Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 1: Synthesis and Characterization



Abstract
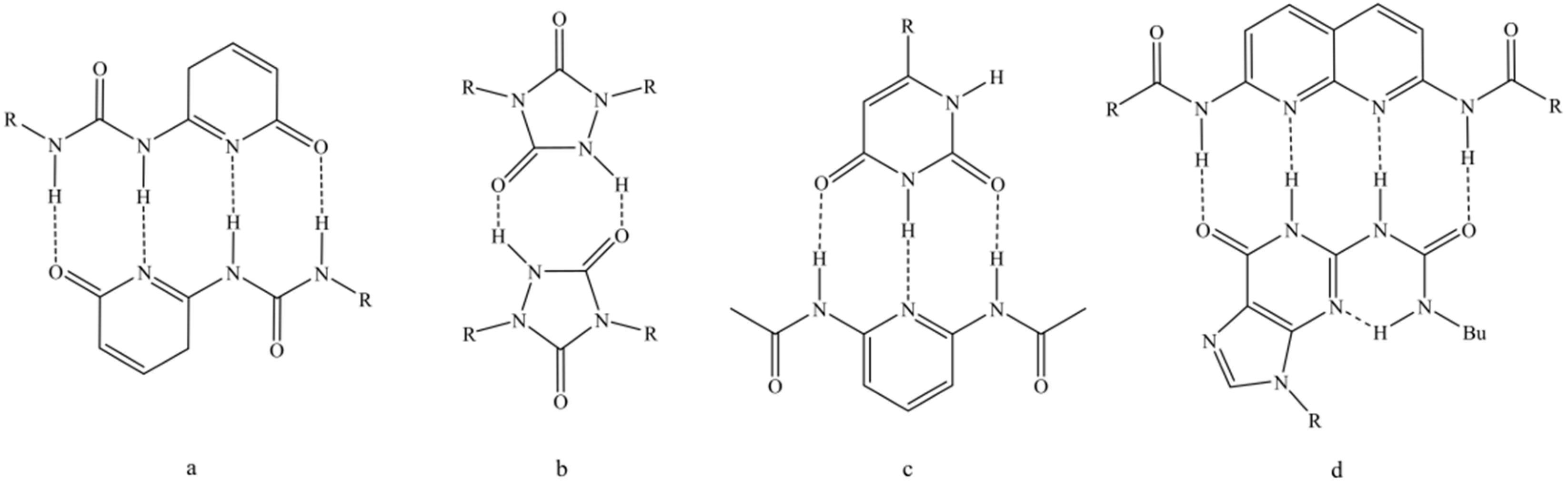
1. Introduction
2. Materials and Methods
2.1. Materials
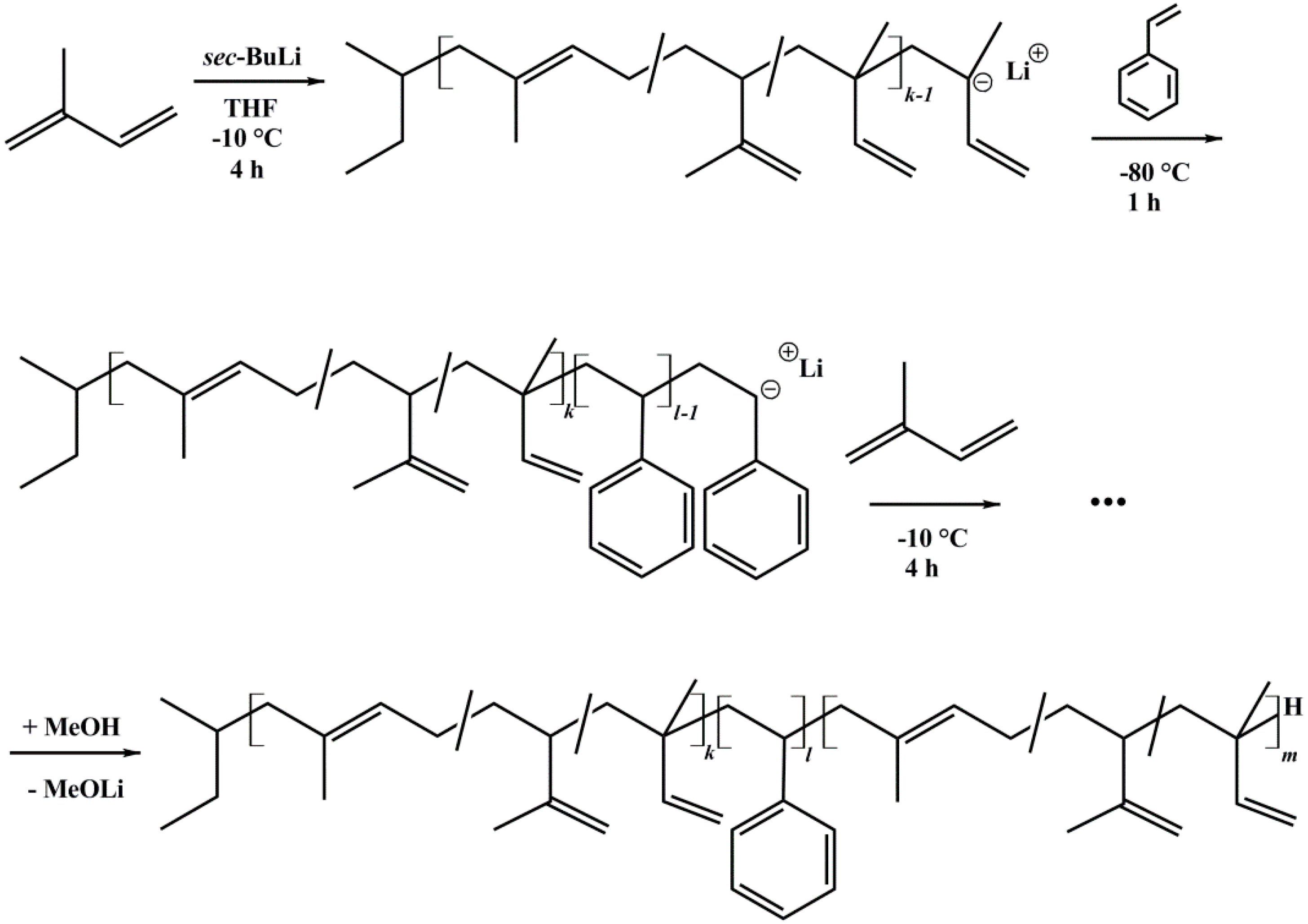
2.2. Anionic Polymerization
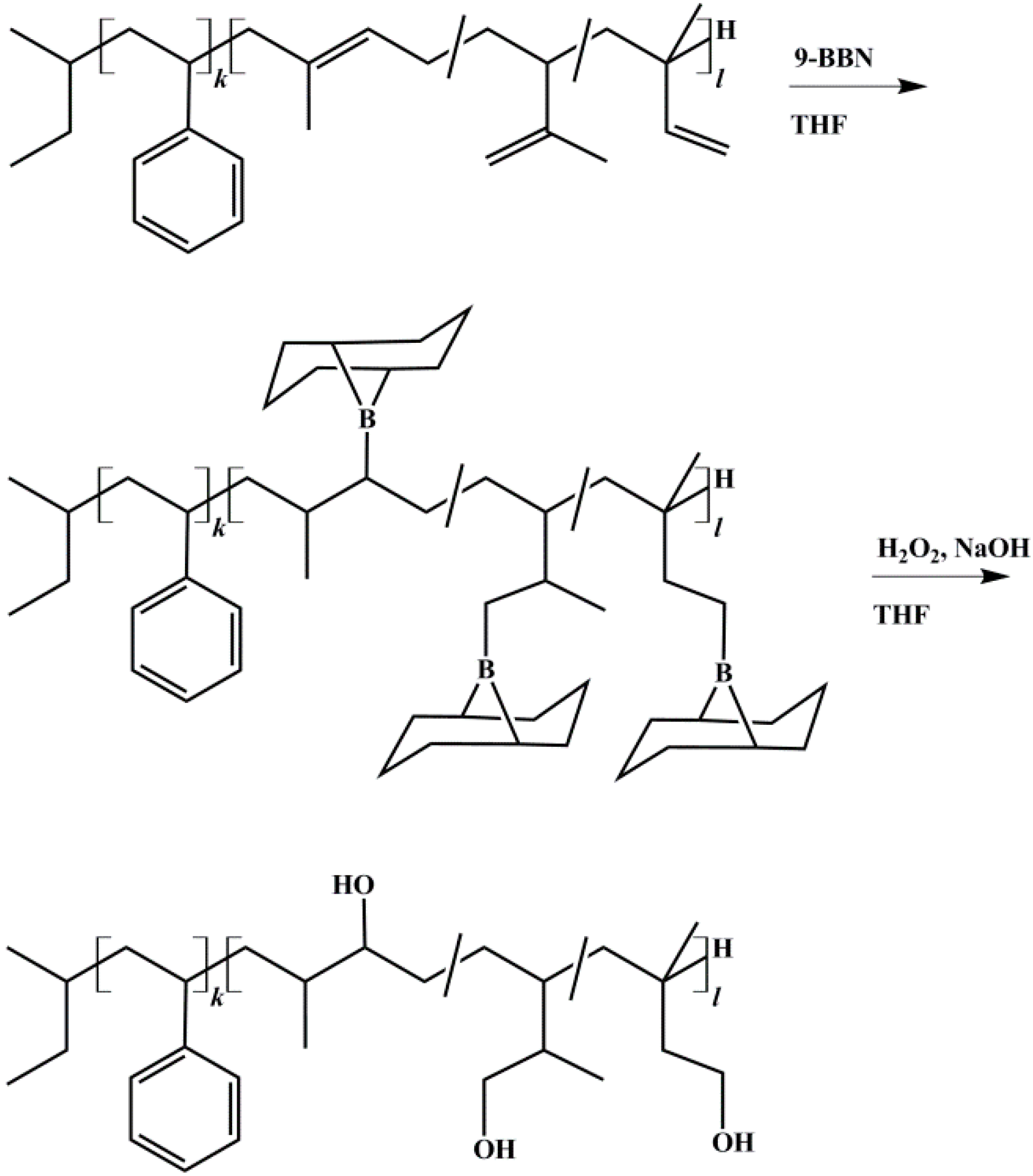
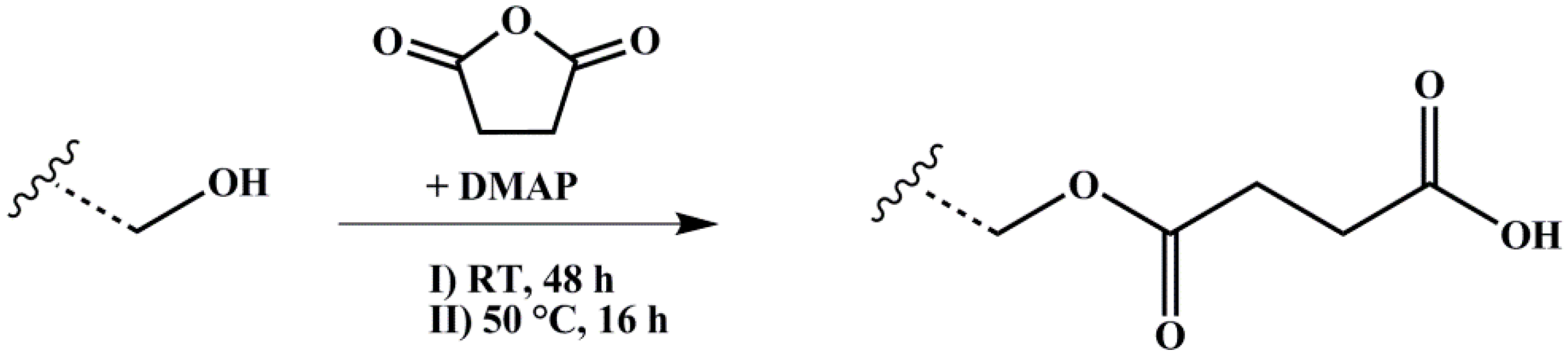
2.3. End-Block Modification of Block Copolymers
2.3.1. Hydroxylation
2.3.2. Carboxylation
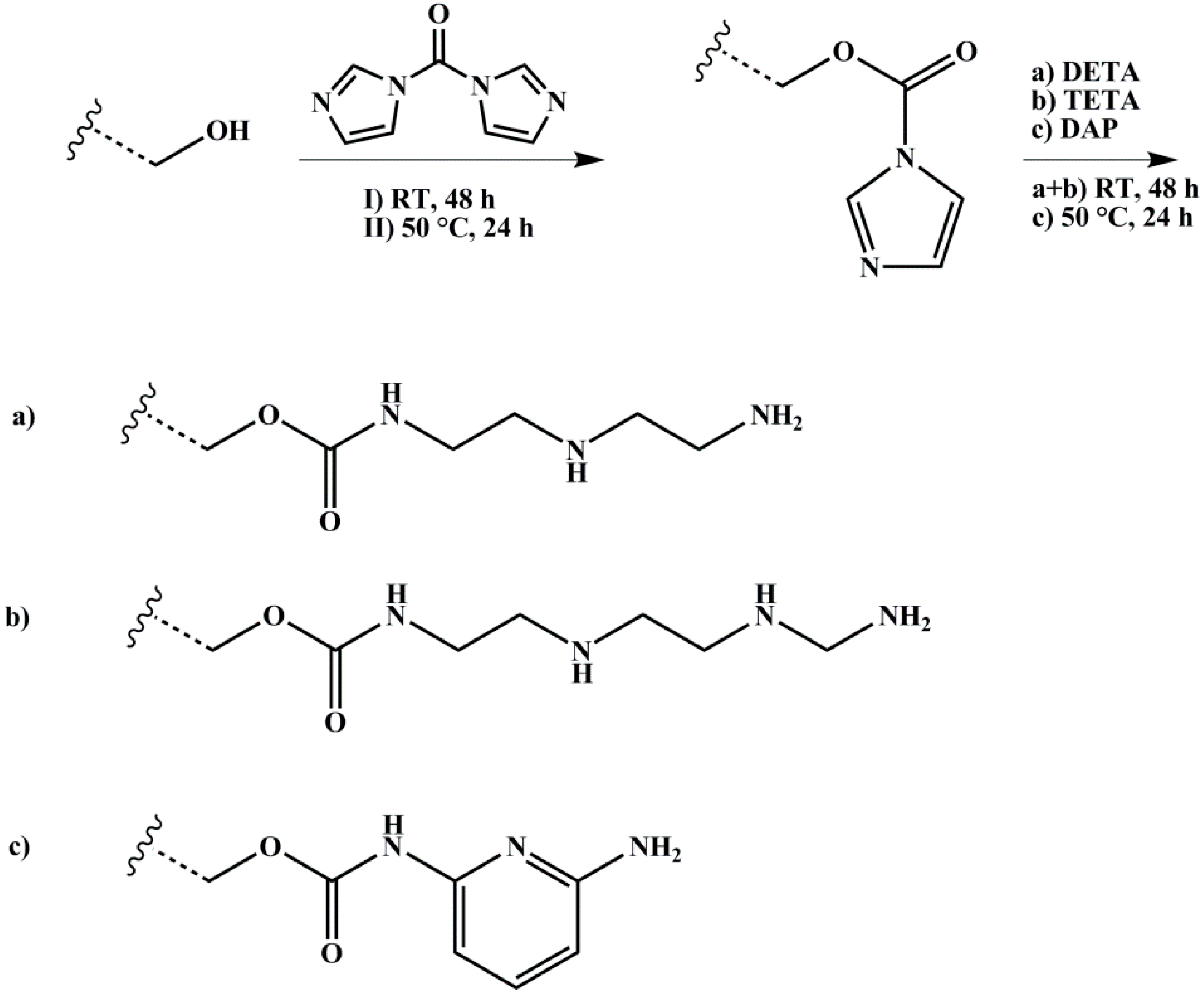
2.3.3. Amination
2.4. Characterization
2.4.1. Size Exclusion Chromatography
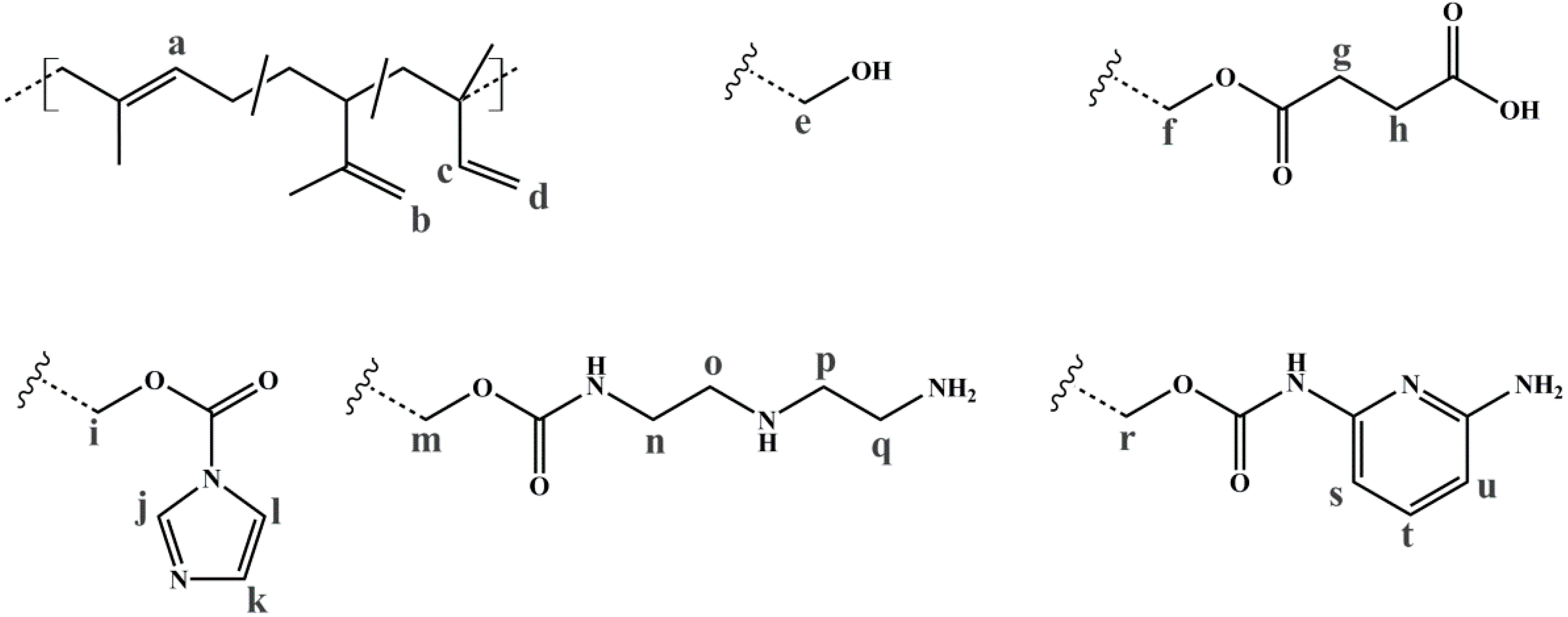
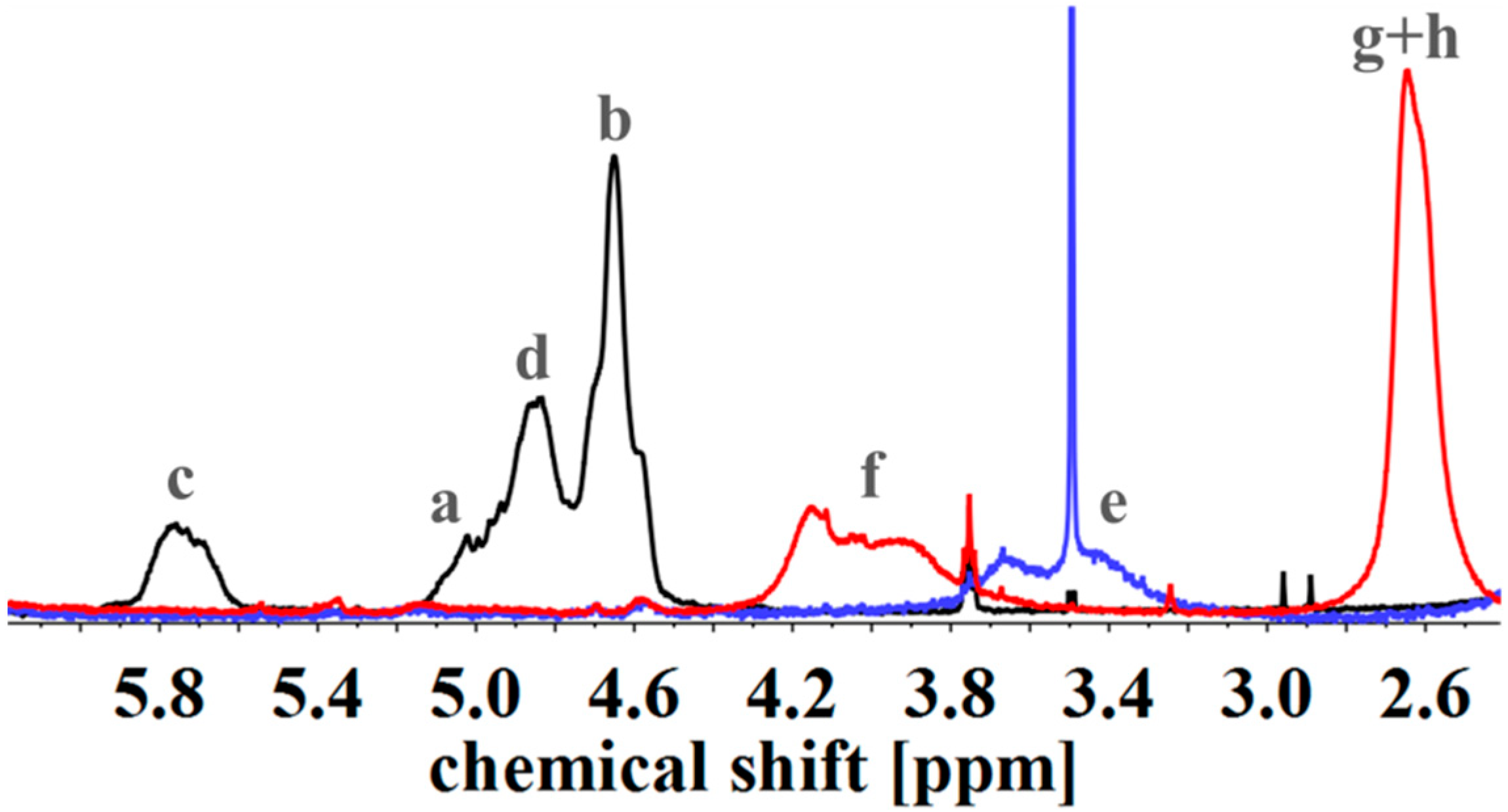
2.4.2. Nuclear Magnetic Resonance Spectroscopy
2.4.3. Fourier Transform Infrared Spectroscopy
2.4.4. Differential Scanning Calorimetry
2.4.5. Thermogravimetric Analysis
2.4.6. Small Angle X-ray Scattering
3. Results and Discussion
3.1. Synthesis of Modified Block Copolymers and the Calculation of Degree of Functionalization Df
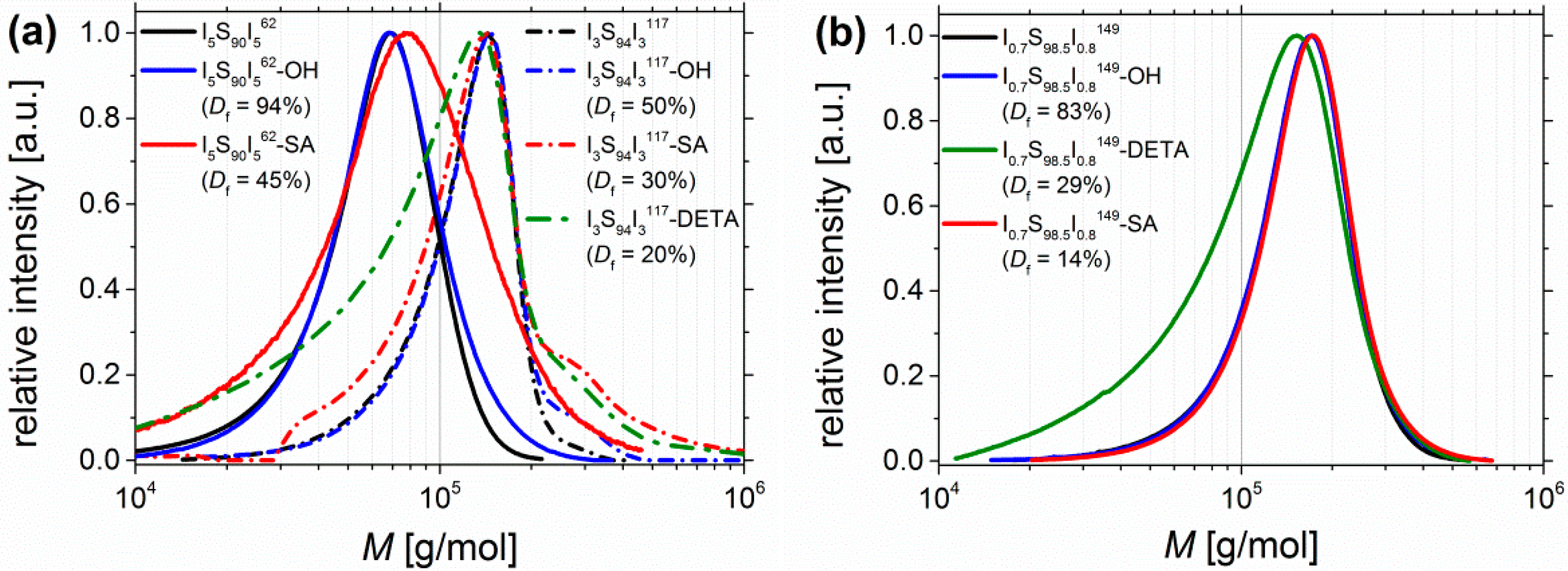
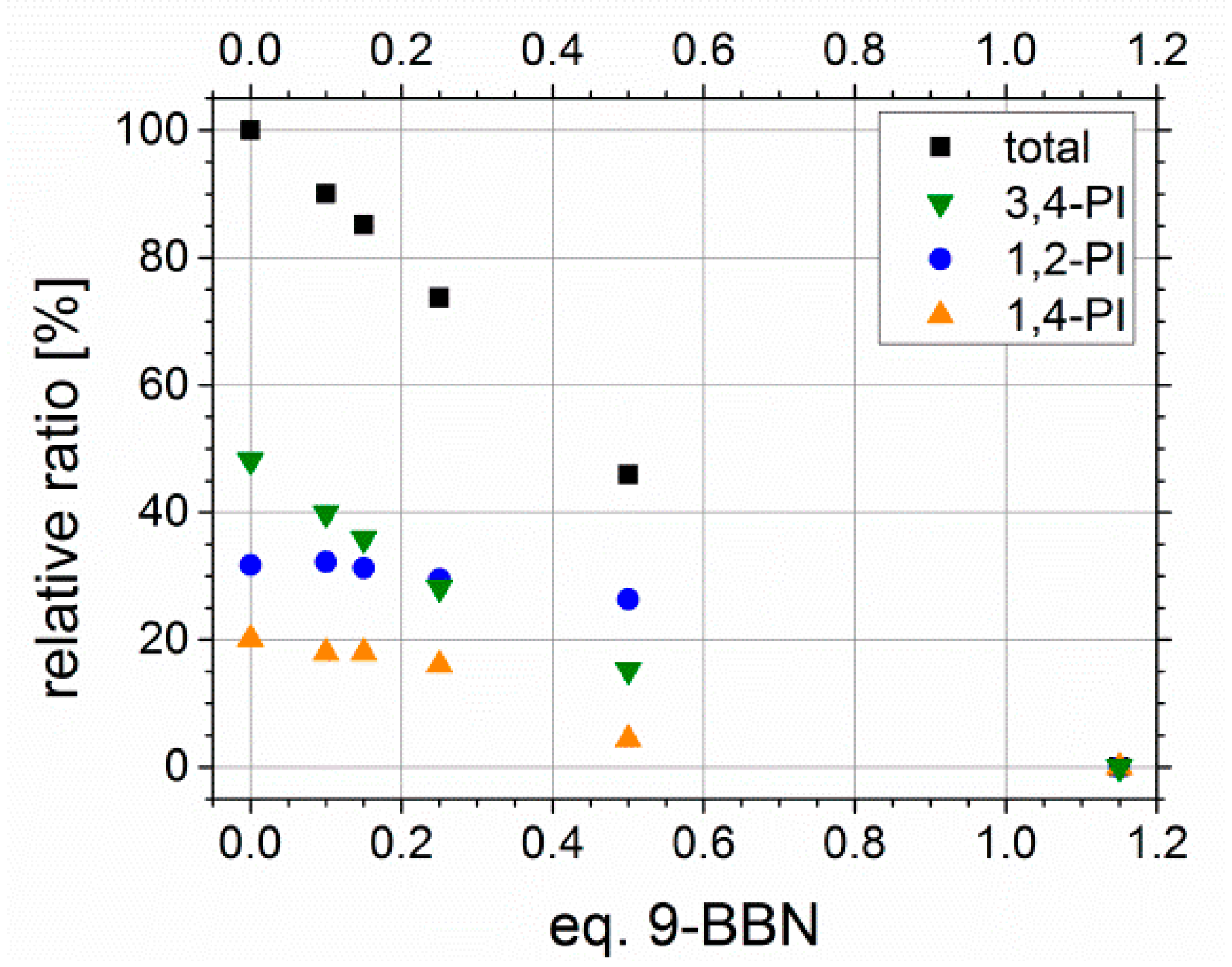
Adjustability of Degree of Functionalization Df
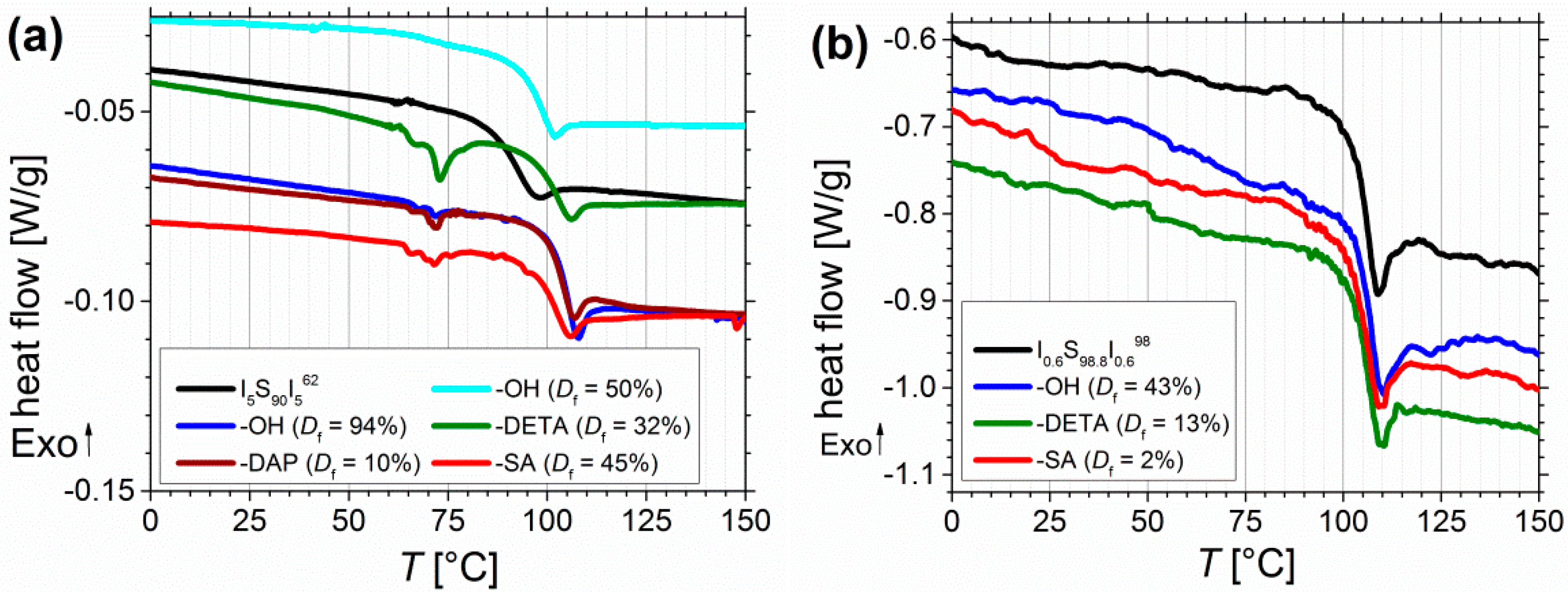
3.2. Thermal Characterization
3.3. Fourier Transform Infrared Spectroscopy
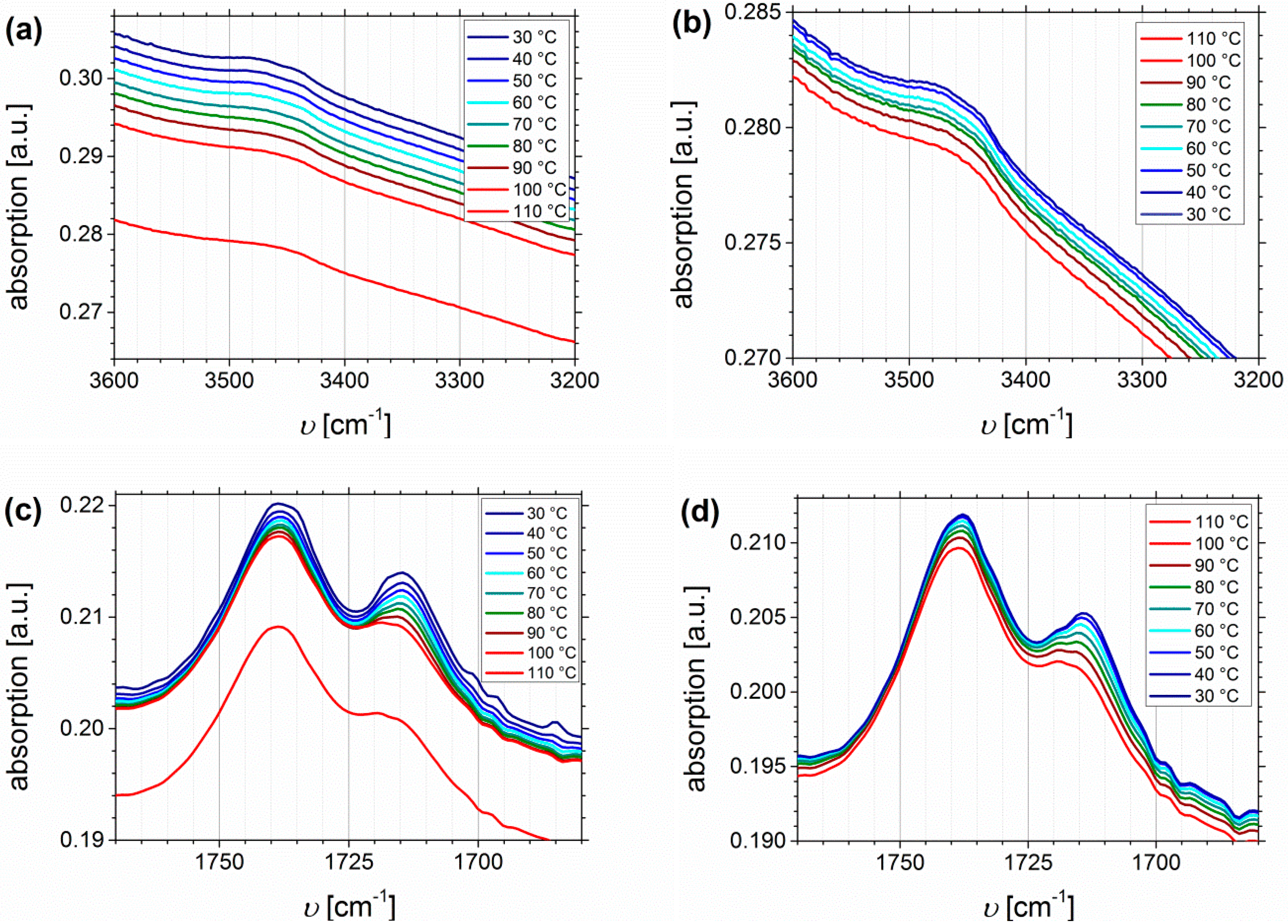
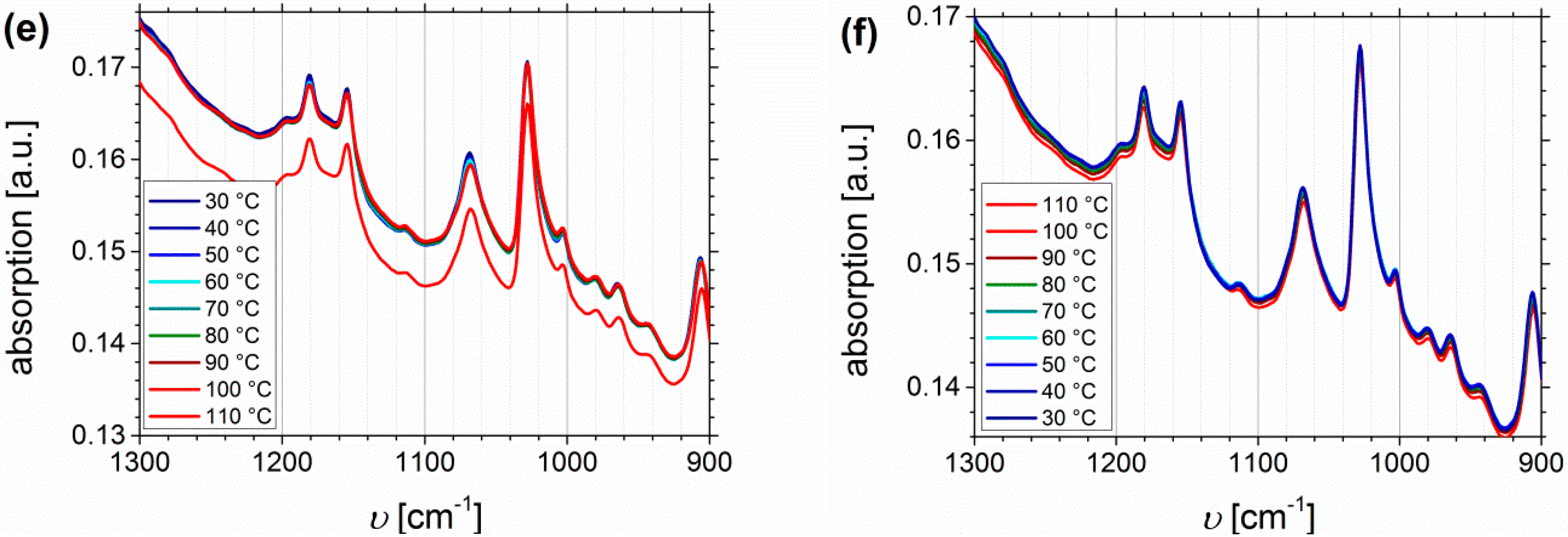
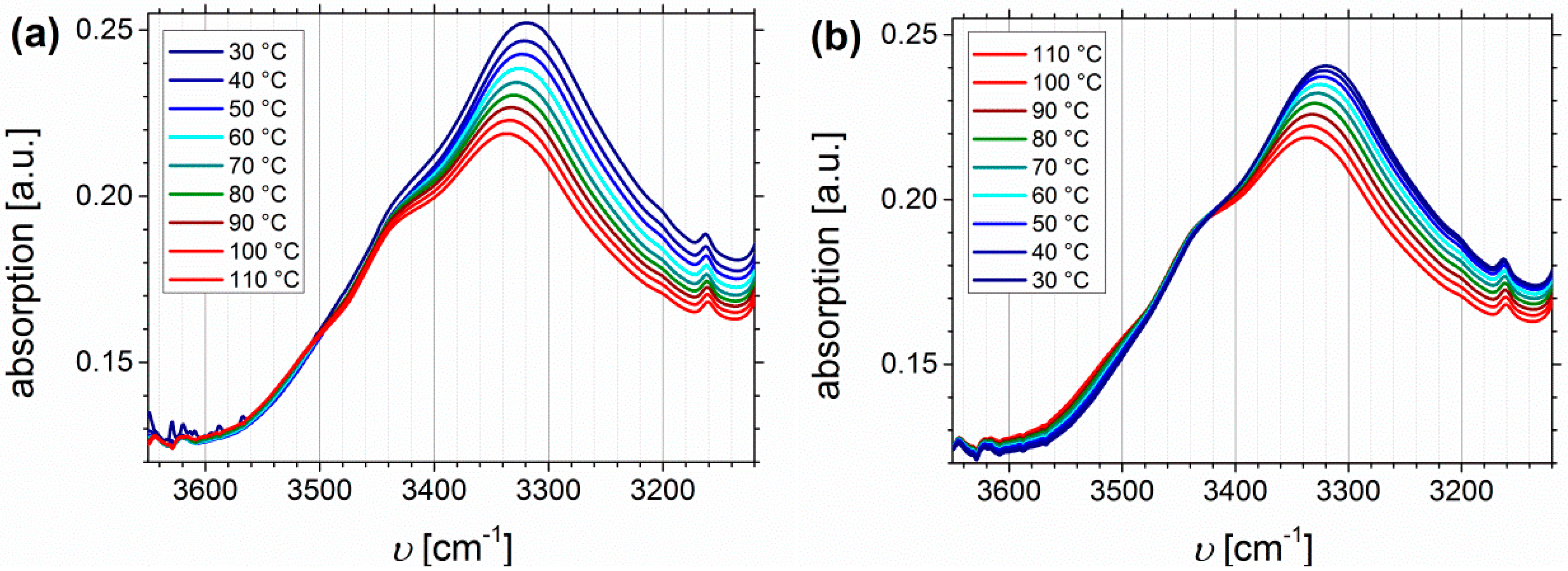
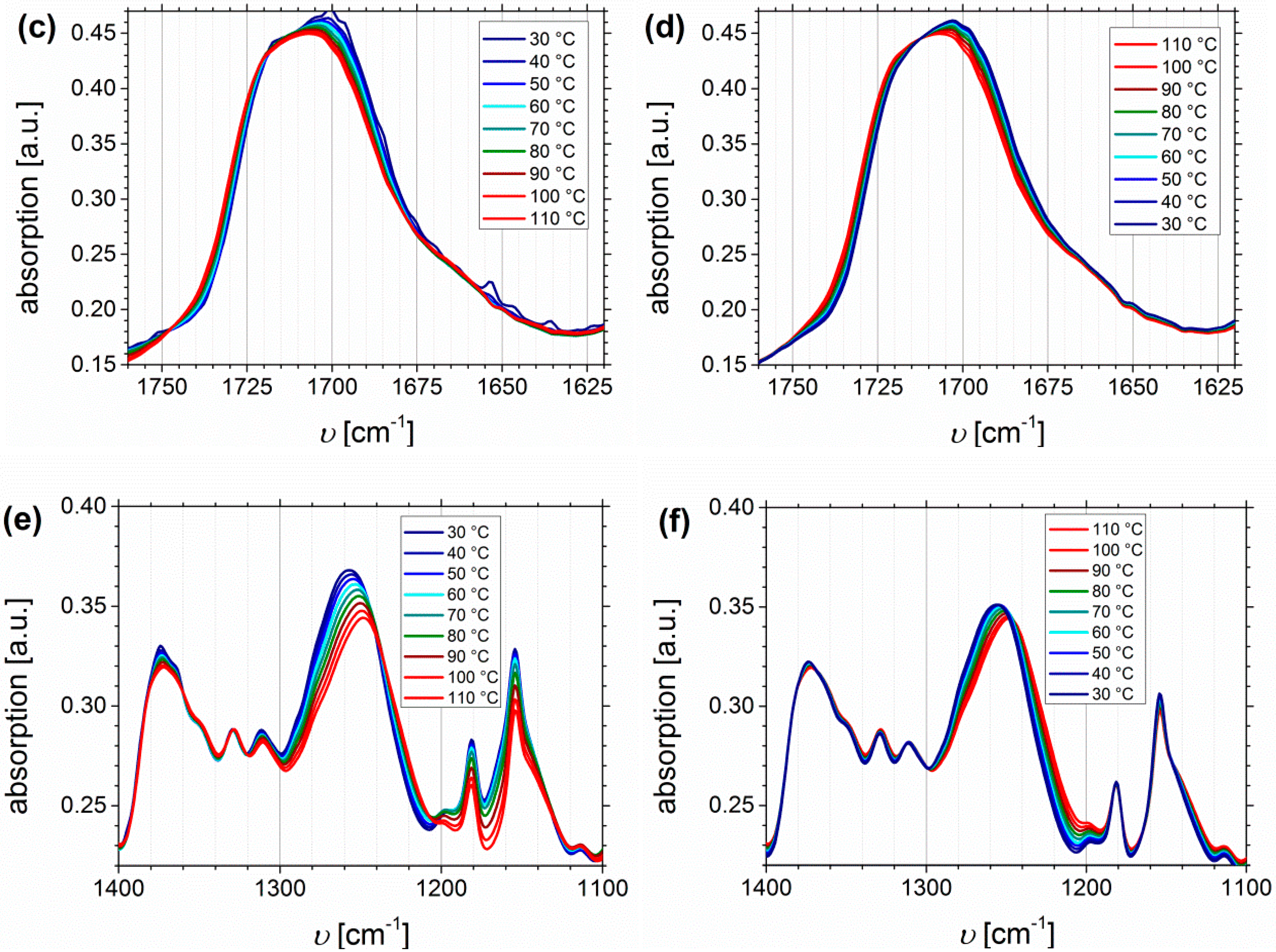
Temperature Dependent FTIR Spectroscopy of I1.5S96.1I2.482-SA and I5S90I562-DETA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Binder, W. Hydrogen Bonded Polymers; Binder, W., Ed.; Springer: Berlin, Germany, 2007; Volume 207, ISBN 9783540685876. [Google Scholar]
- Brunsveld, L.; Folmer, B.J.B.; Meijer, E.W.; Sijbesma, R.P. Supramolecular Polymers. Chem. Rev. 2001, 101, 4071–4098. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, S.; Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev. 2012, 41, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.; Lortie, F.; Bernard, J. Routes to Hydrogen Bonding Chain-End Functionalized Polymers. Macromol. Rapid Commun. 2012, 33, 2062–2091. [Google Scholar] [CrossRef] [PubMed]
- Hilger, C.; Stadler, R. New multiphase thermoplastic elastomers by combination of covalent and association-chain structures. Die Makromol. Chem. 1990, 191, 1347–1361. [Google Scholar] [CrossRef]
