Tunable Bandgap and Optical Properties of Black Phosphorene Nanotubes


Abstract
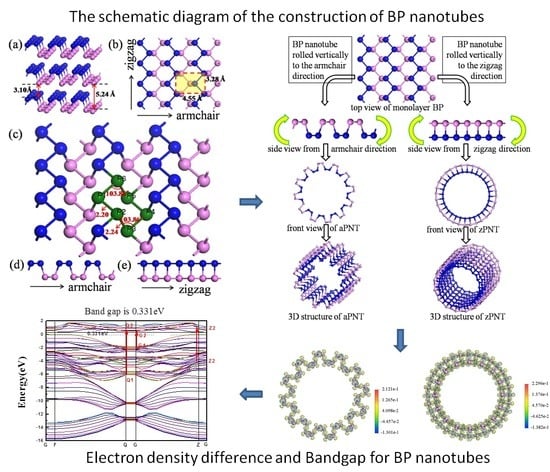
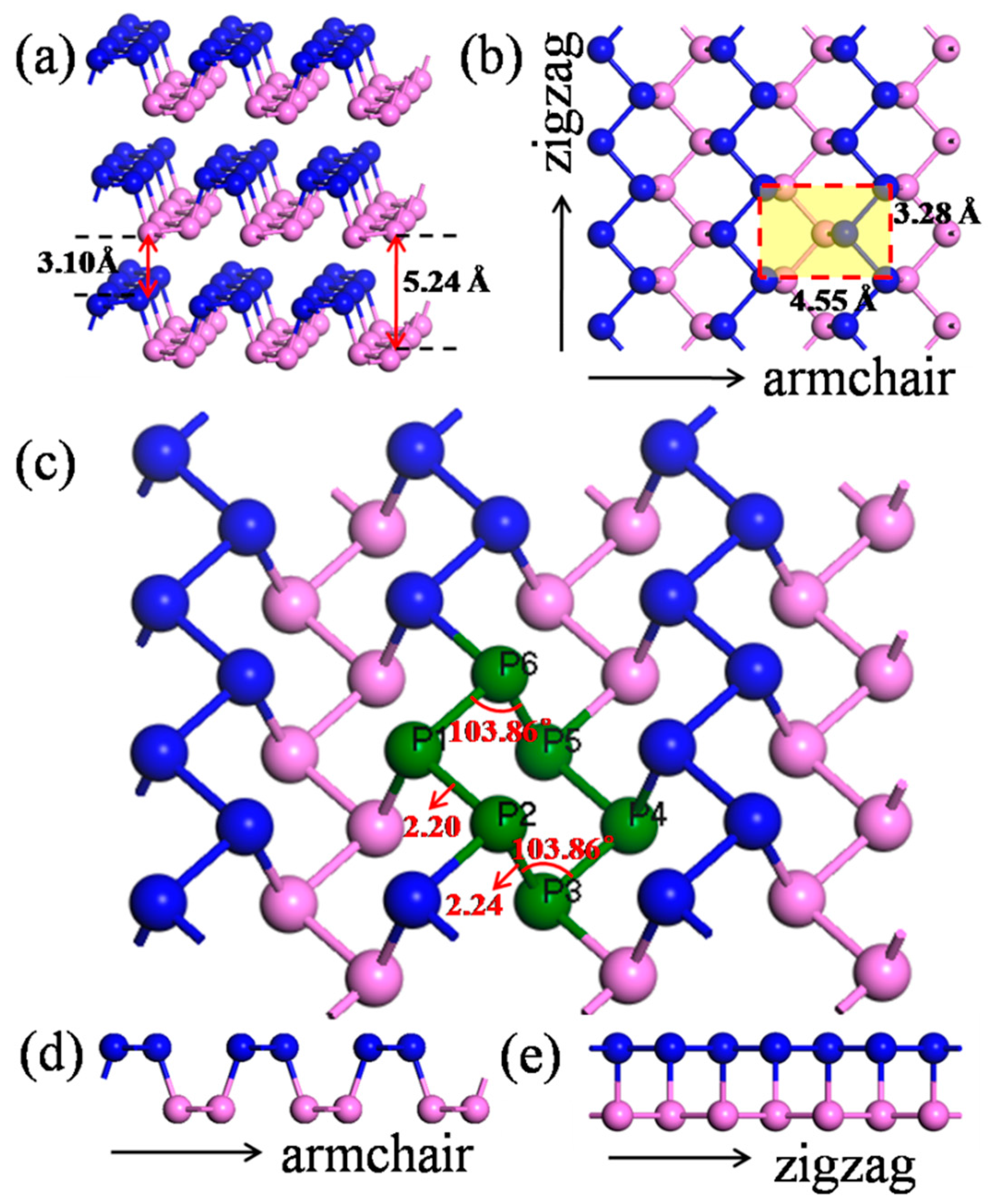
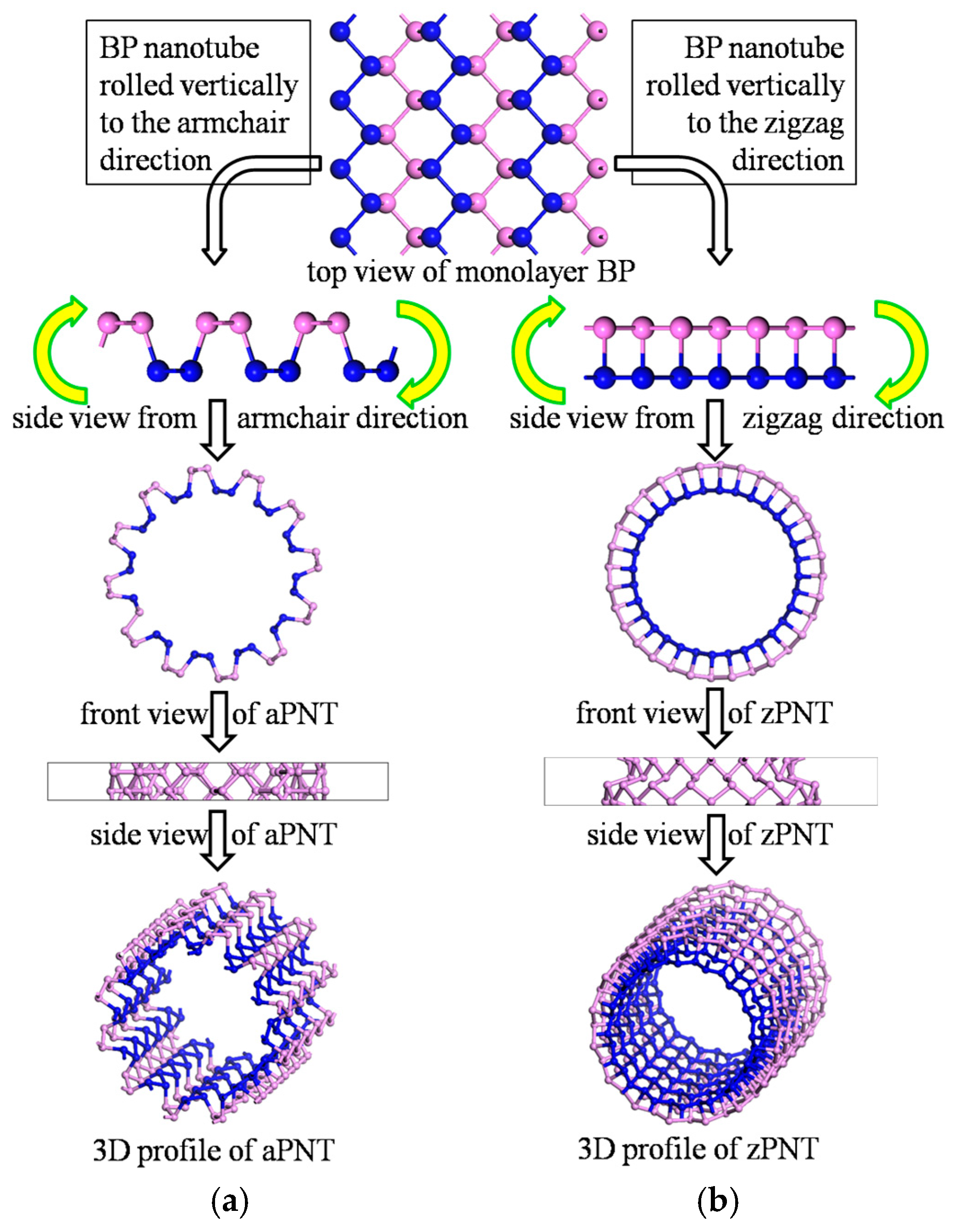
