First-Principles Investigation on the Electronic and Mechanical Properties of Cs-Doped CH3NH3PbI3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Calculation Method and Model
3. Results and Discussion
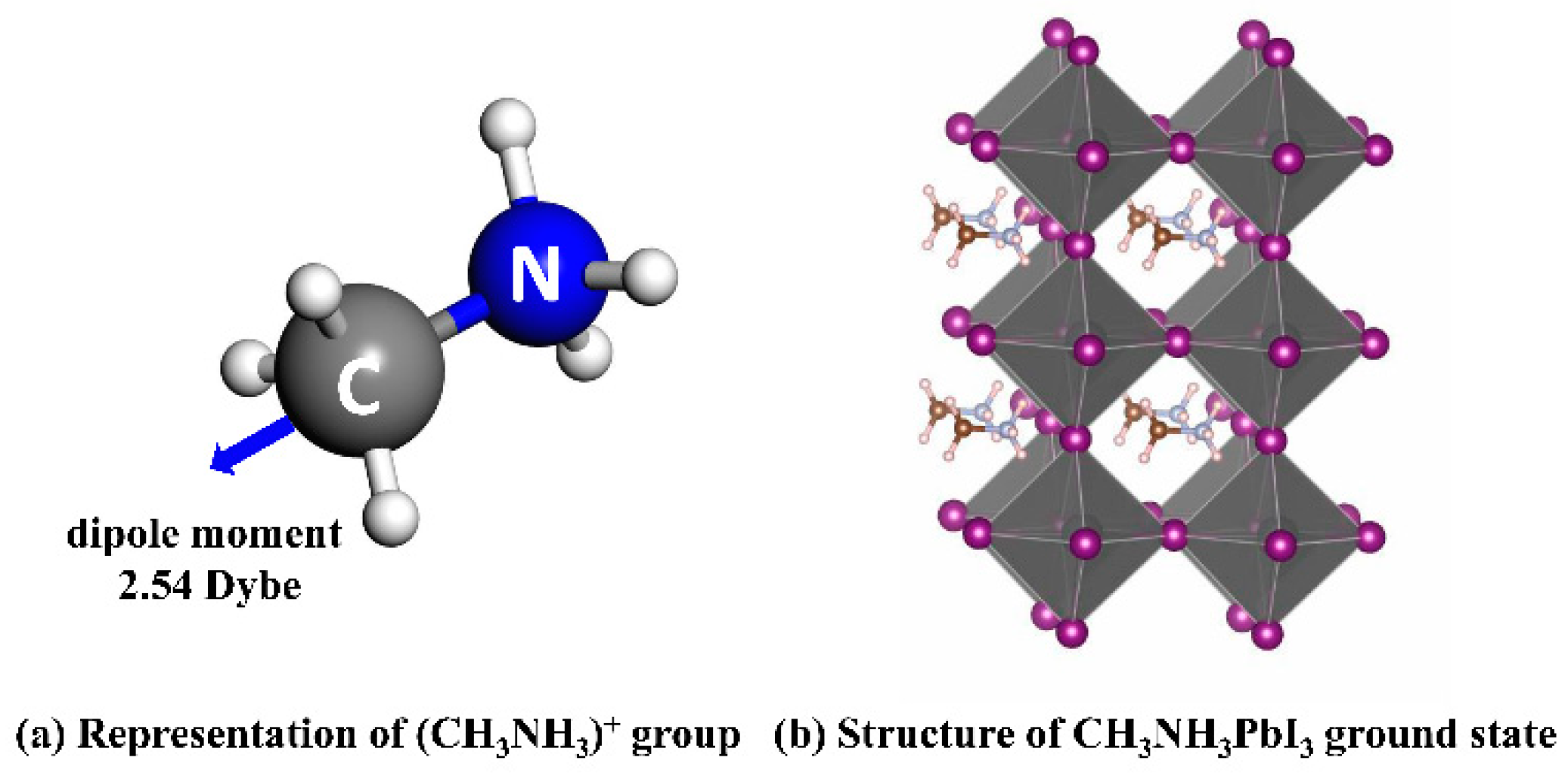
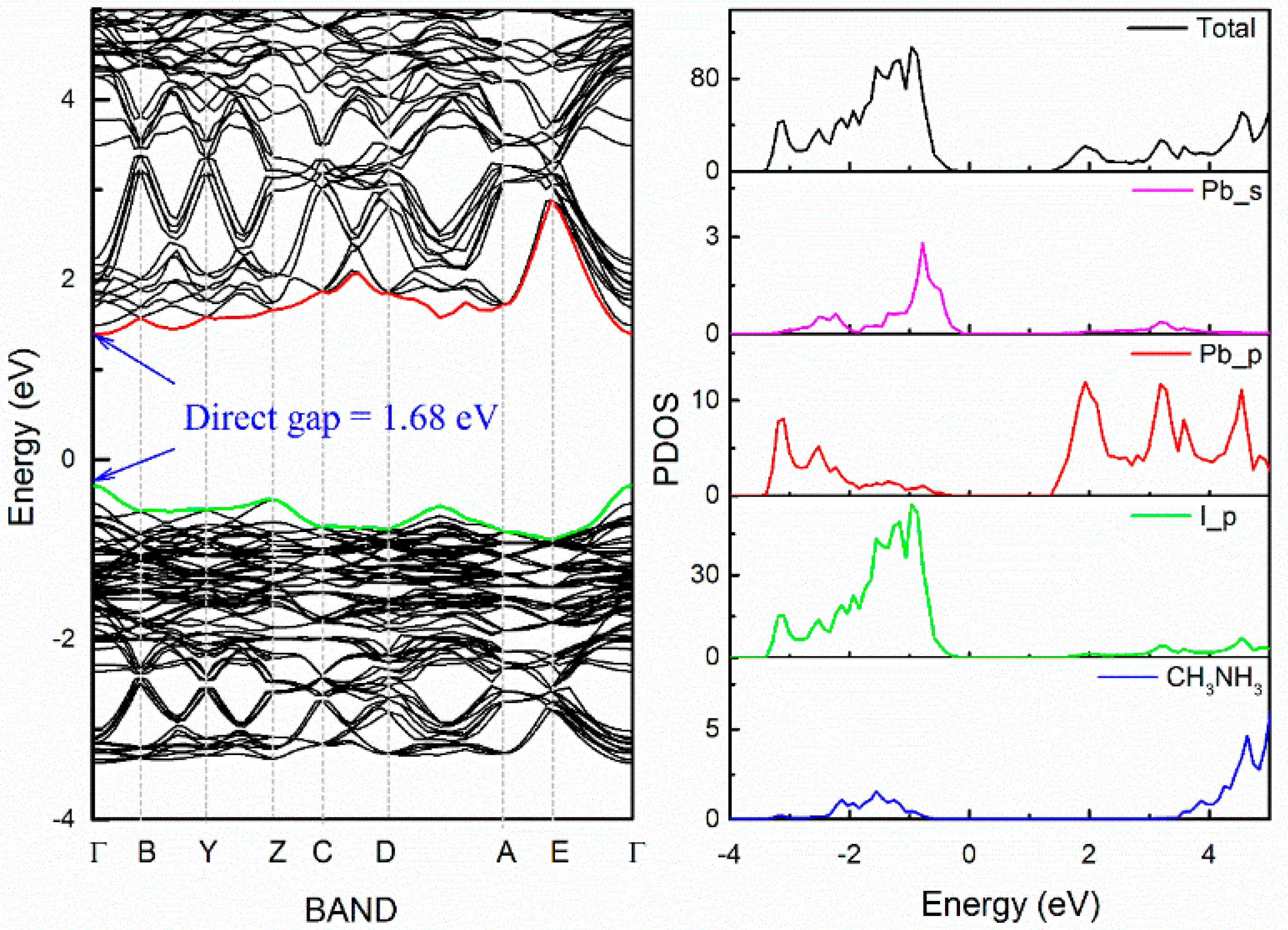
3.1. Intrinsic Properties of CH3NH3PbI3: Structure and Band
