Geometrical Structures and Electronic Properties of Ga6 and Ga5X (X = B, C, N, O, F, Al, Si, P, S, Cl) Clusters


Abstract
1. Introduction
2. Computational Detail
3. Results and Discussion
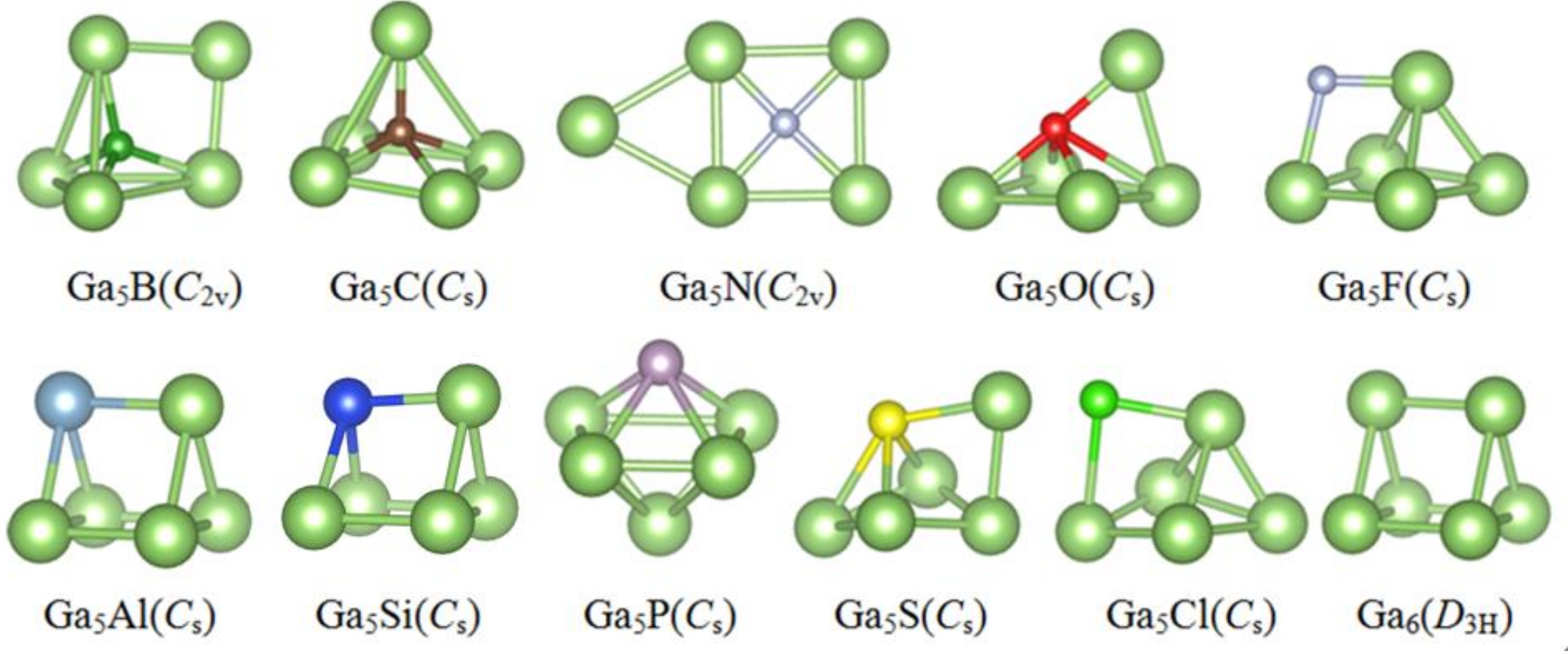
3.1. Lowest Energy Structures and Growth Pattern
3.2. Relative Stability
3.3. The HOMO-LUMO Gaps
3.4. Charge Transfer in the Ga5X Cluster
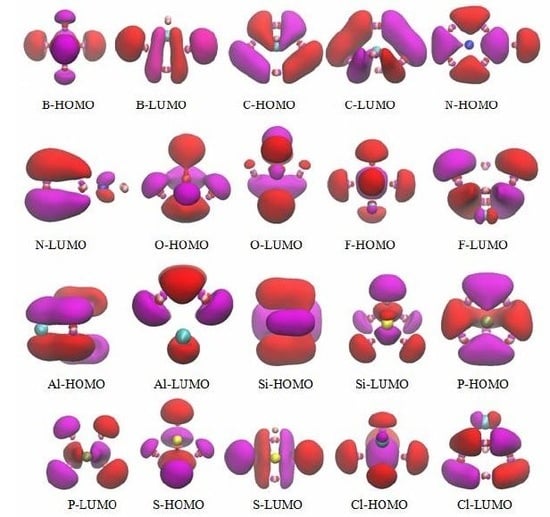
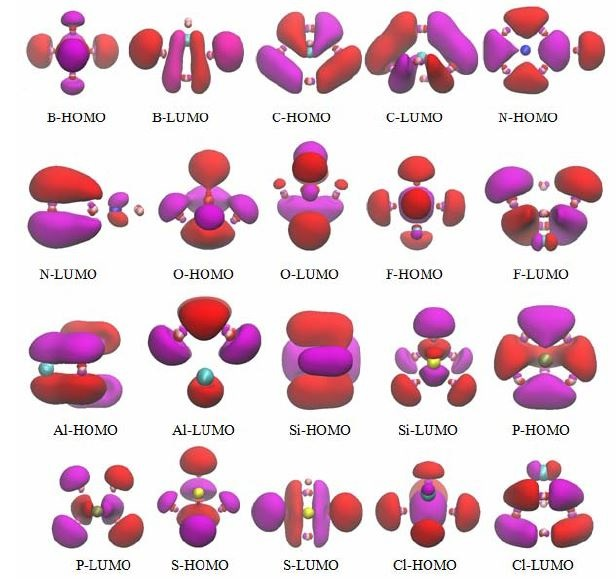
3.5. The Molecular Orbitals
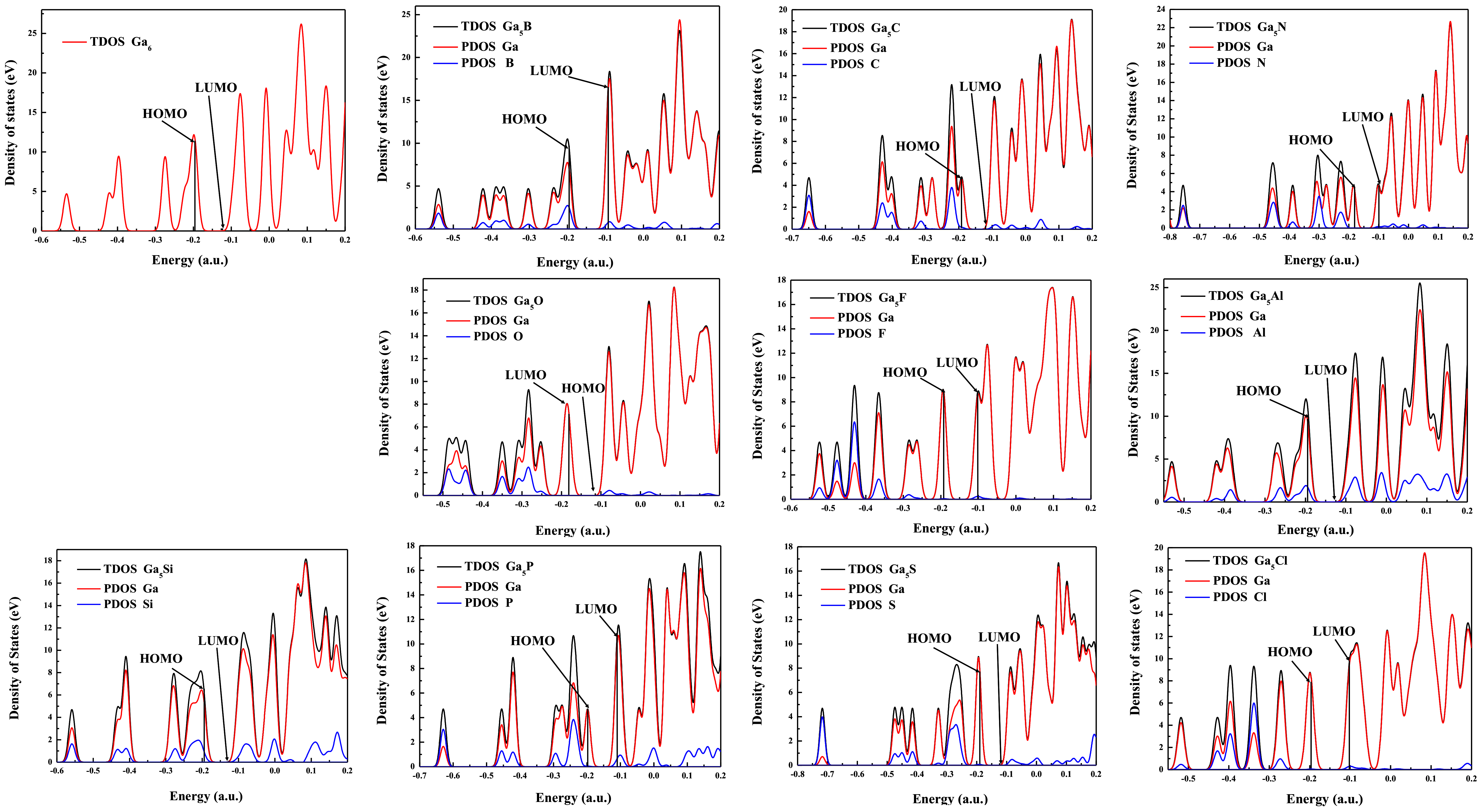
3.6. The Density of States
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Donohue, J. The Structure of the Element; Wiley: New York, NY, USA, 1974. [Google Scholar]
- Stroud, J.D.; Stott, M.J. The electronic structure and Knight shift of β-Ga. J. Phys. F Metal Phys. 1975, 5, 1667–1675. [Google Scholar] [CrossRef]
- Young, D.A. Phase Diagrams of the Elements; University of California Press: Berkeley, CA, USA, 1991. [Google Scholar]
- Song, B.; Cao, P.L. Evolution of the geometrical and electronic structures of Gan (n = 2–26) clusters: A density-functional theory study. J. Chem. Phys. 2005, 123, 144–312. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, K.; Feng, P.Y. The electronic states of Ga3. Chem. Phys. Lett. 1988, 146, 155–161. [Google Scholar] [CrossRef]
- Gong, X.G.; Tosatti, E. Structure of small gallium clusters. Phys. Lett. A 1992, 166, 369–372. [Google Scholar] [CrossRef]
- Yi, J.Y. Stability, structural transformation, and reactivity of Ga13 clusters. Phys. Rev. B 2000, 61, 7277–7279. [Google Scholar] [CrossRef]
- Belbruno, J.J. Bonding and energetics in small clusters of gallium and arsenic. Heteroat. Chem. 2003, 14, 189. [Google Scholar] [CrossRef]
- Jones, R.O. Simulated annealing study of neutral and charged clusters: Aln and Gan. J. Chem. Phys. 1993, 99, 1194–1206. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, W.G.; Li, Q.S.; Xie, Y.M.; Schaefer, H.F. Gallium clusters Gan (n = 1–6): Structures, thermochemistry, and electron affinities. J. Phys. Chem. A 2004, 108, 7448–7459. [Google Scholar] [CrossRef]