:
1. Introduction
2. Computational Models and Method
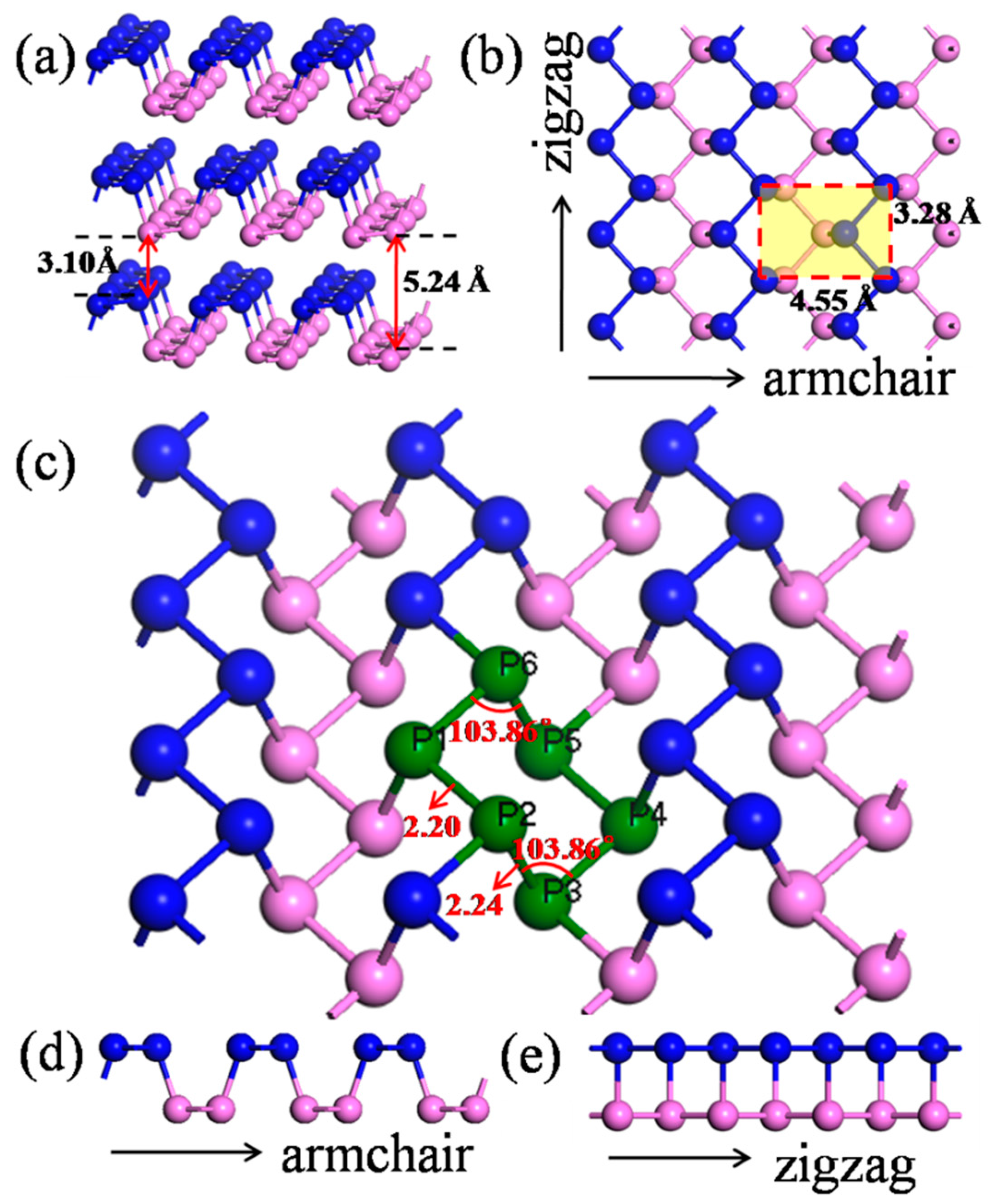