- Lange, R.F.M.; Van Gurp, M.; Meijer, E.W. Hydrogen-bonded supramolecular polymer networks. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 3657–3670. [Google Scholar] [CrossRef]
- Gloe, K.; Heßke, H.; Lindoy, L. Supramolekulare Chemie: Vom Einzelmolekül zur komplexen Funktionseinheit. Wissenschaftliche Z. Tech. Univ. Dresden 2007, 56, 32–38. [Google Scholar]
- Yang, S.K.; Zimmerman, S.C. Hydrogen bonding modules for use in supramolecular polymers. Isr. J. Chem. 2013, 53, 511–520. [Google Scholar] [CrossRef]
- Sijbesma, R.P.; Meijer, E.W. Quadruple hydrogen bonded systems. Chem. Commun. 2003, 0, 5–16. [Google Scholar] [CrossRef]
- Sijbesma, R.P.; Beijer, F.H.; Brunsveld, L.; Folmer, B.J.B.; Hirschberg, J.H.K.K.; Lange, R.F.M.; Lowe, J.K.L.; Meijer, E.W. Reversible Polymers Formed from Self-Complementary Monomers Using Quadruple Hydrogen Bonding. Science 1997, 278, 1601–1604. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, D.J.M.; Spiering, A.J.H.; Peters, G.W.M.; Te Nijenhuis, K.; Sijbesma, R.P. Unidirectional dimerization and stacking of ureidopyrimidinone end groups in polycaprolactone supramolecular polymers. Macromolecules 2007, 40, 8464–8475. [Google Scholar] [CrossRef]
- Dankers, P.Y.W.; Zhang, Z.; Wisse, E.; Grijpma, D.W.; Sijbesma, R.P.; Feijen, J.; Meijer, E.W. Oligo(trimethylene carbonate)-Based Supramolecular Biomaterials. Macromolecules 2006, 39, 8763–8771. [Google Scholar] [CrossRef]
- Hilger, C.; Stadler, R. New multiphase architecture from statistical copolymers by cooperative hydrogen bond formation. Macromolecules 1990, 23, 2095–2097. [Google Scholar] [CrossRef]
- Stadler, R.; de Lucca Freitas, L. Thermoplastic elastomers by hydrogen bonding. Polym. Bull. 1986, 15, 173–179. [Google Scholar] [CrossRef]
- Seidel, U.; Schollmeyer, D.; Stadler, R. Two-dimensional assembly of functionalized 4-aryl-1-alkenyl-3,5-dioxo-1,2,4-triazolidines. Supramol. Chem. 1995, 2, 45–50. [Google Scholar] [CrossRef]
- De Lucca Freitas, L.; Auschra, C.; Abetz, V.; Stadler, R. Hydrogen bonds in unpolar matrix—Comparison of complexation in polymeric and low molecular-weight systems. Colloid Polym. Sci. 1991, 269, 566–575. [Google Scholar] [CrossRef]