- O’Brien, S.C.; Liu, Y.; Zhang, Q.; Heath, J.R.; Tittel, F.K.; Curl, R.F.; Smalley, R.E. Supersonic cluster beams of III-V semiconductors: GaxAsy. J. Chem. Phys. 1986, 84, 4074–4079. [Google Scholar] [CrossRef]
- Cha, C.; Gantefor, G.; Eberhardt, W. The development of the 3p and 4p valence band of small aluminum and gallium clusters. J. Chem. Phys. 1994, 100, 995–1010. [Google Scholar] [CrossRef]
- And, X.T.; Dagdigian, P.J. Electronic spectrum of the gallium dimer. J. Phys. Chem. A 2003, 107, 2642–2649. [Google Scholar]
- Himmel, H.J.; Gaertner, B. Characterization of isolated Ga2 molecules by resonance Raman spectroscopy and variations of Ga-Ga bonding. Chem. Eur. J. 2004, 10, 5936–5941. [Google Scholar] [CrossRef] [PubMed]
- Balducci, G.; Gigli, G.; Meloni, G. Dissociation energies of the Ga2, In2, and GaIn molecules. J. Chem. Phys. 1998, 109, 4384–4388. [Google Scholar] [CrossRef]
- Costales, A.; Kandalam, A.K.; Pandey, R. Theoretical study of neutral and anionic group III nitride clusters: MnNn (M = Al, Ga, and In.; n = 4–6). J. Phys. Chem. B. 2003, 107, 4508–4514. [Google Scholar] [CrossRef]
- Costales, A.; Pandey, R. Density functional calculations of small anionic clusters of group III nitrides. J. Phys. Chem. A 2003, 107, 191–197. [Google Scholar] [CrossRef][Green Version]
- Song, B.; Cao, P.L. Theoretical study of structures of Ga3N3 cluster. Phys. Lett. A 2002, 300, 485–490. [Google Scholar] [CrossRef]
- Song, B.; Cao, P.L. Theoretical study of structures of Ga5N5 cluster. Phys. Lett. A 2002, 306, 57–61. [Google Scholar] [CrossRef]
- Song, B.; Cao, P.L.; Li, B.X. Theoretical study of the structure of a Ga6N6 cluster. Phys. Lett. A 2003, 315, 308–312. [Google Scholar] [CrossRef]
- Neal, C.M.; Starace, A.K.; Jarrold, M.F. Melting of alloy clusters: Effects of aluminum doping on gallium cluster melting. J. Phys. Chem. A 2007, 111, 8056–8061. [Google Scholar] [CrossRef] [PubMed]
