Nano-Graphene Oxide Functionalized Bioactive Poly(lactic acid) and Poly(ε-caprolactone) Nanofibrous Scaffolds



Abstract
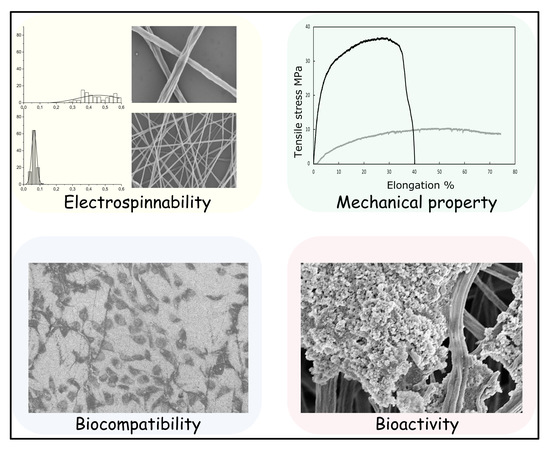
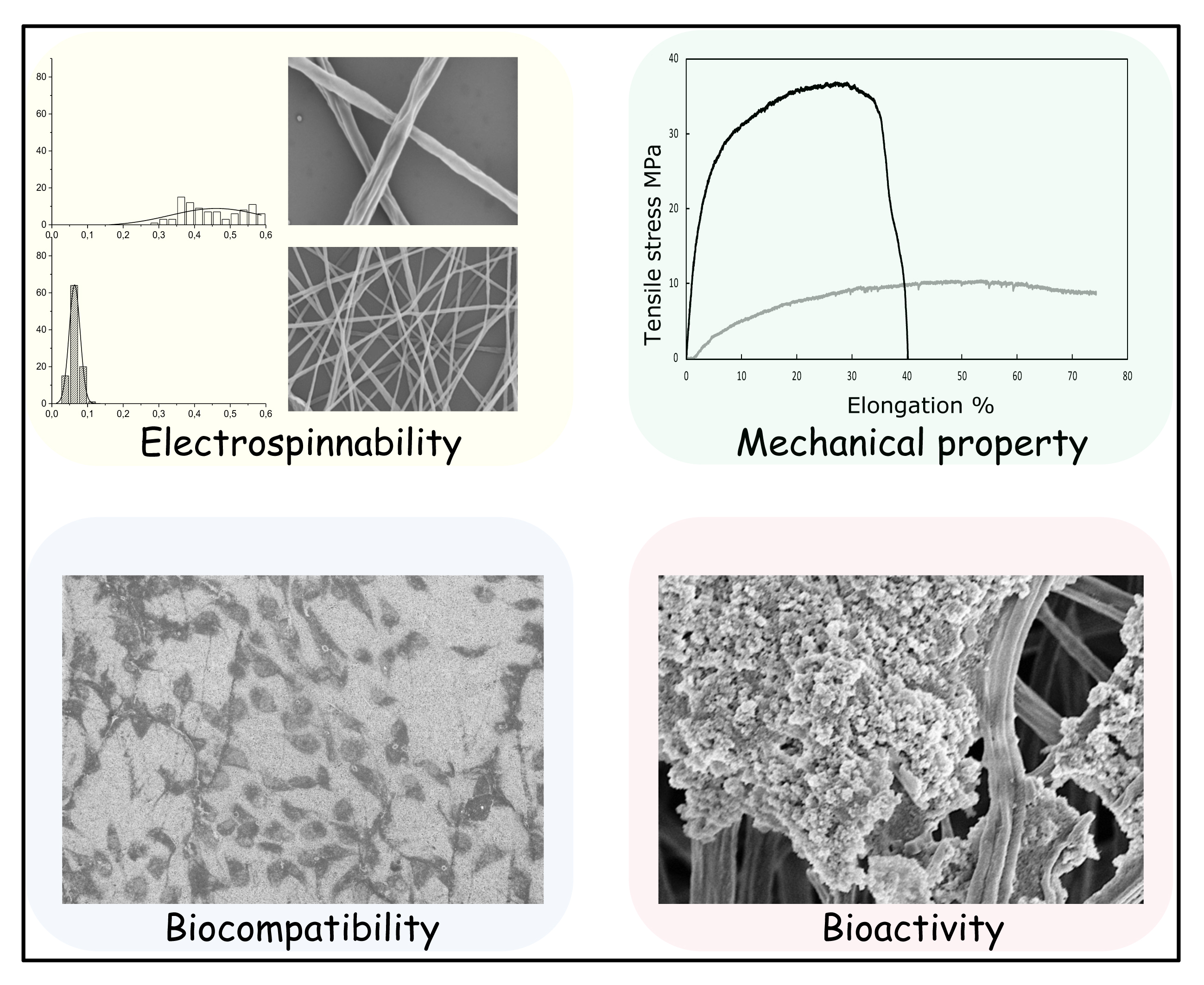{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Electrospinning
2.3. Characterizations
2.3.1. Transmission Electron Microscopy (TEM)
2.3.2. Fourier Transform Infrared Spectroscopy (FTIR)
2.3.3. Scanning Electron Microscopy (SEM)
2.3.4. X-ray Diffraction (XRD)
2.3.5. Micromechanical Testing
2.3.6. Water Contact Angle Measurements
2.3.7. Cell Viability Test
2.3.8. Mineralization Test
3. Results and Discussion
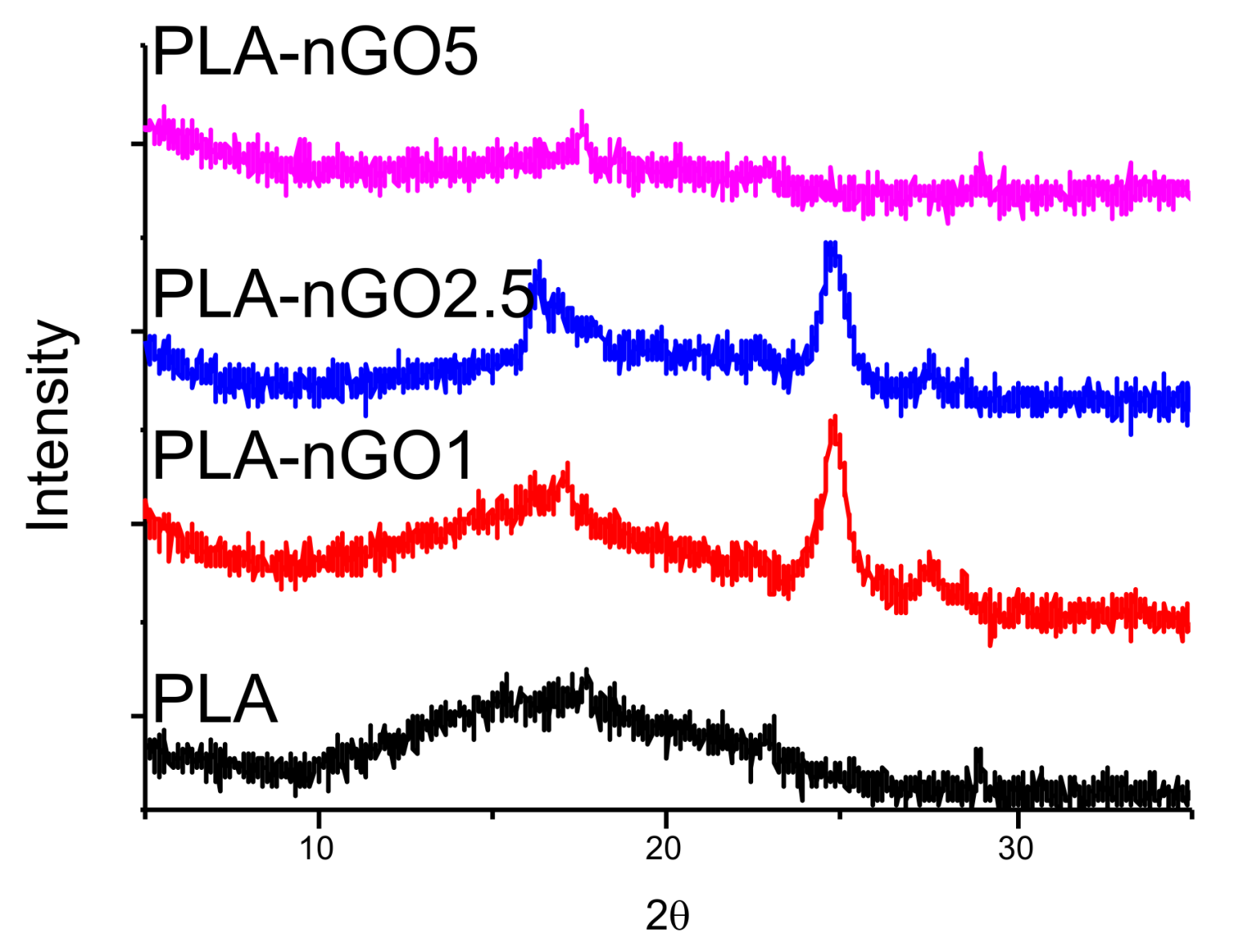
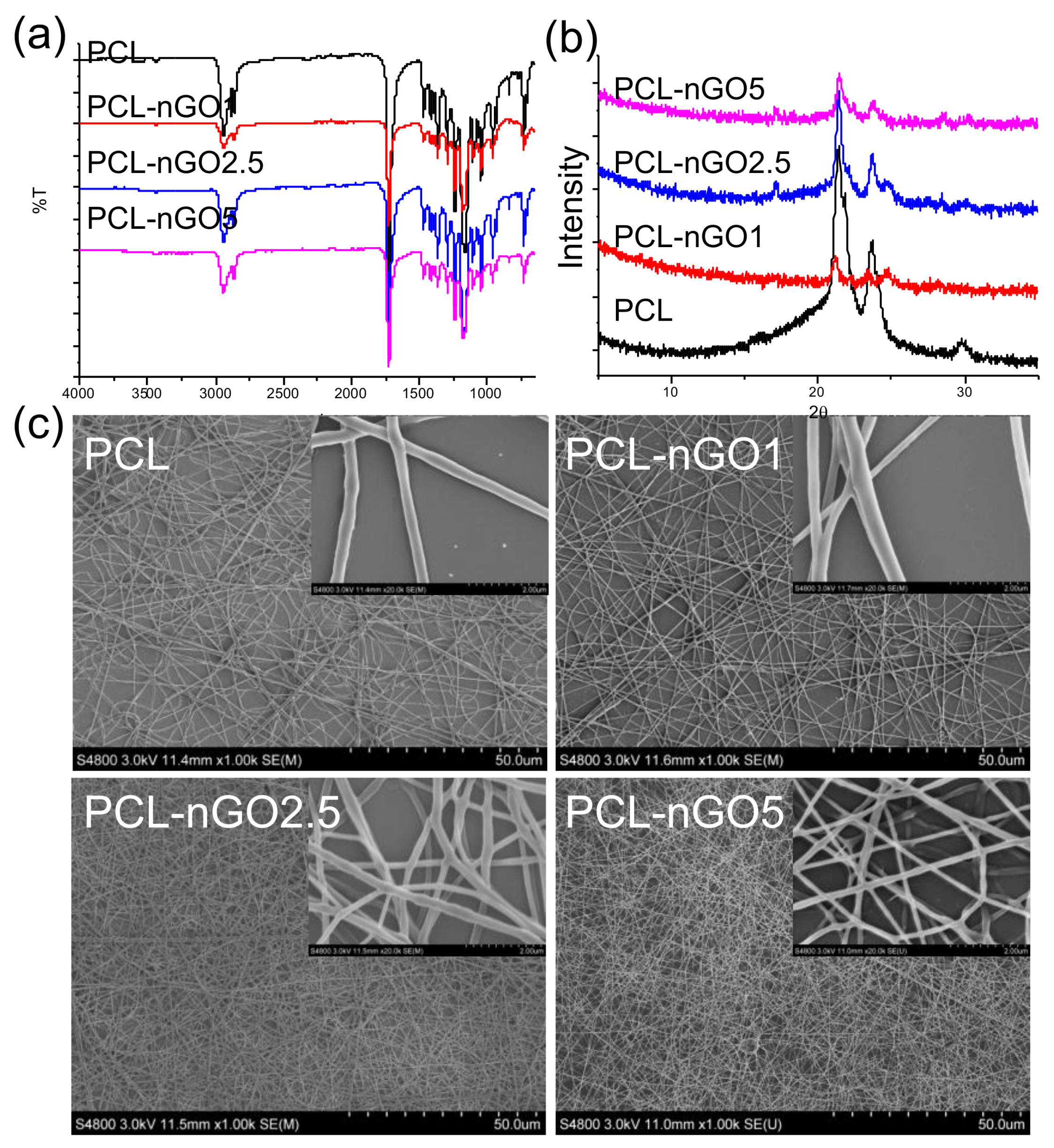
3.1. Interaction between PLA and nGO
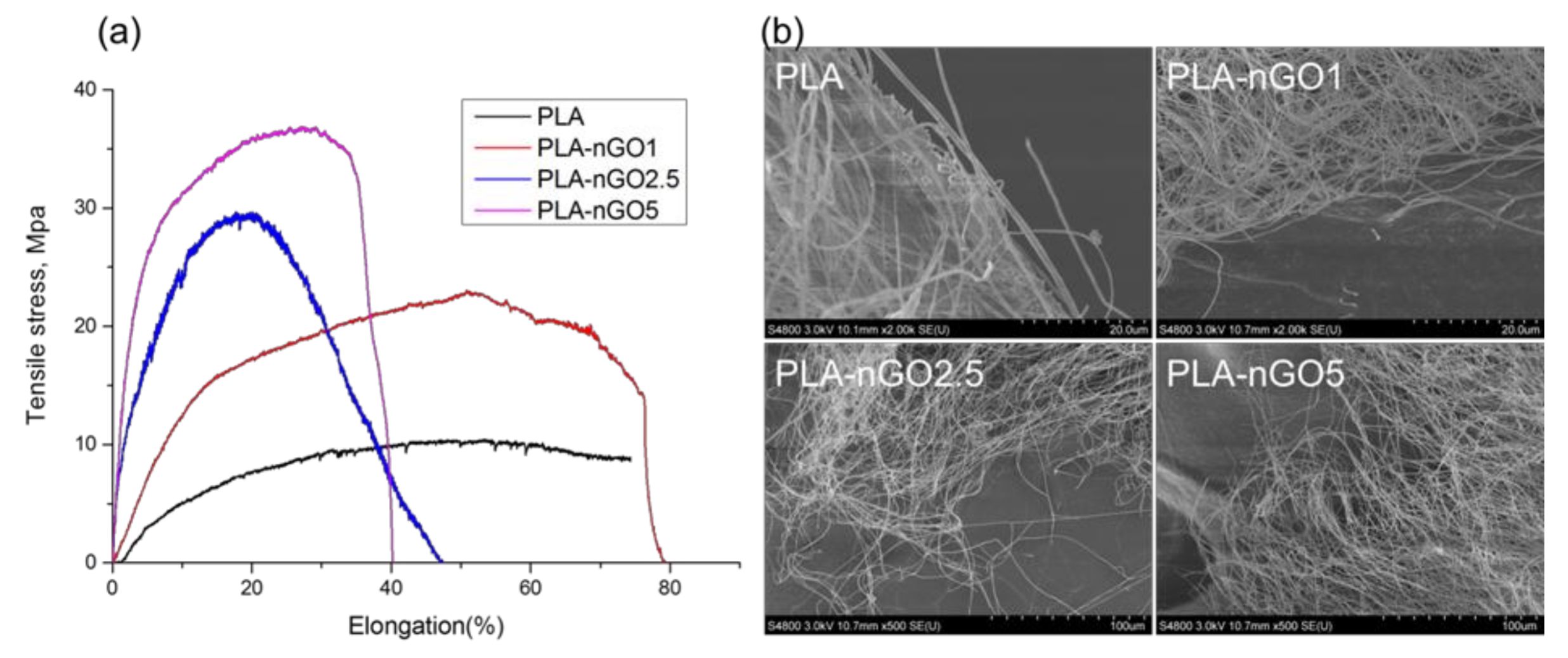
3.2. Fiber Morphology
3.3. Fiber Microstructure
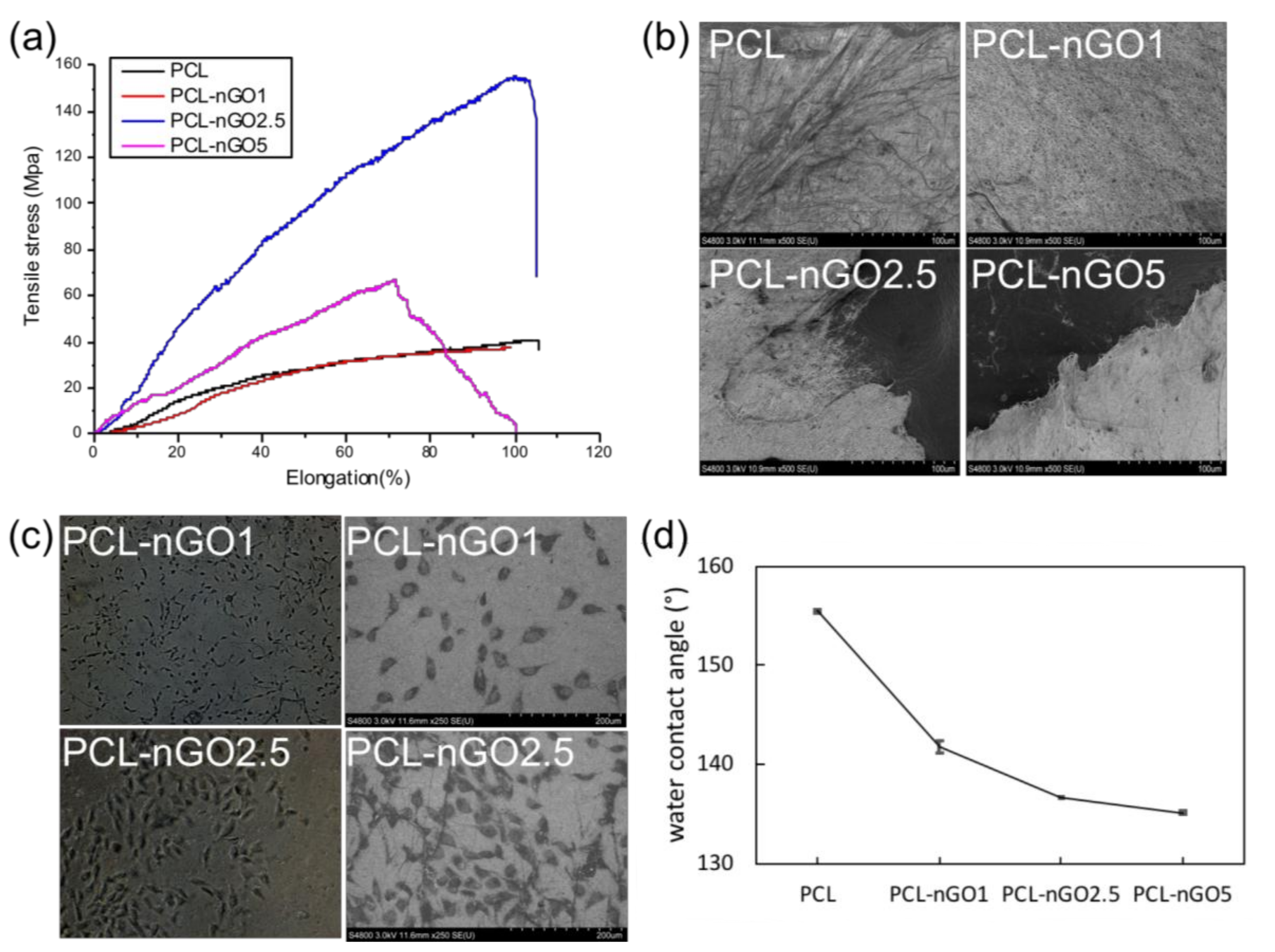
3.4. Mechanical Properties
3.5. Biocompatibility Test
3.6. Biomineralization in SBF
3.7. PCL-nGO Electrospun Fibers
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Langer, R.; Vacanti, J. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.X. Biomimetic materials for tissue engineering. Adv. Drug Deliv. Rev. 2008, 60, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, Z.; Najeeb, S.; Khurshid, Z.; Verma, V.; Rashid, H.; Glogauer, M. Biodegradable materials for bone repair and tissue engineering applications. Materials 2015, 8, 5744–5794. [Google Scholar] [CrossRef] [PubMed]