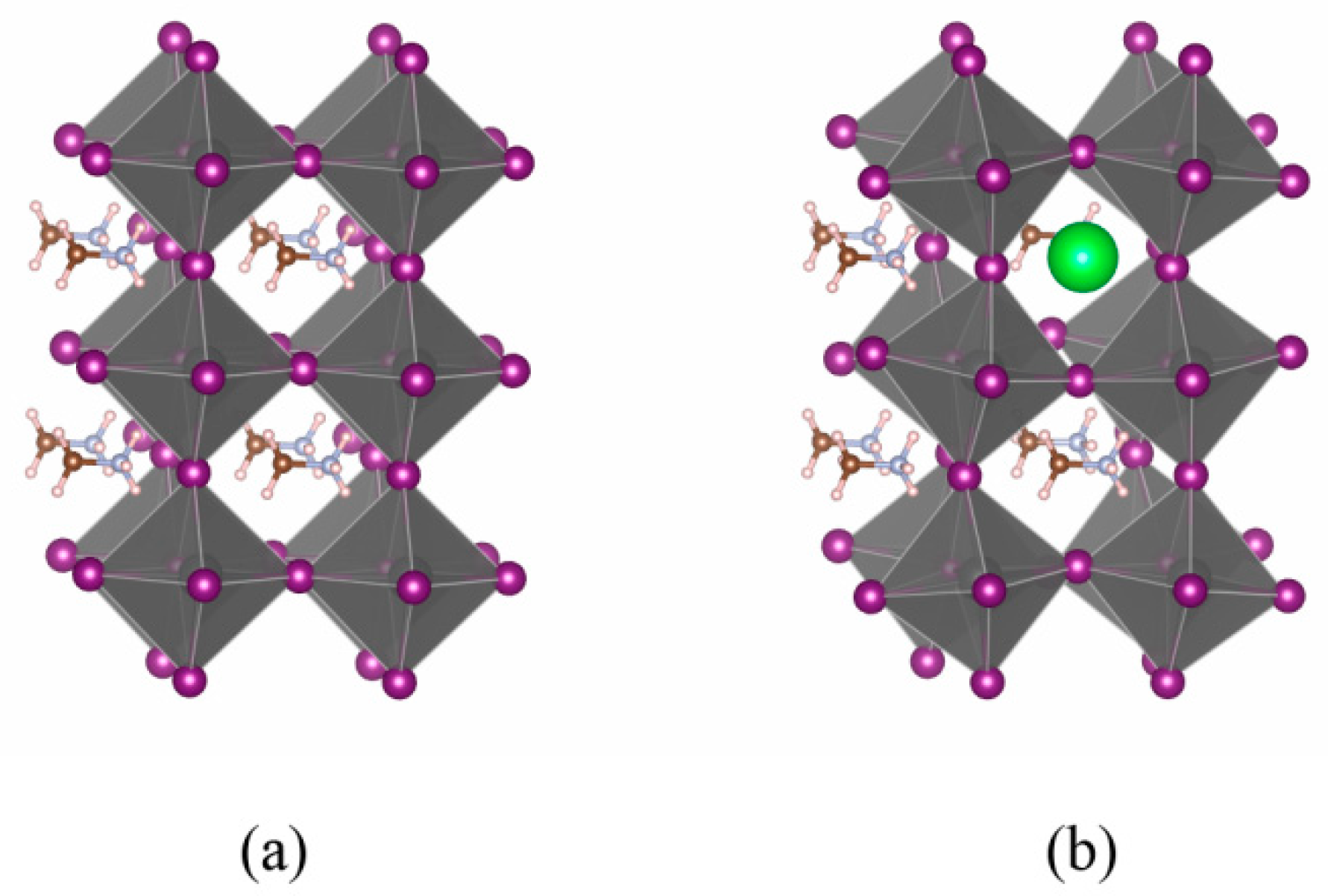
3.2. Cs-Doped CH3NH3PbI3: Stability and Electronic Properties
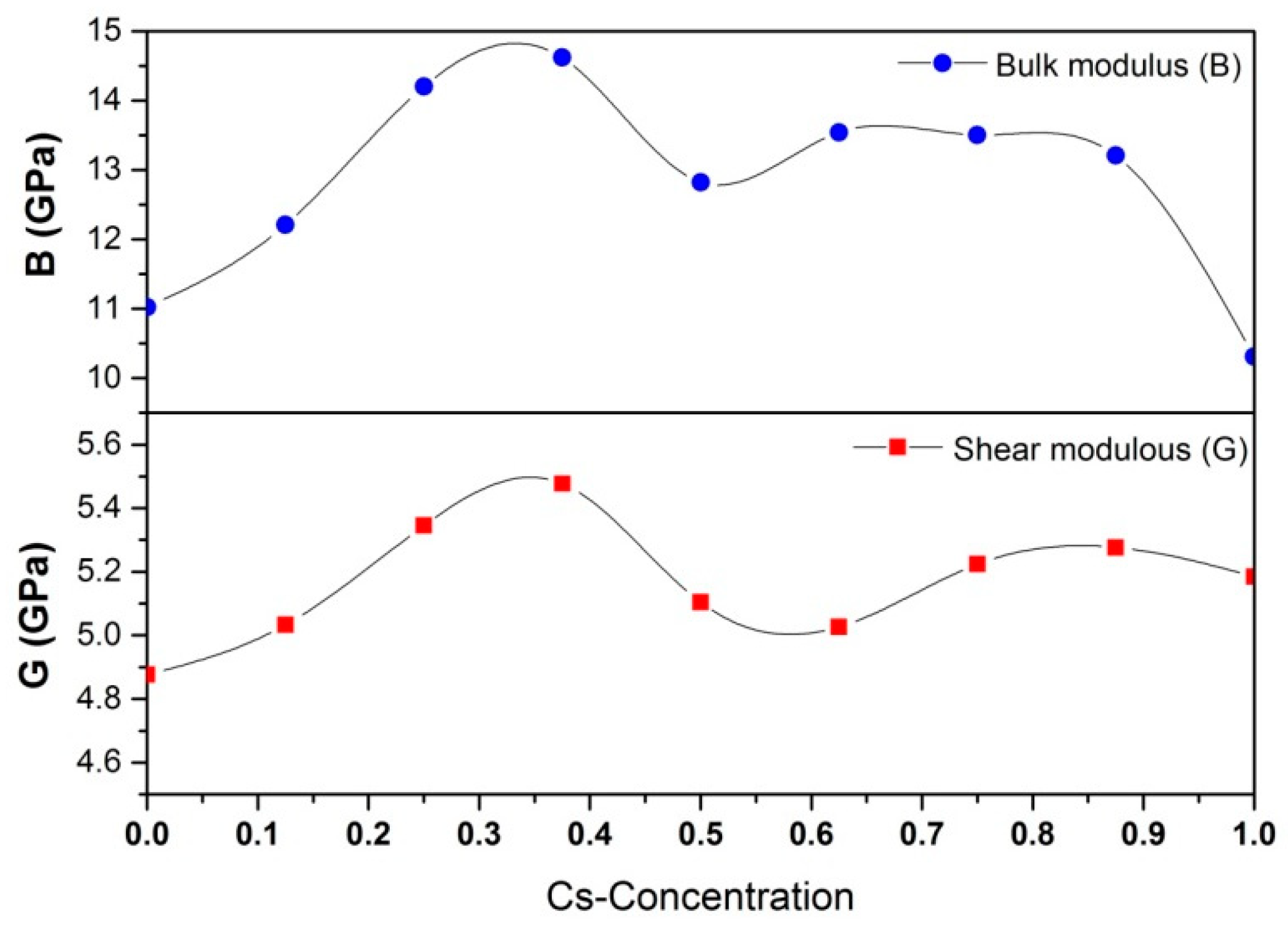
3.3. Cs-Doped CH3NH3PbI3: Mechanical Properties
4. Conclusions
- (1)
- The difference in orientation energy of (CH3NH3)+ is comparable to the thermal power at room temperature, which causes a random orientation of (CH3NH3)+ group in the perovskite lattice.
- (2)
- The local ordered arrangement of (CH3NH3)+ is energetic favorable that facilitates the formation of the electronic dipole domain, which helps to improve the separation and lifetime of photo-generated carriers.
- (3)
- The band edge states are dominated by (PbI6) anion group in CH3NH3PbI3. A-site (CH3NH3)+ or Cs+ does not directly participate in the construction of the band edge states, but indirectly influences the structural stability and electronic level through Jahn–Teller effect.
- (4)
- It has been demonstrated that the suitable concentration of Cs can enhance both thermodynamic and mechanical stability of CH3NH3PbI3 without deteriorating the conversion efficiency.