- Guo, L. Computational investigation of GanAl (n = 1–15) clusters by the density-functional theory. Comp. Mater. Sci. 2009, 45, 951–958. [Google Scholar] [CrossRef]
- Deshpande, M.; Kanhere, D.G.; Pandey, R. Structural and electronic properties of neutral and ionic GanOn clusters with n = 4–7. J. Phys. Chem. A 2006, 37, 3814–3819. [Google Scholar] [CrossRef] [PubMed]
- Li, E.L.; Luo, X.M.; Shi, W.; Wang, X.W. Study of geometric structure, electronic state and stability of GanPm clusters. J. Mol. Struct. 2005, 723, 79–84. [Google Scholar] [CrossRef]
- Zhu, X.L. Electronic states of Ga3Si, GaSi3, and their ions. Spectrochim. Acta A 2005, 62, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.F.; Gao, T. Laser cooling of BH and GaF: Insights from an ab initio study. Phys. Chem. Chem. Phys. 2015, 17, 10830–10837. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Yao, C.H.; Cao, P.L. Density-functional study of structural and electronic properties of GanN (n = 1–19) clusters. Phys. Rev. B 2006, 74, 035306. [Google Scholar] [CrossRef]
- Reber, A.C.; Gamboa, G.U.; Khanna, S.N. The oblate structure and unexpected resistance in reactivity of Ag15+ with O2. J. Phys. Conf. Ser. 2013, 438, 012002. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lv, J.; Zhu, L.; Ma, Y.M. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef]
- Wang, Y.; Miao, M.; Lv, J.; Zhu, L.; Yin, K.; Liu, H.; Ma, Y. An effective structure prediction method for layered materials based on 2D particle swarm optimization algorithm. J. Chem. Phys. 2012, 137, 224108. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Hermann, A.; Kuang, X.; Ju, M.; Lu, C.; Jin, Y.; Xia, X.; Maroulis, G. Insights into the geometries, electronic and magnetic properties of neutral and charged palladium clusters. Sci. Rep. 2016, 6, 19656. [Google Scholar] [CrossRef] [PubMed]
- Li, C.G.; Zhang, J.; Yuan, Y.Q.; Tang, Y.N.; Ren, B.Z. Geometries, stabilities and electronic properties of copper and selenium doped copper clusters: Density functional theory study. Phys. E 2017, 86, 303–310. [Google Scholar] [CrossRef]
