare Chemie: Vom Einzelmolekül zur komplexen Funktionseinheit. Wiss. Z. Tech. Univ. Dresd. 2007, 56, 32–38. [Google Scholar]
- Hoeben, F.J.M.; Jonkheijm, P.; Meijer, E.W.; Schenning, A.P.H.J. About supramolecular assemblies of pi-conjugated systems. Chem. Rev. 2005, 105, 1491–1546. [Google Scholar] [CrossRef] [PubMed]
- Lange, R.F.M.; Van Gurp, M.; Meijer, E.W. Hydrogen-bonded supramolecular polymer networks. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 3657–3670. [Google Scholar] [CrossRef]
- Bertrand, A.; Lortie, F.; Bernard, J. Routes to hydrogen bonding chain-end functionalized polymers. Macromol. Rapid Commun. 2012, 2062–2091. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Urban, M.W. Self-healing polymeric materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Dardin, A.; Seidel, U.; Balsamo, V.; Iván, B.; Spiess, H.W.; Stadler, R. Junction dynamics in telechelic hydrogen bonded polyisobutylene networks. Macromolecules 1996, 29, 2577–2583. [Google Scholar] [CrossRef]
- Engle, L.P.; Wagener, K.B. A Review of thermally controlled covalent bond formation in polymer chemistry. J. Macromol. Sci. Part C Polym. Rev. 1993, 33, 239–257. [Google Scholar] [CrossRef]
- Balkenende, D.W.R.; Olson, R.A.; Balog, S.; Weder, C.; Montero de Espinosa, L. Epoxy resin-inspired reconfigurable supramolecular networks. Macromolecules 2016, 49, 7877–7885. [Google Scholar] [CrossRef]
- Ruiz De Luzuriaga, A.; Martin, R.; Markaide, N.; Rekondo, A.; Cabañero, G.; Rodríguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horiz. 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Denissen, W.; Droesbeke, M.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Chemical control of the viscoelastic properties of vinylogous urethane vitrimers. Nat. Commun. 2017, 8, 14857. [Google Scholar] [CrossRef] [PubMed]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Chujo, Y.; Sada, K.; Saegusa, T. Reversible gelation of polyoxazoline by means of Diels−Alder reaction. Macromolecules 1990, 23, 2636–2641. [Google Scholar] [CrossRef]
- Goussé, C.; Gandini, A.; Hodge, P. Application of the Diels−Alder reaction to polymers bearing furan moieties. 2. Diels−Alder and Retro−Diels−Alder reactions involving furan rings in some styrene copolymers. Macromolecules 1998, 31, 314–321. [Google Scholar] [CrossRef]
- Rahmstorf, E.; Abetz, V. Supramolecular networks from block copolymers based on styrene and isoprene using hydrogen bonding motifs—Part 1: Synthesis and characterization. Materials 2018, 11, 1608. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.K.; Zimmerman, S.C. Hydrogen bonding modules for use in supramolecular polymers. Isr. J. Chem. 2013, 53, 511–520. [Google Scholar] [CrossRef]
- Sijbesma, R.P.; Meijer, E.W. Quadruple hydrogen bonded systems. Chem. Commun. 2003, 5–16. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Pranata, J. Importance of secondary interactions in triply hydrogen bonded complexes: Guanine-cytosine vs. uracil-2,6-diaminopyridine. J. Am. Chem. Soc. 1990, 112, 2008–2010. [Google Scholar] [CrossRef]
- Sijbesma, R.P.; Beijer, F.H.; Brunsveld, L.; Folmer, B.J.B.; Hirschberg, J.H.K.K.; Lange, R.F.M.; Lowe, J.K.L.; Meijer, E.W. Reversible polymers formed from self-complementary monomers using quadruple hydrogen bonding. Science 1997, 278, 1601–1604. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, D.J.M.; Spiering, A.J.H.; Peters, G.W.M.; Te Nijenhuis, K.; Sijbesma, R.P. Unidirectional dimerization and stacking of ureidopyrimidinone end groups in polycaprolactone supramolecular polymers. Macromolecules 2007, 40, 8464–8475. [Google Scholar] [CrossRef]