- Fouquey, C.; Lehn, J.; Levelut, A. Molecular recognition directed self-assembly of supramolecular liquid crystalline polymers from complementary chiral components. Adv. Mater. 1990, 2, 254–257. [Google Scholar] [CrossRef]
- Park, T.; Zimmerman, S.C.; Nakashima, S. A highly stable quadruply hydrogen-bonded heterocomplex useful for supramolecular polymer blends. J. Am. Chem. Soc. 2005, 127, 6520–6521. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Urban, M.W. Self-healing polymeric materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef] [PubMed]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [PubMed]
- De Luzuriaga, A.R.; Martin, R.; Markaide, N.; Rekondo, A.; Cabañero, G.; Rodríguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horiz. 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Balkenende, D.W.R.; Olson, R.A.; Balog, S.; Weder, C.; Montero de Espinosa, L. Epoxy Resin-Inspired Reconfigurable Supramolecular Networks. Macromolecules 2016, 49, 7877–7885. [Google Scholar] [CrossRef]
- Denissen, W.; Droesbeke, M.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Chemical control of the viscoelastic properties of vinylogous urethane vitrimers. Nat. Commun. 2017, 8, 14857. [Google Scholar] [CrossRef] [PubMed]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Engle, L.P.; Wagener, K.B. A Review of Thermally Controlled Covalent Bond Formation in Polymer Chemistry. J. Macromol. Sci. Part C Polym. Rev. 1993, 33, 239–257. [Google Scholar] [CrossRef]
- Chujo, Y.; Sada, K.; Saegusa, T. Reversible gelation of polyoxazoline by means of Diels-Alder reaction. Macromolecules 1990, 23, 2636–2641. [Google Scholar] [CrossRef]
- Goussé, C.; Gandini, A.; Hodge, P. Application of the Diels−Alder Reaction to Polymers Bearing Furan Moieties. 2. Diels−Alder and Retro-Diels−Alder Reactions Involving Furan Rings in Some Styrene Copolymers. Macromolecules 1998, 31, 314–321. [Google Scholar] [CrossRef]
- Bayer, U.; Stadler, R. Synthesis and properties of amphiphilic “dumbbell”-shaped grafted block copolymers, 1. Anionic synthesis via a polyfunctional initiator. Macromol. Chem. Phys. 1994, 195, 2709–2722. [Google Scholar] [CrossRef]