- Li, C.G.; Shen, Z.G.; Hu, Y.F.; Tang, Y.N.; Chen, W.G.; Ren, B.Z. Insights into the structures and electronic properties of Cun+1u and CunSu (n = 1–12; u = 0, ±1) clusters. Sci. Rep. 2017, 7, 1345. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Henry, D.J. Structures and stability of doped gallium nanoclusters. J. Phys. Chem. C 2012, 116, 24814–24823. [Google Scholar] [CrossRef]
- Froben, F.W.; Schulze, W.; Kloss, U. Raman spectra of matrix-isolated group IIIA dimmers: Ga2, In2, Tl2. Chem. Phys. Lett. 1983, 99, 500–502. [Google Scholar] [CrossRef]
- Yang, A.P.; Wen, J.Q.; Guo, P. First principles study on geometries and stabilities of small GaSin (n = 1–6) clusters. J. At. Mol. Phys. 2017, 34, 61–70. [Google Scholar]
- Drebov, N.; Weigend, F.; Ahlrichs, R. Structures and properties of neutral gallium clusters: A theoretical investigation. J. Chem. Phys. 2011, 135, 044314. [Google Scholar] [CrossRef] [PubMed]
- Li, E.L.; Chen, G.C.; Wang, X.W.; Ma, D.M.; Ma, H.; Ying, X. Ab initio investigation of structures and stability of GanNm Clusters. J. At. Mol. Phys. 2007, 24, 477–485. [Google Scholar]
- Pearson, R.G. Chemical Hardness; Applications from Molecules to Solids; Wiley-VCH: Weinheim, Germany, 1997. [Google Scholar]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford Press: Oxford, UK, 1989. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.D.; Duan, L.Y. Structural Chemistry Basis; Peking University Press: Beijing, China, 2002. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isomer | State | Sym | VIP | VEA | η | Egaps | Frequencies | |
|---|---|---|---|---|---|---|---|---|
| Lowest Frequency | Intensity Vibration | |||||||
| Ga5B | 1A1 | C2v | 6.76 | 1.16 | 5.60 | 2.879 | 36(a2) | 543(a1) |
| Ga5C | 2A″ | Cs | 6.67 | 1.95 | 4.72 | 1.934 | 15(a″) | 547(a′) |
| Ga5N | 1A1 | C2v | 6.44 | 1.56 | 4.88 | 2.241 | 27(b1) | 631(a1) |