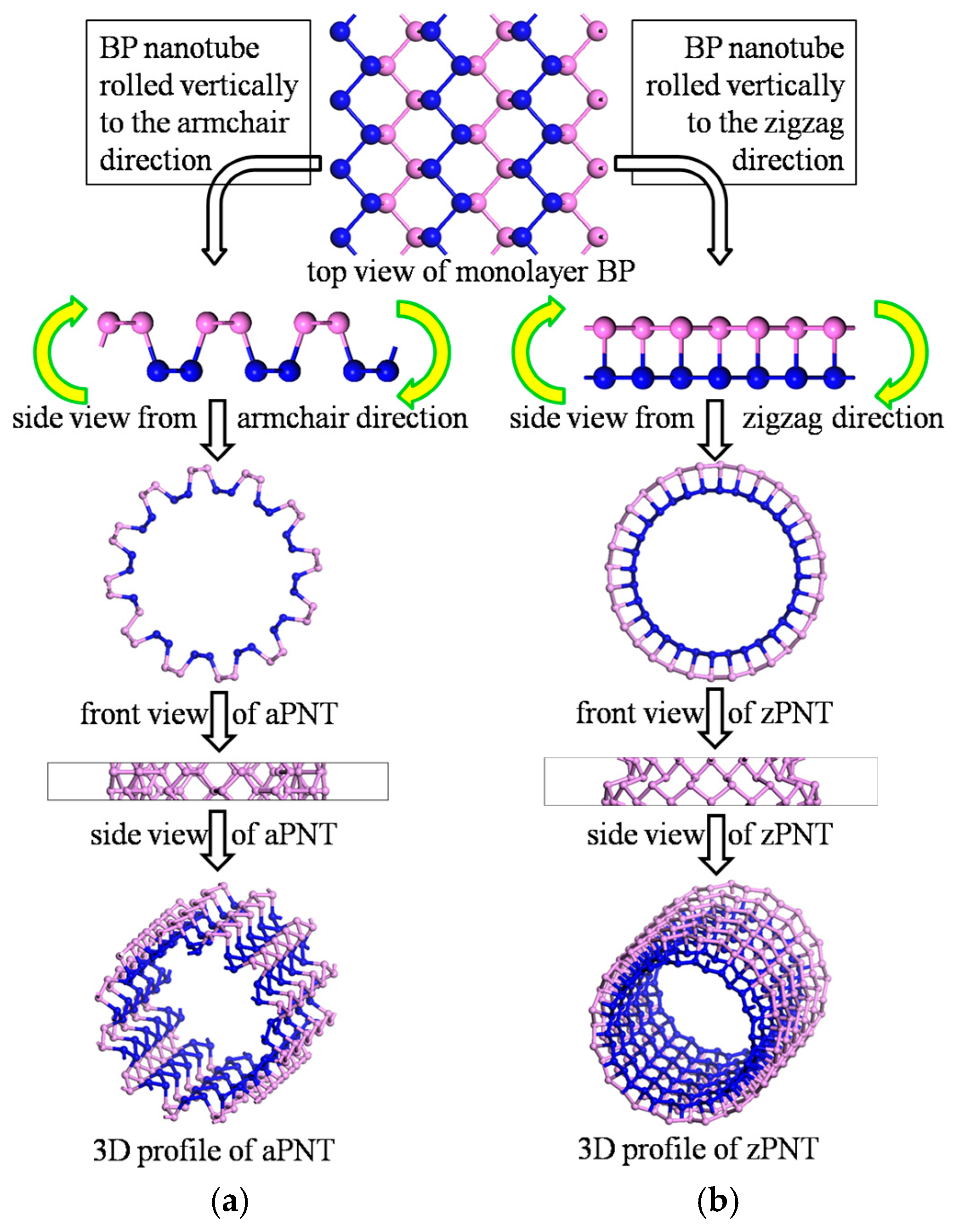
2.1. Model Construction
2.2. Energy Calculation Method