- Nejatian, T.; Khurshid, Z.; Zafar, M.S.; Najeeb, S.; Zohaib, S.; Mazafari, M.; Hopkinson, L.; Sefat, F. 5—dental biocomposites. In Biomaterials for Oral and Dental Tissue Engineering; Woodhead Publishing: Cambridge, UK, 2017; pp. 65–84. [Google Scholar]
- Wang, J.; Valmikinathan, C.M.; Liu, W.; Laurencin, C.T.; Yu, X. Spiral-structured, nanofibrous, 3d scaffolds for bone tissue engineering. J. Biomed. Mater. Res. Part A 2010, 93, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.W. Scaffolds in tissue engineering bone and cartilage. Biomaterials 2000, 21, 2529–2543. [Google Scholar] [CrossRef]
- Holzwarth, J.M.; Ma, P.X. 3d nanofibrous scaffolds for tissue engineering. J. Mater. Chem. 2011, 21, 10243–10251. [Google Scholar] [CrossRef]
- Wang, X.; Ding, B.; Li, B. Biomimetic electrospun nanofibrous structures for tissue engineering. Mater. Today 2013, 16, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.; Najeeb, S.; Khurshid, Z.; Vazirzadeh, M.; Zohaib, S.; Najeeb, B.; Sefat, F. Potential of electrospun nanofibers for biomedical and dental applications. Materials 2016, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- Qasim, S.; Zafar, M.; Najeeb, S.; Khurshid, Z.; Shah, A.; Husain, S.; Rehman, I. Electrospinning of chitosan-based solutions for tissue engineering and regenerative medicine. Int. J. Mol. Sci. 2018, 19, 407. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Yu, M.; Zong, X.; Chiu, J.; Fang, D.; Seo, Y.-S.; Hsiao, B.S.; Chu, B.; Hadjiargyrou, M. Control of degradation rate and hydrophilicity in electrospun non-woven poly (d, l-lactide) nanofiber scaffolds for biomedical applications. Biomaterials 2003, 24, 4977–4985. [Google Scholar] [CrossRef]