- (5)
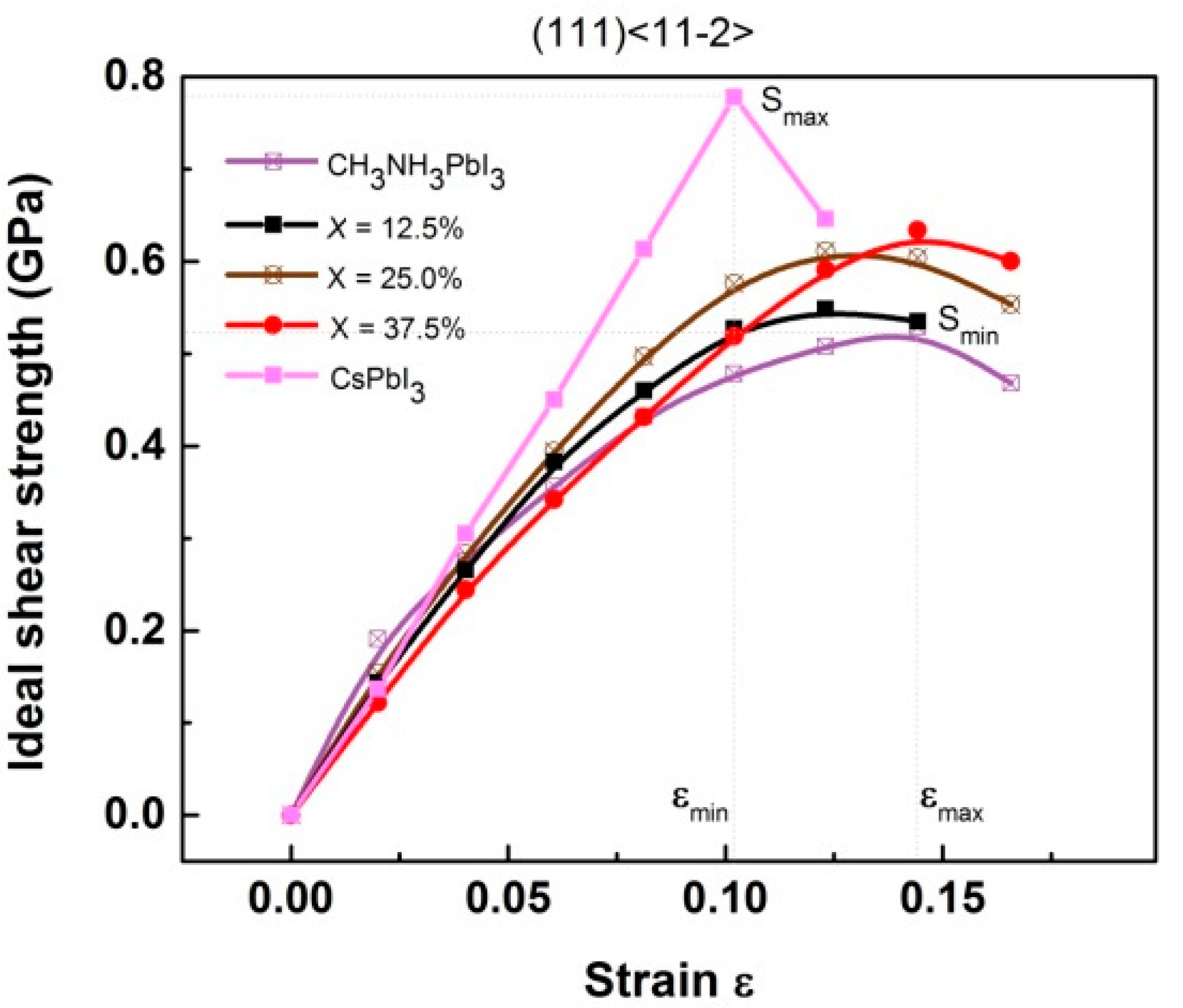
- Goldschmidt’s tolerance factor suggests that the Cs-concentration should be less than 62.5 at.%, while mechanical performance indicates that the optimal Cs-concentration should be less than 37.5%. Below this mark, the mechanical properties including stability, hardness, strength, and ductility will continuously rise with the Cs-concentration.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Science News. Newcomer Juices up the race to harness sunlight. Science 2013, 342, 1438–1439. [Google Scholar]
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.M.; Teuscher, J.; Miyasaka, T.; Murakami, T.N.; Snaith, H.J. Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science 2012, 338, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.Z.; Johnston, M.B.; Snaith, H.J. Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature 2013, 501, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Best Research-Cell Efficiencies (National Renewable Energy Laboratory, 2018). Available online: https://www.nrel.gov/pv/assets/images/efficiency-chart.png (accessed on 26 June 2018).