3. Results and Discussion
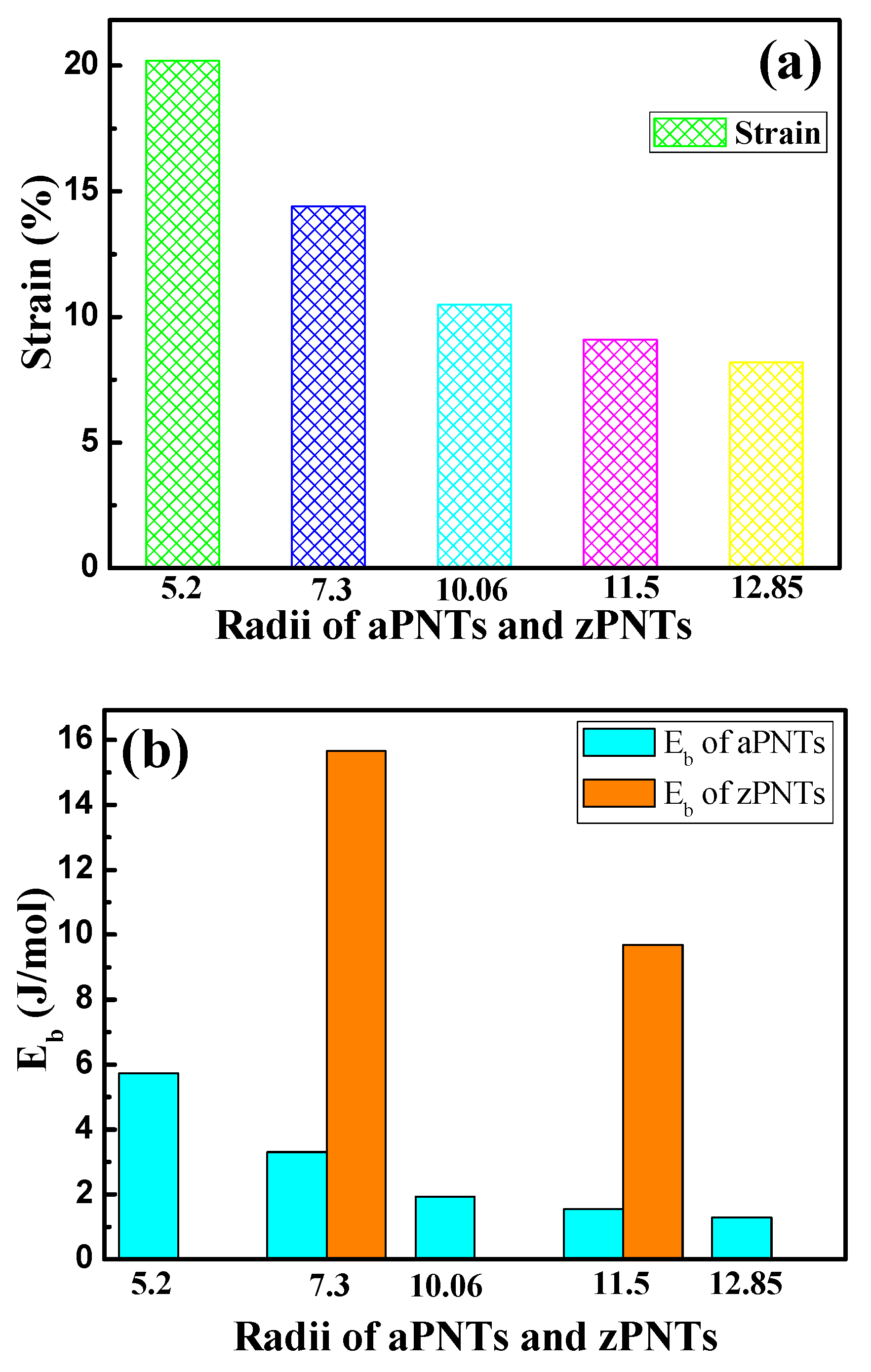
3.1. Bending Strain Energy