- Lee, K.; Kim, H.; Khil, M.; Ra, Y.; Lee, D. Characterization of nano-structured poly (ε-caprolactone) nonwoven mats via electrospinning. Polymer 2003, 44, 1287–1294. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, J.; Gao, W.; Liang, H.; Wang, H.; Li, J. Preparation of chitosan/pla blend micro/nanofibers by electrospinning. Mater. Lett. 2009, 63, 658–660. [Google Scholar] [CrossRef]
- Xu, X.; Yang, Q.; Wang, Y.; Yu, H.; Chen, X.; Jing, X. Biodegradable electrospun poly (l-lactide) fibers containing antibacterial silver nanoparticles. Eur. Polym. J. 2006, 42, 2081–2087. [Google Scholar] [CrossRef]
- Yoshimoto, H.; Shin, Y.; Terai, H.; Vacanti, J. A biodegradable nanofiber scaffold by electrospinning and its potential for bone tissue engineering. Biomaterials 2003, 24, 2077–2082. [Google Scholar] [CrossRef]
- Zhang, Y.; Ouyang, H.; Lim, C.T.; Ramakrishna, S.; Huang, Z.M. Electrospinning of gelatin fibers and gelatin/pcl composite fibrous scaffolds. J. Biomed. Mater. Res. Part B Appl. Biomater. 2005, 72, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.X.; Zheng, W.; Li, L.; Zheng, Y.F. Fabrication and characterization of three-dimensional nanofiber membrance of pcl–mwcnts by electrospinning. Mater. Sci. Eng. C 2010, 30, 1014–1021. [Google Scholar] [CrossRef]
- Gonçalves, C.; Gonçalves, I.; Magalhães, F.; Pinto, A. Poly(lactic acid) composites containing carbon-based nanomaterials: A review. Polymers 2017, 9, 269. [Google Scholar] [CrossRef]