- Mao, G.; Wang, J.; Clingman, S.R.; Ober, C.K.; Chen, J.T.; Thomas, E.L. Molecular Design, Synthesis, and Characterization of Liquid Crystal−Coil Diblock Copolymers with Azobenzene Side Groups. Macromolecules 1997, 30, 2556–2567. [Google Scholar] [CrossRef]
- Georgopanos, P.; Filiz, V.; Handge, U.A.; Abetz, V. Chemical Modification, Thermal Characterization and Dielectric Spectroscopy of Polystyrene-block-Polyisoprene Diblock Copolymers. Macromol. Chem. Phys. 2016, 217, 1–12. [Google Scholar] [CrossRef]
- Schmidtke, C.; Pöselt, E.; Ostermann, J.; Pietsch, A.; Kloust, H.; Tran, H.; Schotten, T.; Bastús, N.G.; Eggers, R.; Weller, H. Amphiphilic, cross-linkable diblock copolymers for multifunctionalized nanoparticles as biological probes. Nanoscale 2013, 5, 7433–7444. [Google Scholar] [CrossRef] [PubMed]
- Böhme, F.; Kunert, C.; Komber, H.; Voigt, D.; Friedel, P.; Khodja, M.; Wilde, H. Polymeric and macrocyclic ureas based on meta-substituted aromatic diamines. Macromolecules 2002, 35, 4233–4237. [Google Scholar] [CrossRef]
- Uhrig, D.; Mays, J.W. Experimental techniques in high-vacuum anionic polymerization. J. Polym. Sci. Part. A Polym. Chem. 2005, 43, 6179–6222. [Google Scholar] [CrossRef]
- Rembaum, A.; Siao, S.-P.; Indictor, N. Decomposition of ethyllithium in tetrahydrofuran. J. Polym. Sci. 1962, 56, S17–S19. [Google Scholar] [CrossRef]
- Auschra, C.; Stadler, R. Synthesis of block copolymers with poly(methyl methacrylate): P(B-b-MMA), P(EB-b-MMA), P(S-b-B-b-MMA) and P(S-b-EB-b-MMA). Polym. Bull. 1993, 30, 257–264. [Google Scholar] [CrossRef]
- Breiner, U.; Krappe, U.; Abetz, V.; Stadler, R. Cylindrical morphologies in asymmetric ABC triblock copolymers. Macromol. Chem. Phys. 1997, 198, 1051–1083. [Google Scholar] [CrossRef]
- Lee, K.M.; Han, C.D. Order-Disorder Transition Induced by the Hydroxylation of Homogeneous Polystyrene-block- polyisoprene Copolymer. Macromolecules 2002, 35, 760–769. [Google Scholar] [CrossRef]
- Dhillon, R.S. Hydroboration and Organic Synthesis; Springer: Berlin, Germany, 2007; ISBN 978-3-540-49075-3. [Google Scholar]
- Chung, T.C.; Raate, M.; Berluche, E.; Schulz, D.N. Synthesis of Functional Hydrocarbon Polymers with Well-Defined Molecular Structures. Macromolecules 1988, 21, 1903–1907. [Google Scholar] [CrossRef]