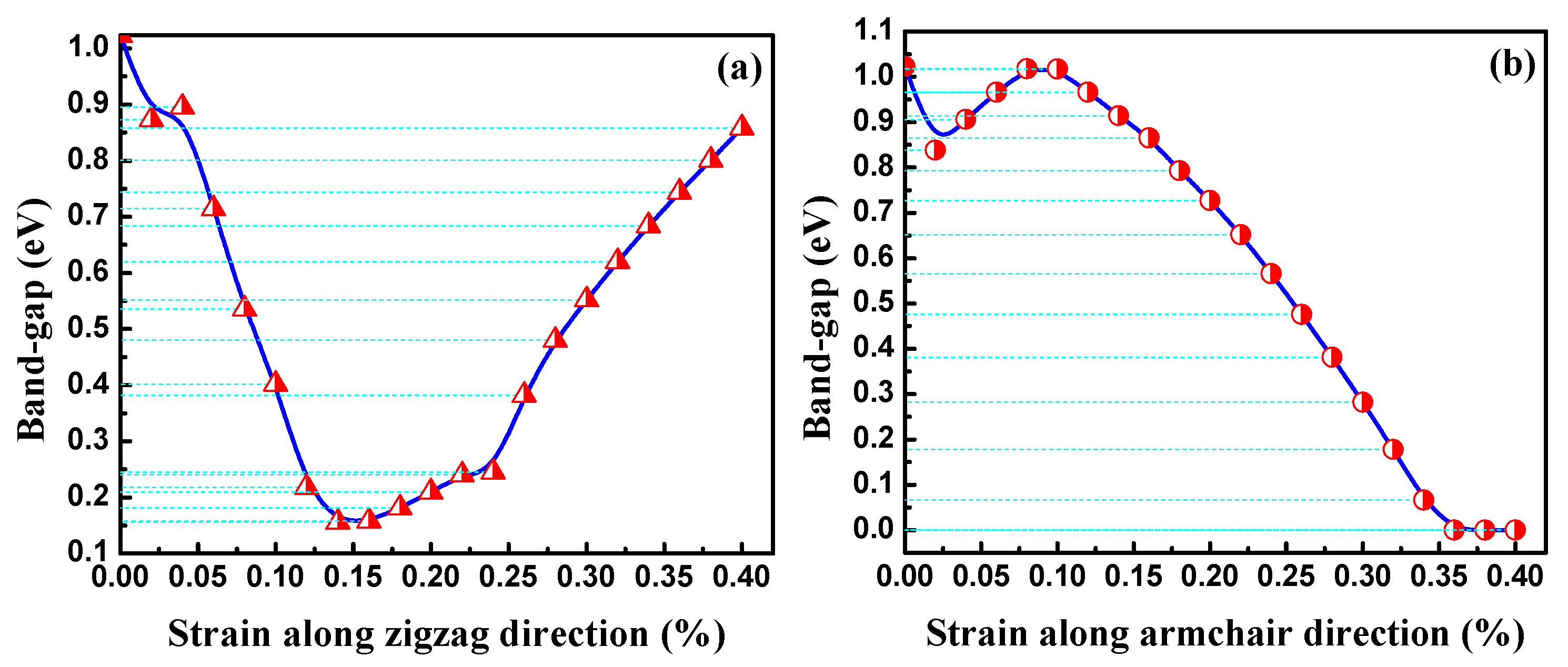
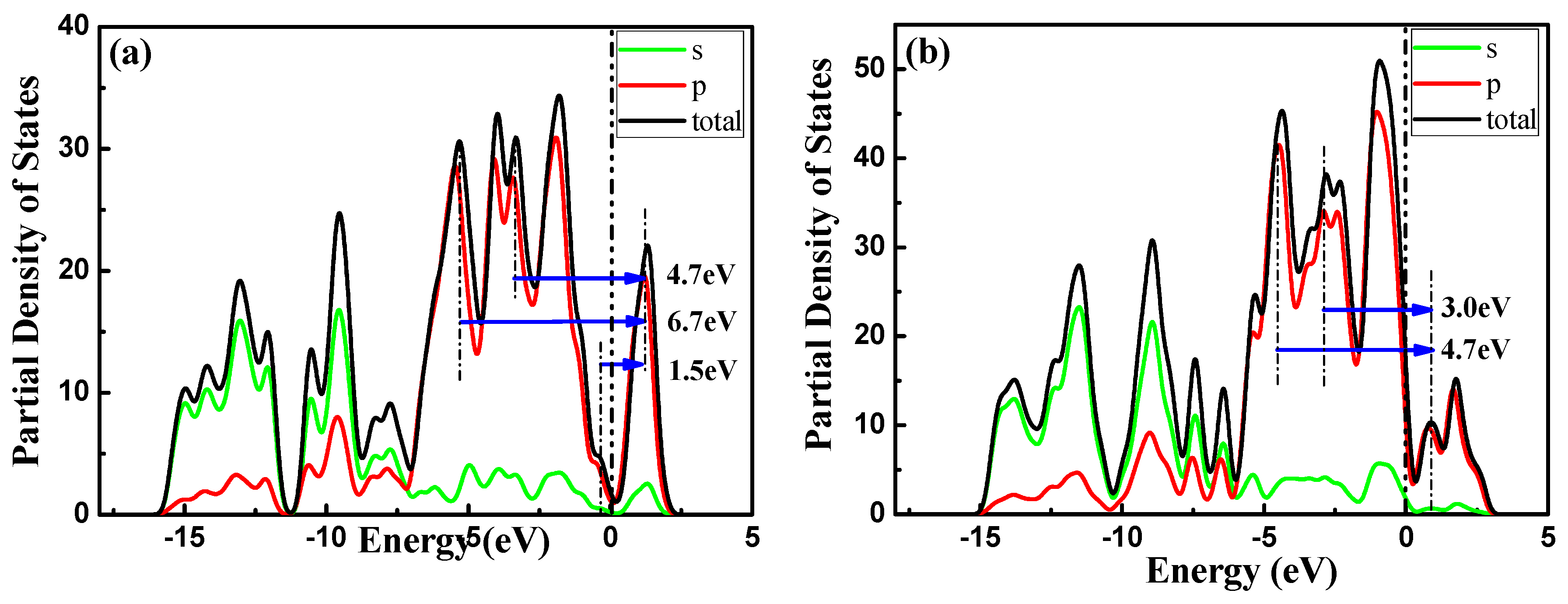