- Dankers, P.Y.W.; Zhang, Z.; Wisse, E.; Grijpma, D.W.; Sijbesma, R.P.; Feijen, J.; Meijer, E.W. Oligo(trimethylene carbonate)-based supramolecular biomaterials. Macromolecules 2006, 39, 8763–8771. [Google Scholar] [CrossRef]
- Hilger, C.; Stadler, R. New multiphase architecture from statistical copolymers by cooperative hydrogen bond formation. Macromolecules 1990, 23, 2095–2097. [Google Scholar] [CrossRef]
- Stadler, R.; de Lucca Freitas, L. Thermoplastic elastomers by hydrogen bonding. Polym. Bull. 1986, 15, 173–179. [Google Scholar] [CrossRef]
- Seidel, U.; Schollmeyer, D.; Stadler, R. Two-dimensional assembly of functionalized 4-aryl-1-alkenyl-3,5-dioxo-1,2,4-triazolidines. Supramol. Chem. 1995, 2, 45–50. [Google Scholar] [CrossRef]
- De Lucca Freitas, L.; Auschra, C.; Abetz, V.; Stadler, R. Hydrogen bonds in unpolar matrix—Comparison of complexation in polymeric and low molecular-weight systems. Colloid Polym. Sci. 1991, 269, 566–575. [Google Scholar] [CrossRef]
- Hilger, C.; Stadler, R. New multiphase thermoplastic elastomers by combination of covalent and association-chain structures. Die Makromol. Chem. 1990, 191, 1347–1361. [Google Scholar] [CrossRef]
- Geiger, T.; Stadler, R. Investigations of Cluster Formation in Thermoreversible Networks. In The Wiley Polymer Networks Group Review, Volume 1, Chemical and Physical Networks: Formation and Control of Properties; te Nijenhuis, K., Mijs, W.J., Eds.; John Wiley & Sons: New York, NY, USA, 1998; pp. 129–138. ISBN 978-0-471-97344-7. [Google Scholar]
- Bayer, U.; Stadler, R. Synthesis and properties of amphiphilic “dumbbell”-shaped grafted block copolymers, 1. Anionic synthesis via a polyfunctional initiator. Macromol. Chem. Phys. 1994, 195, 2709–2722. [Google Scholar] [CrossRef]
- Stadler, R. Transient networks by hydrogen bond interactions in polybutadiene-melts. Prog. Colloid Polym. Sci. 1987, 75, 140–145. [Google Scholar] [CrossRef]
- Shabbir, A.; Javakhishvili, I.; Cerveny, S.; Hvilsted, S.; Skov, A.L.; Hassager, O.; Alvarez, N.J. Linear viscoelastic and dielectric relaxation response of unentangled UPy-based supramolecular networks. Macromolecules 2016, 49, 3899–3910. [Google Scholar] [CrossRef]
- Wittenberg, E.; Meyer, A.; Eggers, S.; Abetz, V. Hydrogen bonding and thermoplastic elastomers—A nice couple with temperature-adjustable mechanical properties. Soft Matter 2018, 14, 2701–2711. [Google Scholar] [CrossRef] [PubMed]
- Pape, A.C.H.; Bastings, M.M.C.; Kieltyka, R.E.; Wyss, H.M.; Voets, I.K.; Meijer, E.W.; Dankers, P.Y.W. Mesoscale characterization of supramolecular transient networks using SAXS and rheology. Int. J. Mol. Sci. 2014, 15, 1096–1111. [Google Scholar] [CrossRef] [PubMed]
- Stadler, R.; de Lucca Freitas, L. Thermoplastic elastomers by hydrogen bonding 1. Rheological properties of modified polybutadiene. Colloid Polym. Sci. 1986, 264, 773–778. [Google Scholar] [CrossRef]
- Breiner, U.; Krappe, U.; Abetz, V.; Stadler, R. Cylindrical morphologies in asymmetric ABC triblock copolymers. Macromol. Chem. Phys. 1997, 198, 1051–1083. [Google Scholar] [CrossRef]
- Georgopanos, P.; Handge, U.A.; Abetz, C.; Abetz, V. Influence of block sequence and molecular weight on morphological, rheological and dielectric properties of weakly and strongly segregated styrene-isoprene triblock copolymers. Polymer 2016, 104, 279–295. [Google Scholar] [CrossRef]