- Wutticharoenmongkol, P.; Sanchavanakit, N.; Pavasant, P.; Supaphol, P. Preparation and characterization of novel bone scaffolds based on electrospun polycaprolactone fibers filled with nanoparticles. Macromol. Biosci. 2006, 6, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, K.; Kotaki, M.; Ramakrishna, S. Guided bone regeneration membrane made of polycaprolactone/calcium carbonate composite nano-fibers. Biomaterials 2005, 26, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.M.; Moreira, S.; Gonçalves, I.C.; Gama, F.M.; Mendes, A.M.; Magalhães, F.D. Biocompatibility of poly(lactic acid) with incorporated graphene-based materials. Colloids Surf. B Biointerfaces 2013, 104, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.M.; Gonçalves, I.C.; Magalhães, F.D. Graphene-based materials biocompatibility: A review. Colloids Surf. B Biointerfaces 2013, 111, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xi, P.; Xie, G.; Shi, Y.; Hou, F.; Huang, L.; Chen, F.; Zeng, Z.; Shao, C.; Wang, J. Simultaneous reduction and surface functionalization of graphene oxide for hydroxyapatite mineralization. J. Phys. Chem. C 2012, 116, 3334–3341. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, J.; Chen, F.; Bai, D.; Shao, C.; Wang, J.; Xi, P.; Zeng, Z. Gelatin functionalized graphene oxide for mineralization of hydroxyapatite: Biomimetic and in vitro evaluation. Nanoscale 2014, 6, 5315–5322. [Google Scholar] [CrossRef] [PubMed]