- Wang, K.K.; Brown, H.C. Hydroboration Kinetics. 2. Improved Procedure for Following the Kinetics for the Reaction of Alkenes with 9-Borabicyclo[3.3.1]nonane. Further Evidence for the Dissociation Mechanism. J. Org. Chem. 1980, 45, 5303–5306. [Google Scholar] [CrossRef]
- Peng, C.-C.; Abetz, V. A Simple Pathway toward Quantitative Modification of Polybutadiene: A New Approach to Thermoreversible Cross-Linking Rubber Comprising Supramolecular Hydrogen-Bonding Networks. Macromolecules 2005, 38, 5575–5580. [Google Scholar] [CrossRef]
- D’Addona, D.; Bochet, C.G. Preparation of carbamates from amines and alcohols under mild conditions. Tetrahedron Lett. 2001, 42, 5227–5229. [Google Scholar] [CrossRef]
- Worsfold, D.J.; Bywater, S. Anionic polymerization of isoprene. Can. J. Chem. 1964, 42, 2884–2892. [Google Scholar] [CrossRef]
- Bywater, S.; Worsfold, D.J. Anionic polymerization of isoprene. Ion and ion-pair contributions to polymerization in tetrahydrofuran. Can. J. Chem. 1967, 45, 1821–1824. [Google Scholar] [CrossRef]
- Sato, H.; Tanaka, Y. 1H-NMR study of polyisoprenes. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 3551–3558. [Google Scholar] [CrossRef]
- Šimák, V.P.; Fahrbach, G. Spektroskopische Untersuchung der Mikrostruktur von Polyisopren, Polybutadien und Isopren-Butadien-Copolymeren. Angew. Makromol. Chem. 1971, 16, 309–324. [Google Scholar] [CrossRef]
- Jang, S.G.; Kramer, E.J.; Hawker, C.J. Controlled Supramolecular Assembly of Micelle-Like Gold Nanoparticles in PS-b-P2VP Diblock Copolymers via Hydrogen Bonding. J. Am. Chem. Soc. 2011, 133, 16986–16996. [Google Scholar] [CrossRef] [PubMed]
- Peng, E.; Ding, J.; Xue, J.M. Succinic Anhydrides Functionalized Alkenoic Ligands: A Facile Route to Synthesize Water Dispersible Nanocrystals. J. Mater. Chem. 2012, 22, 13832–13840. [Google Scholar] [CrossRef]
- He, Y.; Zhu, B.; Inoue, Y. Hydrogen bonds in polymer blends. Prog. Polym. Sci. 2004, 29, 1021–1051. [Google Scholar] [CrossRef]
- Pochan, J.M.; Beatty, C.L.; Pochan, D.F. Different approach for the correlation of the Tg of mixed amorphous systems. Polymer 1979, 20, 879–886. [Google Scholar] [CrossRef]
- Hiemenz, P.C.; Lodge, T.P. Polymer Chemistry, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; ISBN 9781420018271. [Google Scholar]