- Haenelt, T.G.; Georgopanos, P.; Abetz, C.; Rangou, S.; Alisch, D.; Meyer, A.; Handge, U.A.; Abetz, V. Morphology and elasticity of polystyrene-block-polyisoprene diblock copolymers in the melt. Korea-Aust. Rheol. J. 2014, 26, 263–275. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics, 1st ed.; Clarendon Press: Oxford, UK, 1988; ISBN 9780198520337. [Google Scholar]
- Mark, J.E. Physical Properties of Polymers Handbook Edited, 2nd ed.; Springer Science + Business Media, LLC: New York, NY, USA, 2007; ISBN 9780387312354. [Google Scholar]
- Quach, A.; Simha, R. Pressure-volume-temperature properties and transitions of amorphous polymers; Polystyrene and poly (orthomethylstyrene). J. Appl. Phys. 1971, 42, 4592–4606. [Google Scholar] [CrossRef]
- Krishnamoorti, R.; Giannelis, E.P. Rheology of end-tethered polymer layered silicate nanocomposites. Macromolecules 1997, 30, 4097–4102. [Google Scholar] [CrossRef]
- Mezger, T.G. Das Rheologie Handbuch, 5th ed.; Vincentz Network GmbH & Co. KG: Hannover, Germany, 2016; ISBN 9783866306332. [Google Scholar]
- Rosedale, J.H.; Bates, F.S. Rheology of ordered and disordered symmetric poly(ethylenepropylene)-poly(ethylethylene) diblock copolymers. Macromolecules 1990, 23, 2329–2338. [Google Scholar] [CrossRef]
- Eckstein, A.; Suhm, J.; Friedrich, C.; Maier, R.-D.; Sassmannshausen, J.; Bochmann, M.; Mülhaupt, R. Determination of plateau moduli and entanglement molecular weights of isotactic, syndiotactic, and atactic polypropylenes synthesized with metallocene catalysts. Macromolecules 1998, 31, 1335–1340. [Google Scholar] [CrossRef]
- Larson, R.G.; Winey, K.I.; Patel, S.S.; Watanabe, H.; Bruinsma, R. The rheology of layered liquids: Lamellar block copolymers and smectic liquid crystals. Rheol. Acta 1993, 32, 245–253. [Google Scholar] [CrossRef]
- Witten, T.A.; Leibler, L.; Pincus, P.A. Stress relaxation in the lamellar copolymer mesophase. Macromolecules 1990, 23, 824–829. [Google Scholar] [CrossRef]
- Shabbir, A.; Goldansaz, H.; Hassager, O.; van Ruymbeke, E.; Alvarez, N.J. Effect of hydrogen bonding on linear and nonlinear rheology of entangled polymer melts. Macromolecules 2015, 48, 5988–5996. [Google Scholar] [CrossRef]
- Arendt, B.H.; Krishnamoorti, R.; Kannan, R.M.; Seitz, K.; Kornfield, J.A.; Roovers, J. Dynamics of disordered diblocks of polyisoprene and polyvinylethylene. Macromolecules 1997, 30, 1138–1145. [Google Scholar] [CrossRef]
- Hirose, Y.; Urakawa, O.; Adachi, K. Dynamics in disordered block copolymers and miscible blends composed of poly(vinyl ethylene) and polyisoprene. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4084–4094. [Google Scholar] [CrossRef]
- Roberts, M.C.; Hanson, M.C.; Massey, A.P.; Karren, E.A.; Kiser, P.F. Dynamically restructuring hydrogel networks formed with reversible covalent crosslinks. Adv. Mater. 2007, 19, 2503–2507. [Google Scholar] [CrossRef]
- Handge, U.A.; Haenelt, T.G.; Georgopanos, P.; Abetz, C.; Rangou, S.; Alisch, D.; Vainio, U.; Meyer, A.; Abetz, V. Analysis of stability and viscoelastic properties of melts of polystyrene-block-polyisoprene diblock copolymers in oscillatory shear and creep-recovery experiments. AIP Conf. Proc. 2015, 1664, 170001. [Google Scholar] [CrossRef]
- Cangelosi, F.; Shaw, M.T. A review of hydrogen bonding in solid polymers: Structural relationships, analysis, and importance. Polym. Plast. Technol. Eng. 1983, 21, 13–98. [Google Scholar] [CrossRef]
- Hesse, M.; Meier, H.; Zeeh, B. Spektroskopische Methoden in der Organischen Chemie, 6th ed.; Thieme Verlag: Stuttgart, Germany, 2002. [Google Scholar]
- Painter, P.C.; Koenig, J.L. A normal vibrational analysis of syndiotactic polystyrene. J. Polym. Sci. Polym. Phys. Ed. 1977, 15, 1885–1903. [Google Scholar] [CrossRef]