- Depan, D.; Pesacreta, T.C.; Misra, R.D.K. The synergistic effect of a hybrid graphene oxide-chitosan system and biomimetic mineralization on osteoblast functions. Biomater. Sci. 2014, 2, 264–274. [Google Scholar] [CrossRef]
- Adolfsson, K.H.; Hassanzadeh, S.; Hakkarainen, M. Valorization of cellulose and waste paper to graphene oxide quantum dots. RSC Adv. 2015, 5, 26550–26558. [Google Scholar] [CrossRef]
- Hassanzadeh, S.; Adolfsson, K.H.; Hakkarainen, M. Controlling the cooperative self-assembly of graphene oxide quantum dots in aqueous solutions. RSC Adv. 2015, 5, 57425–57432. [Google Scholar] [CrossRef]
- Wu, D.; Xu, H.; Hakkarainen, M. From starch to polylactide and nano-graphene oxide: Fully starch derived high performance composites. RSC Adv. 2016, 6, 54336–54345. [Google Scholar] [CrossRef]
- Wu, D.; Hakkarainen, M. A closed-loop process from microwave-assisted hydrothermal degradation of starch to utilization of the obtained degradation products as starch plasticizers. ACS Sustain. Chem. Eng. 2014, 2, 2172–2181. [Google Scholar] [CrossRef]
- Hassanzadeh, S.; Aminlashgari, N.; Hakkarainen, M. Chemo-selective high yield microwave assisted reaction turns cellulose to green chemicals. Carbohydr. Polym. 2014, 112, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, S.; Aminlashgari, N.; Hakkarainen, M. Microwave-assisted recycling of waste paper to green platform chemicals and carbon nanospheres. ACS Sustain. Chem. Eng. 2015, 3, 177–185. [Google Scholar] [CrossRef]