- Fox, T.G.; Flory, P.J. The glass temperature and related properties of polystyrene. Influence of molecular weight. J. Polym. Sci. 1954, 14, 315–319. [Google Scholar] [CrossRef]
- Schawe, J.; Riesen, R.; Widmann, J.; Schubnell, M.; Jörimann, U. DSC-Kurven interpretieren Teil 1: Dynamische Messungen. UserCom 2000, 1, 1–24. [Google Scholar]
- Widmaier, J.M.; Meyer, G.C. Glass Transition Temperature of Anionic Polyisoprene. Macromolecules 1981, 14, 450–452. [Google Scholar] [CrossRef]
- Avgeropoulos, A.; Paraskeva, S.; Hadjichristidis, N.; Thomas, E.L. Synthesis and microphase separation of linear triblock terpolymers of polystyrene, high 1,4-polybutadiene, and high 3,4-polyisoprene. Macromolecules 2002, 35, 4030–4035. [Google Scholar] [CrossRef]
- Stadler, R.; Burgert, J. Influence of hydrogen bonding on the properties of elastomers and elastomeric blends. Die Makromol. Chem. 1986, 187, 1681–1690. [Google Scholar] [CrossRef]
- Mori, K.; Hasegawa, H.; Hashimoto, T. Small-Angle X-Ray Scattering from Bulk Block Polymers in Disordered State. Estimation of χ-Values from Accidental Thermal Fluctuations. Polym. J. 1985, 17, 799–806. [Google Scholar] [CrossRef]
- Matsen, M.W. Equilibrium behavior of asymmetric ABA triblock copolymer melts. J. Chem. Phys. 2000, 113, 5539–5544. [Google Scholar] [CrossRef]
- Cangelosi, F.; Shaw, M.T. A Review of Hydrogen Bonding in Solid Polymers: Structural Relationships, Analysis, and Importance. Polym. Plast. Technol. Eng. 1983, 21, 13–98. [Google Scholar] [CrossRef]
- Hesse, M.; Meier, H.; Zeeh, B. Spektroskopische Methoden in der Organischen Chemie, 6th ed.; Thieme Verlag: Stuttgart, Germany, 2002; ISBN 978-3135761053. [Google Scholar]
- Park, S.-Y.; Park, M.-H. The preparation and characterization of the cross-linked spherical, cylindrical, and vesicular micelles of poly(styrene-b-isoprene) diblock copolymers. Langmuir 2007, 23, 6788–6795. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.L.; Ransaw, H.C. Analysis Of Polyisoprenes by Infrared Spectroscopy. Anal. Chem. 1957, 29, 503–508. [Google Scholar] [CrossRef]
- Painter, P.C.; Koenig, J.L. A normal vibrational analysis of syndiotactic polystyrene. J. Polym. Sci. Polym. Phys. Ed. 1977, 15, 1885–1903. [Google Scholar] [CrossRef]