| Ga5O | 2A′ | Cs | 6.38 | 1.79 | 4.59 | 1.812 | 46(a′) | 449(a′) |
| Ga5F | 1A′ | Cs | 6.64 | 1.49 | 5.15 | 2.360 | 60(a′) | 424(a′) |
| Ga5Al | 3A″ | Cs | 6.70 | 2.13 | 4.57 | 1.874 | 28(a″) | 170(a′) |
| Ga5Si | 2A′ | Cs | 6.79 | 2.16 | 4.63 | 1.836 | 18(a″) | 207(a″) |
| Ga5P | 1A′ | Cs | 6.88 | 1.68 | 5.2 | 2.387 | 45(a″) | 336(a′) |
| Ga5S | 2A′ | Cs | 6.65 | 2.05 | 4.6 | 1.837 | 30(a″) | 289(a′) |
| Ga5Cl | 1A′ | Cs | 6.74 | 1.45 | 5.29 | 2.536 | 44(a′) | 274(a′) |
| Ga6 | 3A1′ | D3h | 6.70 | 2.12 | 4.58 | 1.889 | 29(a1′) | 115(e′) |
| Methods | B | C | N | O | F | Al | Si | P | S | Cl |
|---|---|---|---|---|---|---|---|---|---|---|
| Hirshfeld | −0.399 | −0.436 | −0.405 | −0.366 | −0.274 | 0.001 | −0.175 | −0.224 | −0.248 | −0.177 |
| Mulliken | −0.861 | −1.350 | −1.150 | −0.822 | −0.471 | 0.254 | 0.023 | −0.061 | −0.156 | −0.262 |
| NPA | −2.865 | −1.395 | −2.179 | −0.789 | −0.755 | 0.189 | −0.332 | −0.921 | −0.503 | −0.485 |
| Bader | −1.282 | −1.816 | −1.673 | −1.343 | −0.773 | 0.350 | −0.371 | −0.857 | −0.974 | −0.591 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Ji, G.; Yao, Y.; Yuan, J.; Xu, W. Geometrical Structures and Electronic Properties of Ga6 and Ga5X (X = B, C, N, O, F, Al, Si, P, S, Cl) Clusters. Materials 2018, 11, 552. https://doi.org/10.3390/ma11040552
Hu Y, Ji G, Yao Y, Yuan J, Xu W. Geometrical Structures and Electronic Properties of Ga6 and Ga5X (X = B, C, N, O, F, Al, Si, P, S, Cl) Clusters. Materials. 2018; 11(4):552. https://doi.org/10.3390/ma11040552
Chicago/Turabian StyleHu, Yanfei, Guangfu Ji, Yachuan Yao, Jiaonan Yuan, and Weisen Xu. 2018. "Geometrical Structures and Electronic Properties of Ga6 and Ga5X (X = B, C, N, O, F, Al, Si, P, S, Cl) Clusters" Materials 11, no. 4: 552. https://doi.org/10.3390/ma11040552
APA StyleHu, Y., Ji, G., Yao, Y., Yuan, J., & Xu, W. (2018). Geometrical Structures and Electronic Properties of Ga6 and Ga5X (X = B, C, N, O, F, Al, Si, P, S, Cl) Clusters. Materials, 11(4), 552. https://doi.org/10.3390/ma11040552