- Velada, J.L.; Cesteros, C.; Madoz, A.; Katime, I. A study of the thermal degradation of poly(mono-n-alkyl itaconates). Macromol. Chem. Phys. 1995, 196, 3171–3185. [Google Scholar] [CrossRef]
- Zou, H.; Yi, C.; Wang, L.; Liu, H.; Xu, W. Thermal degradation of poly(lactic acid) measured by thermogravimetry coupled to Fourier transform infrared spectroscopy. J. Therm. Anal. Calorim. 2009, 97, 929–935. [Google Scholar] [CrossRef]
- Kameno, N.; Yamada, S.; Amimoto, T.; Amimoto, K.; Ikeda, H.; Koga, N. Thermal degradation of poly(lactic acid) oligomer: Reaction mechanism and multistep kinetic behavior. Polym. Degrad. Stab. 2016, 134, 284–295. [Google Scholar] [CrossRef]
- McNeill, I.C.; Leiper, H.A. Degradation studies of some polyesters and polycarbonates—1. Polylactide: General features of the degradation under programmed heating conditions. Polym. Degrad. Stab. 1985, 11, 267–285. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Okamoto, T.; Ishida, A.; Sobue, H. Polymerization of butyl esters of methacrylic acid and hydrolysis of the polymers. J. Polym. Sci. Part A Polym. Chem. 1964, 2, 1105–1114. [Google Scholar] [CrossRef]
- Shaw, J.N.; Marshall, M.C. Infrared spectroscopic studies of polystyrene emulsion polymers: Effect of oxidation during polymerization. J. Polym. Sci. Part A Polym. Chem. 1968, 6, 449–458. [Google Scholar] [CrossRef]
- Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct addition of amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | FGint | Đ | Df (%) | Tg (°C) a | Me (kg/mol) |
|---|---|---|---|---|---|
| I5S90I562 | - | 1.3 | - | 86.0 | 21.1 |
| I5S90I562 | OH | 1.2 | 58 | 93.2 | 13.1 |
| I5S90I562 | OH | 1.3 | 70 | c | 12.5 |
| I5S90I562 | OH | 1.3 | 94 | 101.5 | 11.4 |
| I5S90I562 | DETA | b | 32 | 97.8 | 8.9 |
| I5S90I562 | TETA | 1.4 | 21 | 94.8 | 11.9 |
| I5S90I562 | DAP/CDI | 1.3 | 10/7 | 100.7 | 9.2 |
| I5S90I562 | SA | 2.0 | 45 | 97.3 | 12.2 |
| I1.5S96.1I2.482 | - | 1.2 | - | 95.0 | 17.3 |
| I1.5S96.1I2.482 | OH | 1.2 | 75 | 97.9 | c |
| I1.5S96.1I2.482 | DETA | 1.3 | 48 | 98.9 | 12.1 |
| I1.5S96.1I2.482 | SA | 1.2 | 33 | 100 | 11.1 |
| I3S94I3117 | - | 1.2 | - | 83.7 | 15.3 |
| I3S94I3117 | OH | 1.5 | 50 | 101.1 | 11.0 |
| I3S94I3117 | DETA | 2.3 | 20 | 98.2 | 13.1 |
| I3S94I3117 | SA | 1.7 | 30 | 99.8 | 16.2 |
| S91I967 | - | - | 98.2 | 16.2 | |
| S91I967 | OH | 1.2 | 30 | 99.1 | 16.0 |
| S91I967 | SA | 1.4 | 11 | 100.8 | 15.1 |
| S91I967 | OH | 1.1 | 92 | 97.8 | 9.0 |
| S91I967 | SA | 1.5 | 45 | 99.0 | 11.3 |
| T (°C) | 1st (min) | 2nd (min) | 3rd (min) | 4th (min) |
|---|---|---|---|---|
| 30 | 0 | |||
| 135 | 0 | 7 | 8 | |
| 30 | 0 | 120 | 120 | |
| 120 | 0 | 10 | 30 | 30 |
| 140 | 0 | 10 | 30 | 30 |
| 160 | 0 | 10 | 30 | 30 |
| 180 | 0 | 10 | 30 | 30 |
| 120 | 0 | 10 | 30 | 30 |
| 30 | 0 | 120 | 120 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmstorf, E.; Abetz, V. Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 2: Dynamic Mechanical Analysis. Materials 2018, 11, 1688. https://doi.org/10.3390/ma11091688
Rahmstorf E, Abetz V. Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 2: Dynamic Mechanical Analysis. Materials. 2018; 11(9):1688. https://doi.org/10.3390/ma11091688
Chicago/Turabian StyleRahmstorf, Elaine, and Volker Abetz. 2018. "Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 2: Dynamic Mechanical Analysis" Materials 11, no. 9: 1688. https://doi.org/10.3390/ma11091688
APA StyleRahmstorf, E., & Abetz, V. (2018). Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 2: Dynamic Mechanical Analysis. Materials, 11(9), 1688. https://doi.org/10.3390/ma11091688