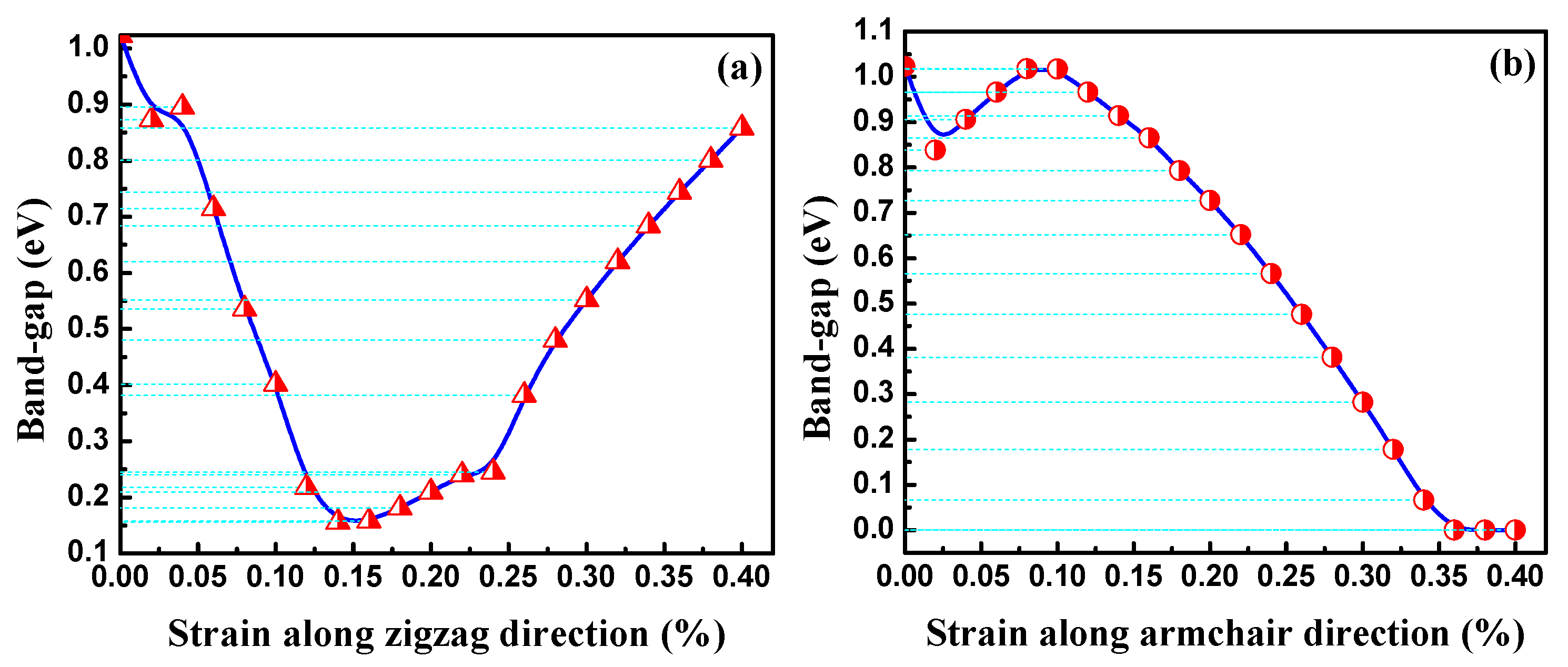
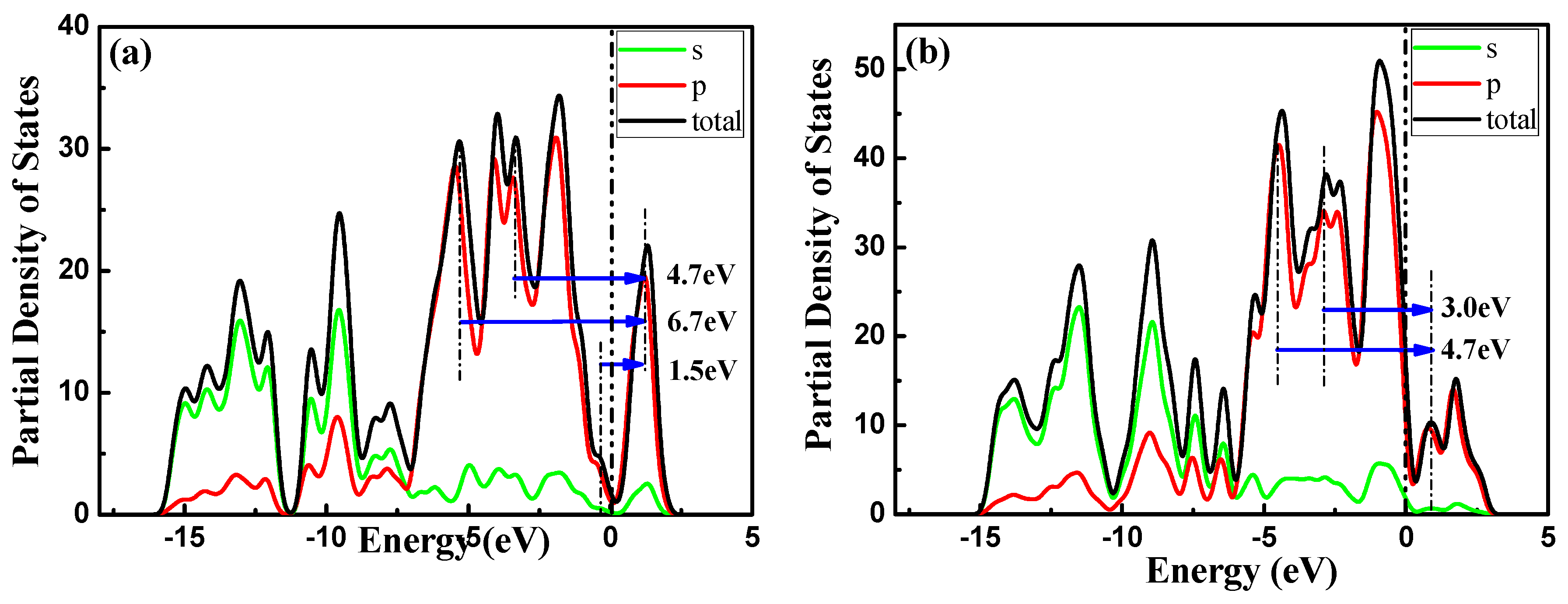
3.2. Electronic Properties