- Conings, B.; Drijkoningen, J.; Gauquelin, N.; Babayigit, A.; D’Haen, J.; D’Olieslaeger, L.; Ethirajan, A.; Verbeeck, J.; Manca, J.; Mosconi, E. Intrinsic thermal instability of methylammonium lead trihalide perovskite. Adv. Energy Mater. 2015, 5. [Google Scholar] [CrossRef]
- Christians, J.A.; Herrera, P.A.M.; Kamat, P.V. Transformation of the excited state and photovoltaic efficiency of CH3NH3PbI3 perovskite upon controlled exposure to humidified air. J. Am. Chem. Soc. 2015, 137, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Graetzel, M.; Janssen, R.A.J.; Mitzi, D.B.; Sargent, E.H. Materials interface engineering for solution-processed photovoltaics. Nature 2012, 488, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.J.; Eperon, G.E.; Hopkinson, P.E.; Habisreutinger, S.N.; Wang, J.T.; Snaith, H.J.; Greenham, N.C. Oxygen Degradation in Mesoporous Al2O3/CH3NH3PbI3−xClx Perovskite Solar Cells: Kinetics and Mechanisms. Adv. Energy Mater. 2016, 6. [Google Scholar] [CrossRef]
- Alsari, M.; Pearson, A.J.; Wang, J.T.W.; Wang, Z.; Montisci, A.; Greenham, N.C.; Snaith, H.J.; Lilliu, S.; Friend, R.H. Degradation Kinetics of Inverted Perovskite Solar Cells. Sci. Rep. 2018, 8, 5977. [Google Scholar] [CrossRef] [PubMed]
- Alsari, M.; Bikondoa, O.; Bishop, J.; Abdi-Jalebi, M.; Ozer, L.Y.; Hampton, M.; Thompson, P.; Hörantner, M.T.; Mahesh, S.; Greenland, C.; et al. In situ simultaneous photovoltaic and structural evolution of perovskite solar cells during film formation. Energy Environ. Sci. 2018, 11, 383–393. [Google Scholar] [CrossRef]
- Mosconi, E.; Quarti, C.; Ivanovska, T.; Ruani, G.; Angelis, F.D. Structural and electronic properties of organo-halide lead perovskites: A combined IR-spectroscopy and ab initio molecular dynamics investigation. Phys. Chem. Chem. Phys. 2014, 16, 16137–16144. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.H.; Im, S.H.; Heo, J.H.; Mandal, T.N.; Seok, S.I. Chemical management for colorful, efficient, and stable inorganic–organic hybrid nanostructured solar cells. Nano Lett. 2013, 13, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Hoke, E.T.; Slotcavage, D.J.; Dohner, E.R.; Bowring, A.R.; Karunadas, H.I.; McGehee, M.D. Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics. Chem. Sci. 2015, 6, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Navas, J.; Sánchez-Coronilla, A.; Gallardo, J.J.; Hernández, N.C.; Piñero, J.C.; Alcántara, R.; Lorenzo, C.F.; De los Santos, D.M.; Aguilar, T.; Calleja, J.M. New insights into organic–inorganic hybrid perovskite CH3NH3PbI3 nanoparticles. An experimental and theoretical study of doping in Pb2+sites with Sn2+, Sr2+, Cd2+ and Ca2+. Nanoscale 2015, 7, 6216–6229. [Google Scholar] [CrossRef] [PubMed]
- Ogomi, Y.; Morita, A.; Tsukamoto, S.; Saitho, T.; Fujikawa, N.; Shen, Q.; Toyoda, T.; Yoshino, K.; Pandey, S.; Ma, T.; et al. CH3NH3SnxPb(1−x)I3 perovskite solar cells covering up to 1060 nm. J. Phys. Chem. Lett. 2014, 5, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Eperon, G.E.; Paternò, G.M.; Sutton, R.J.; Zampetti, A.; Haghighirad, A.A.; Cacialli, F.; Snaith, H.J. Inorganic caesium lead iodide perovskite solar cells. J. Mater. Chem. A 2015, 3, 19688–19695. [Google Scholar] [CrossRef]
- Ahmad, M.; Rehman, G.; Ali, L.; Shafiq, M.; Iqbal, R.; Ahmad, R.; Khan, T.; Asadabad, S.J.; Maqbool, M.; Ahmad, I. Structural, electronic and optical properties of CsPbX3 (X = Cl, Br, I) for energy storage and hybrid solar cell applications. J. Alloy Compd. 2017, 705, 828–839. [Google Scholar] [CrossRef]
- Protesescu, L.; Yakunin, S.; Bodnarchuk, M.I.; Krieg, F.; Caputo, R.; Hendon, C.H.; Yang, R.X.; Walsh, A.; Kovalenko, M.V. Nanocrystals of cesium lead halide perovskites (CsPbX3, X = Cl, Br, and I): Novel optoelectronic materials showing bright emission with wide color gamut. Nano Lett. 2015, 15, 3692–3696. [Google Scholar] [CrossRef] [PubMed]