- Hassanzadeh, S.; Adolfsson, K.H.; Wu, D.; Hakkarainen, M. Supramolecular assembly of biobased graphene oxide quantum dots controls the morphology of and induces mineralization on poly(ε-caprolactone) films. Biomacromolecules 2016, 17, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Bäckström, E.; Hakkarainen, M. Starch derived nanosized graphene oxide functionalized bioactive porous starch scaffolds. Macromol. Biosci. 2017, 17, 1600397. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Samanta, A.; Srivastava, R.K.; Hakkarainen, M. Starch-derived nanographene oxide paves the way for electrospinnable and bioactive starch scaffolds for bone tissue engineering. Biomacromolecules 2017, 18, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Oyane, A.; Kim, H.-M.; Furuya, T.; Kokubo, T.; Miyazaki, T.; Nakamura, T. Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. Part A 2003, 65A, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Chaoying, W.; Biqiong, C. Poly(ε-caprolactone)/graphene oxide biocomposites: Mechanical properties and bioactivity. Biomed. Mater. 2011, 6, 055010. [Google Scholar]
- Xu, H.; Adolfsson, K.H.; Xie, L.; Hassanzadeh, S.; Pettersson, T.; Hakkarainen, M. Zero-dimensional and highly oxygenated graphene oxide for multifunctional poly (lactic acid) bionanocomposites. ACS Sustain. Chem. Eng. 2016, 4, 5618–5631. [Google Scholar] [CrossRef]
- Yoon, O.J.; Jung, C.Y.; Sohn, I.Y.; Kim, H.J.; Hong, B.; Jhon, M.S.; Lee, N.-E. Nanocomposite nanofibers of poly(d, l-lactic-co-glycolic acid) and graphene oxide nanosheets. Compos. Part A Appl. Sci. Manuf. 2011, 42, 1978–1984. [Google Scholar] [CrossRef]