- Jiang, S.; Göpfert, A.; Abetz, V. Novel Morphologies of Block Copolymer Blends via Hydrogen Bonding. Macromolecules 2003, 36, 6171–6177. [Google Scholar] [CrossRef]
- Earnest, T.R.; MacKnight, W.J. Infrared Studies of Hydrogen Bonding in Ethylene-Methacrylic Acid Copolymers and Ionomers. Macromolecules 1980, 13, 844–849. [Google Scholar] [CrossRef]
- Velada, J.L.; Cesteros, C.; Madoz, A.; Katime, I. A study of the thermal degradation of poly(mono-n-alkyl itaconates). Macromol. Chem. Phys. 1995, 196, 3171–3185. [Google Scholar] [CrossRef]
- Leiserowitz, L. Molecular packing modes. Carboxylic acids. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1976, 32, 775–802. [Google Scholar] [CrossRef]
- Sieffert, M.A. Wasserstoffbrücken-gebundene Netzwerke. Ph.D. Thesis, Johannes Gutenberg-Universität, Mainz, Germany, 2000. [Google Scholar]
- Wittenberg, E.; Abetz, V. New post modification route for styrene butadiene copolymers leading to supramolecular hydrogen bonded networks—Synthesis and thermodynamic analysis of complexation. Polymer 2017, 121, 304–311. [Google Scholar] [CrossRef]
- Burattini, S.; Greenland, B.W.; Merino, D.H.; Weng, W.; Seppala, J.; Colquhoun, H.M.; Hayes, W.; Mackay, M.E.; Hamley, I.W.; Rowan, S.J. A Healable Supramolecular Polymer Blend Based on Aromatic π−π Stacking and Hydrogen-Bonding Interactions. J. Am. Chem. Soc. 2010, 132, 12051–12058. [Google Scholar] [CrossRef] [PubMed]
- Seymour, R.W.; Estes, G.M.; Cooper, S.L. Infrared Studies of Segmented Polyurethan Elastomers. Macromolecules 1970, 3, 579–583. [Google Scholar] [CrossRef]
- Chen, T.C.S.; Butler, G.B. Chemical Reactions on Polymers. III. Modification of Diene Polymers via the Ene Reaction with 4-Substituted-1,2,4-triazoline-3,5-diones. J. Macromol. Sci. Part A Chem. 1981, 16, 757–768. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | ĐS-Pre | ĐSI | Pn (S/I) | Polymer | ĐIS-Pre | ĐISI | Pn (I/S/I) |
|---|---|---|---|---|---|---|---|
| S91I967 | 1.1 | 1.1 | 585/89 | I5S90I562 | 1.2 | 1.3 | 46/536/46 |
| S85I1551 | 1.4 | 1.4 | 416/112 | I3S94I3117 | 1.2 | 1.2 | 52/1056/52 |
| S41I5931 | 1.1 | 1.1 | 123/273 | I1.5S96.1I2.482 | 1.2 | 1.2 | 18/757/29 |
| S51I495 | 1.3 | 1.1 | 24/35 | I0.6S98.8I0.698 | 1.2 | 1.2 | 9/935/9 |
| I0.7S98.5I0.8149 | 1.2 | 1.4 | 10/932/11 |