- Trots, D.M.; Myagkota, S.V. High-temperature structural evolution of caesium and rubidium triiodoplumbates. J. Phys. Chem. Solids 2008, 69, 2520–2526. [Google Scholar] [CrossRef]
- Eperon, G.E.; Beck, C.E.; Snaith, H.J. Cation exchange for thin film lead iodide perovskite interconversion. Mater. Horiz. 2016, 3, 63–71. [Google Scholar] [CrossRef]
- Jeon, N.J.; Noh, J.H.; Yang, W.S.; Kim, Y.C.; Ryu, S.; Seo, J.; Seok, S., II. Compositional engineering of perovskite materials for high-performance solar cells. Nature 2015, 517, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Binek, A.; Hanusch, F.C.; Docampo, P.; Bein, T. Stabilization of the trigonal high-temperature phase of formamidinium lead iodide. J. Phys. Chem. Lett. 2015, 6, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Luo, J.; Meloni, S.; Boziki, A.; Astani, N.A.; Gratzel, C.; Zakeeruddin, S.M.; Rothlisberger, U.; Gratzel, M. Entropic stabilization of mixed A-cation ABX3 metal halide perovskites for high performance perovskite solar cells. Energy Environ. Sci. 2016, 9, 656–662. [Google Scholar] [CrossRef]
- Li, Z.; Yang, M.; Park, J.S.; Wei, S.H.; Berry, J.J.; Zhu, K. Stabilizing perovskite structures by tuning tolerance factor: Form ation of formamidinium and cesium lead iodide solid-state alloys. Chem. Mater. 2016, 28, 284–292. [Google Scholar] [CrossRef]
- Kulbak, M.; Cahen, D.; Hodes, G. How important is the organic part of lead halide perovskite photovoltaic cells? Efficient CsPbBr3 cells. J. Phys. Chem. Lett. 2015, 6, 2452–2456. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 98; Gaussian Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Bakulin, A.A.; Selig, O.; Bakker, H.J.; Rezus, Y.L.; Muller, C.; Glaser, T.; Lovrincic, R.; Sun, Z.; Chen, Z.; Walsh, A.; et al. Real-time observation of organic cation reorientation in methylammonium lead iodide perovskites. J. Phys. Chem. Lett. 2015, 6, 3663–3669. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.M.; Butler, K.T.; Brivio, F.; Hendon, C.H.; Schilfgaarde, M.V.; Walsh, A. Atomistic origins of high-performance in hybrid halide perovskite solar cells. Nano Lett. 2014, 14, 2584–2590. [Google Scholar] [CrossRef] [PubMed]
- La-o-vorakiat, C.; Salim, T.; Kadro, J.; Khuc, M.; Haselsberger, R.; Cheng, L.; Xia, H.; Gurzadyan, G.G.; Su, H.; Lam, Y.M.; et al. Elucidating the role of disorder and free-carrier recombination kinetics in CH3NH3PbI3 perovskite films. Nat. Commun. 2015, 6, 7903. [Google Scholar] [CrossRef] [PubMed]
- Stroppa, A.; Quarti, C.; De Angelis, F.; Picozzi, S. Ferroelectric polarization of CH3NH3PbI3: A detailed study based on density functional theory and symmetry mode analysis. J. Phys. Chem. Lett. 2015, 6, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Berdiyorov, G.R.; Madjet, M.E.; El-Mellouhi, F.; Peeters, F.M. Effect of crystal structure on the electronic transport properties of the organometallic perovskite CH3NH3PbI3. Sol. Energy Mater. Sol. Cells 2016, 148, 60–66. [Google Scholar] [CrossRef]
- Frost, J.M.; Walsh, A. What is moving in hybrid halide Perovskite solar cells? Acc. Chem. Res. 2016, 49, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Kutes, Y.; Ye, L.; Zhou, Y.; Pang, S.; Huey, B.D.; Padture, N.P. Direct observation of ferroelectric domains in solution-processed CH3NH3PbI3 Perovskite Thin Films. J. Phys. Chem. Lett. 2014, 5, 3335–3339. [Google Scholar] [CrossRef] [PubMed]