- Wang, G.; Wang, B.; Park, J.; Yang, J.; Shen, X.; Yao, J. Synthesis of enhanced hydrophilic and hydrophobic graphene oxide nanosheets by a solvothermal method. Carbon 2009, 47, 68–72. [Google Scholar] [CrossRef]
- Mit-uppatham, C.; Nithitanakul, M.; Supaphol, P. Ultrafine electrospun polyamide-6 fibers: Effect of solution conditions on morphology and average fiber diameter. Macromol. Chem. Phys. 2004, 205, 2327–2338. [Google Scholar] [CrossRef]
- Singh, R.; Pantarotto, D.; Lacerda, L.; Pastorin, G.; Klumpp, C.; Prato, M.; Bianco, A.; Kostarelos, K. Tissue biodistribution and blood clearance rates of intravenously administered carbon nanotube radiotracers. Proc. Natl. Acad. Sci. USA 2006, 103, 3357–3362. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Samulski, E.T. Synthesis of water soluble graphene. Nano Lett. 2008, 8, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Schadler, L.S.; Giannaris, S.C.; Ajayan, P.M. Load transfer in carbon nanotube epoxy composites. Appl. Phys. Lett. 1998, 73, 3842–3844. [Google Scholar] [CrossRef]
- Tang, Y.; Ye, L.; Zhang, Z.; Friedrich, K. Interlaminar fracture toughness and cai strength of fibre-reinforced composites with nanoparticles—A review. Compos. Sci. Technol. 2013, 86, 26–37. [Google Scholar] [CrossRef]
- Erdal, N.B.; Adolfsson, K.H.; Pettersson, T.R.; Hakkarainen, M. Green strategy to reduced nanographene oxide through microwave assisted transformation of cellulose. ACS Sustain. Chem. Eng. 2018, 6, 1246–1255. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, J.; Chen, F.; Hou, F.; Bai, D.; Xi, P.; Zeng, Z. Biomimetic and cell-mediated mineralization of hydroxyapatite by carrageenan functionalized graphene oxide. ACS Appl. Mater. Interfaces 2014, 6, 3132–3140. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ouyang, Z.; Ren, Z.; Li, J.; Zhang, P.; Wei, G.; Su, Z. Self-assembled peptide nanofibers on graphene oxide as a novel nanohybrid for biomimetic mineralization of hydroxyapatite. Carbon 2015, 89, 20–30. [Google Scholar] [CrossRef]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, D.; Samanta, A.; Srivastava, R.K.; Hakkarainen, M. Nano-Graphene Oxide Functionalized Bioactive Poly(lactic acid) and Poly(ε-caprolactone) Nanofibrous Scaffolds. Materials 2018, 11, 566. https://doi.org/10.3390/ma11040566
Wu D, Samanta A, Srivastava RK, Hakkarainen M. Nano-Graphene Oxide Functionalized Bioactive Poly(lactic acid) and Poly(ε-caprolactone) Nanofibrous Scaffolds. Materials. 2018; 11(4):566. https://doi.org/10.3390/ma11040566
Chicago/Turabian StyleWu, Duo, Archana Samanta, Rajiv K. Srivastava, and Minna Hakkarainen. 2018. "Nano-Graphene Oxide Functionalized Bioactive Poly(lactic acid) and Poly(ε-caprolactone) Nanofibrous Scaffolds" Materials 11, no. 4: 566. https://doi.org/10.3390/ma11040566
APA StyleWu, D., Samanta, A., Srivastava, R. K., & Hakkarainen, M. (2018). Nano-Graphene Oxide Functionalized Bioactive Poly(lactic acid) and Poly(ε-caprolactone) Nanofibrous Scaffolds. Materials, 11(4), 566. https://doi.org/10.3390/ma11040566