| Polymer | FGint | Đ | Df [%] | Tg [°C] a |
|---|---|---|---|---|
| I5S90I562 | - | 1.3 | - | 86.0 |
| I5S90I562 | OH | 1.2 | 58 | 93.2 |
| I5S90I562 | OH | 1.3 | 70 | c |
| I5S90I562 | OH | 1.3 | 94 | 101.5 |
| I5S90I562 | DETA | b | 32 | 97.8 |
| I5S90I562 | TETA | 1.4 | 21 | 94.8 |
| I5S90I562 | DAP/CDI | 1.3 | 10/7 | 100.7 |
| I5S90I562 | SA | 2.0 | 45 | 97.3 |
| I1.5S96.1I2.482 | – | 1.2 | – | 95.0 |
| I1.5S96.1I2.482 | OH | 1.2 | 75 | 97.9 |
| I1.5S96.1I2.482 | DETA | 1.3 | 48 | 98.9 |
| I1.5S96.1I2.482 | SA | 1.2 | 33 | 100 |
| I0.6S98.8I0.698 | – | 1.2 | – | 101.8 |
| I0.6S98.8I0.698 | OH | 1.2 | 43 | 102.5 |
| I0.6S98.8I0.698 | DETA | 1.3 | 13 | 102.0 |
| I0.6S98.8I0.698 | SA | 1.2 | 2 | 104.0 |
| I3S94I3117 | – | 1.2 | – | 83.7 |
| I3S94I3117 | OH | 1.5 | 50 | 101.1 |
| I3S94I3117 | DETA | 2.3 | 20 | 98.2 |
| I3S94I3117 | SA | 1.7 | 30 | 99.8 |
| I0.7S98.5I0.8149 | – | 1.2 | – | 99.6 |
| I0.7S98.5I0.8149 | OH | 1.2 | 83 | 103.0 |
| I0.7S98.5I0.8149 | DETA | 1.5 | 29 | 102.0 |
| I0.7S98.5I0.8149 | SA | 1.2 | 14 | 102.0 |
| S85I1551 | – | 1.4 | – | c |
| S85I1551 | OH | 1.5 | 27 | c |
| S85I1551 | CDI | 1.5 | 11 | c |
| S85I1551 | DETA | 2.4 | 12 | c |
| S85I1551 | OH | 1.8 | 100 | c |
| S85I1551 | CDI | c | 18 | c |
| S85I1551 | DAP | 1.6 | 4 | c |
| S91I967 | – | – | – | 98.2 |
| S91I967 | OH | 1.2 | 30 | 99.1 |
| S91I967 | SA | 1.4 | 11 | 100.8 |
| S91I967 | OH | 1.1 | 92 | 97.8 |
| S91I967 | SA | 1.5 | 45 | 99.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmstorf, E.; Abetz, V. Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 1: Synthesis and Characterization. Materials 2018, 11, 1608. https://doi.org/10.3390/ma11091608
Rahmstorf E, Abetz V. Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 1: Synthesis and Characterization. Materials. 2018; 11(9):1608. https://doi.org/10.3390/ma11091608
Chicago/Turabian StyleRahmstorf, Elaine, and Volker Abetz. 2018. "Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 1: Synthesis and Characterization" Materials 11, no. 9: 1608. https://doi.org/10.3390/ma11091608
APA StyleRahmstorf, E., & Abetz, V. (2018). Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 1: Synthesis and Characterization. Materials, 11(9), 1608. https://doi.org/10.3390/ma11091608