- Jeng, J.Y.; Chiang, Y.F.; Lee, M.H.; Peng, S.R.; Guo, T.F.; Chen, P.; Wen, T.C. CH3NH3PbI3 perovskite/fullerene planar heterojunction hybrid solar cells. Adv. Mater. 2013, 25, 3727–3732. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Qiu, Y.; Yan, K.; Zhong, M.; Mu, C.; Yan, H.; Yang, S. All-solid-state hybrid solar cells based on a new organometal halideperovskite sensitizer and one-dimensional TiO2 nanowire arrays. Nanoscale 2013, 5, 3245–3248. [Google Scholar] [CrossRef] [PubMed]
- Umari, P.; Mosconi, E.; Filippo, D. Relativistic GW calculations on CH3NH3PbI3 and CH3NH3SnI3 perovskites for solar cell applications. Sci. Rep. 2014, 4, 4467. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, E.; Amat, A.; Nazeeruddin, M.; Gratzel, M.; Angelis, F.D. First-principles modeling of mixed halide organometal perovskites for photovoltaic applications. J. Phys. Chem. C 2013, 117, 13902–13913. [Google Scholar] [CrossRef]
- Umebayashi, T.; Asai, K.; Kondo, T.; Nakao, A. Electronic structures of lead iodide based low-dimensional crystals. Phys. Rev. B 2003, 67, 155405. [Google Scholar] [CrossRef]
- Brivio, F.; Walker, A.B.; Walsh, A. Structural and electronic properties of hybrid perovskites for high-efficiency thin-film photovoltaics from first-principles. APL Mater. 2013, 1, 042111. [Google Scholar] [CrossRef]
- Meng, X.Y.; Liu, D.Y.; Qin, G.W. Band engineering of multicomponent semiconductors: A general theoretical model on the anion group. Energy Environ. Sci. 2018, 11, 692–701. [Google Scholar] [CrossRef]
- Goldschmidt, V.M. Crystal structure and chemical correlation. Ber. Dtsch. Chem. Ges. 1927, 60, 1263–1268. [Google Scholar] [CrossRef]
- Goldschmidt, V.M. Die Gesetze der Krystallochemie. Naturwissenschaften 1926, 14, 477–485. [Google Scholar] [CrossRef]
- Kieslich, G.; Sun, S.; Cheetham, A.K. Solid-state principles applied to organic–inorganic perovskites: New tricks for an old dog. Chem. Sci. 2014, 5, 4712–4715. [Google Scholar] [CrossRef]
- Stoumpos, C.C.; Kanatzidis, M.G. The renaissance of halide perovskites and their evolution as emerging semiconductors. Acc. Chem. Res. 2015, 48, 2791–2802. [Google Scholar] [CrossRef] [PubMed]
- Niemann, R.G.; Gouda, L.; Hu, J.; Tirosh, S.; Gottesman, R.; Cameron, P.J.; Zaban, A. Cs+ incorporation into CH3NH3PbI3perovskite: Substitution limit and stability enhancement. J. Mater. Chem. A 2016, 4, 17819–17827. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Clarendon Press: Oxford, UK, 1954. [Google Scholar]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Beecher, A.N.; Semonin, O.E.; Skelton, J.M.; Frost, J.M.; Terban, M.W.; Zhai, H.W.; Alatas, A.; Owen, J.S.; Walsh, A.; Billinge, S.J.L. Direct observation of dynamic symmetry breaking above room temperature in methylammonium lead iodide perovskite. ACS Energy Lett. 2016, 1, 880–887. [Google Scholar] [CrossRef]
- Rakita, Y.; Cohen, S.R.; Kedem, N.K.; Hodes, G.; Cahen, D. Mechanical properties of APbX3 (A = Cs or CH3NH3; X = I or Br) perovskite single crystals. MRS Commun. 2015, 5, 623–629. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. Ser. 1954, 45, 823–843. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Li, S.; Bian, F.; Meng, X. First-Principles Investigation on the Electronic and Mechanical Properties of Cs-Doped CH3NH3PbI3. Materials 2018, 11, 1141. https://doi.org/10.3390/ma11071141
Liu D, Li S, Bian F, Meng X. First-Principles Investigation on the Electronic and Mechanical Properties of Cs-Doped CH3NH3PbI3. Materials. 2018; 11(7):1141. https://doi.org/10.3390/ma11071141
Chicago/Turabian StyleLiu, Dongyan, Shanshan Li, Fang Bian, and Xiangying Meng. 2018. "First-Principles Investigation on the Electronic and Mechanical Properties of Cs-Doped CH3NH3PbI3" Materials 11, no. 7: 1141. https://doi.org/10.3390/ma11071141
APA StyleLiu, D., Li, S., Bian, F., & Meng, X. (2018). First-Principles Investigation on the Electronic and Mechanical Properties of Cs-Doped CH3NH3PbI3. Materials, 11(7), 1141. https://doi.org/10.3390/ma11071141