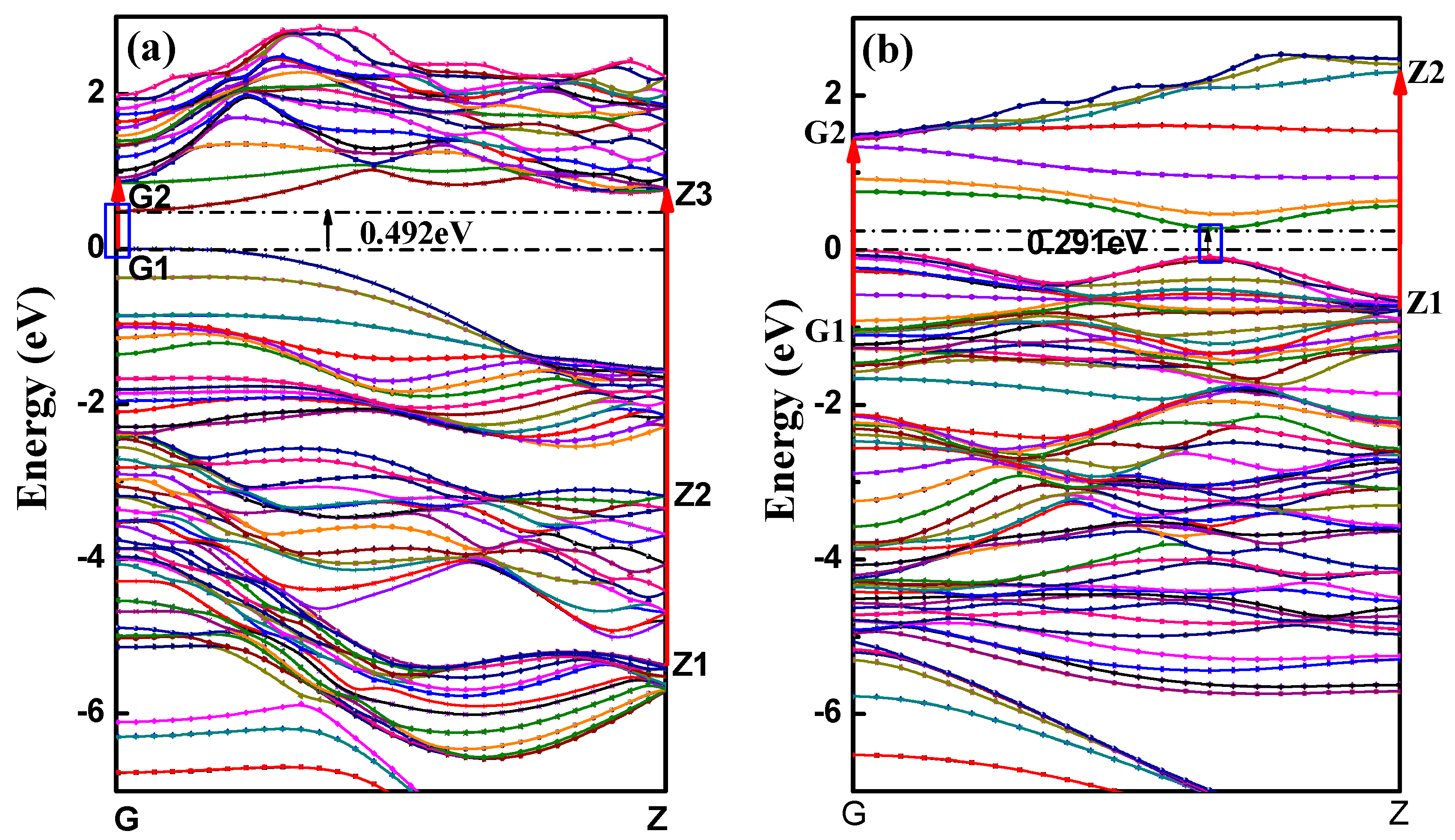
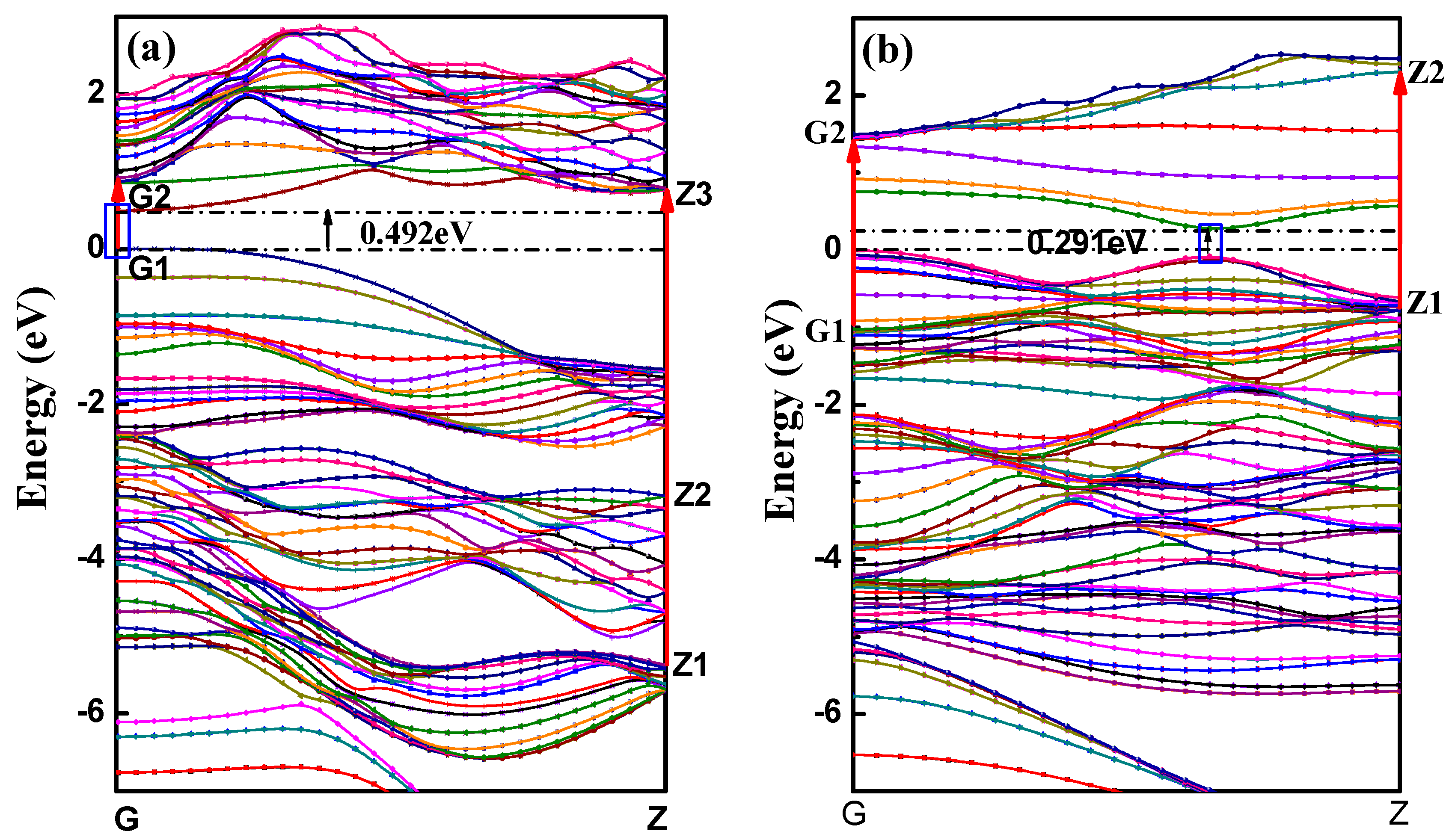
3.2.1. Bands Structure
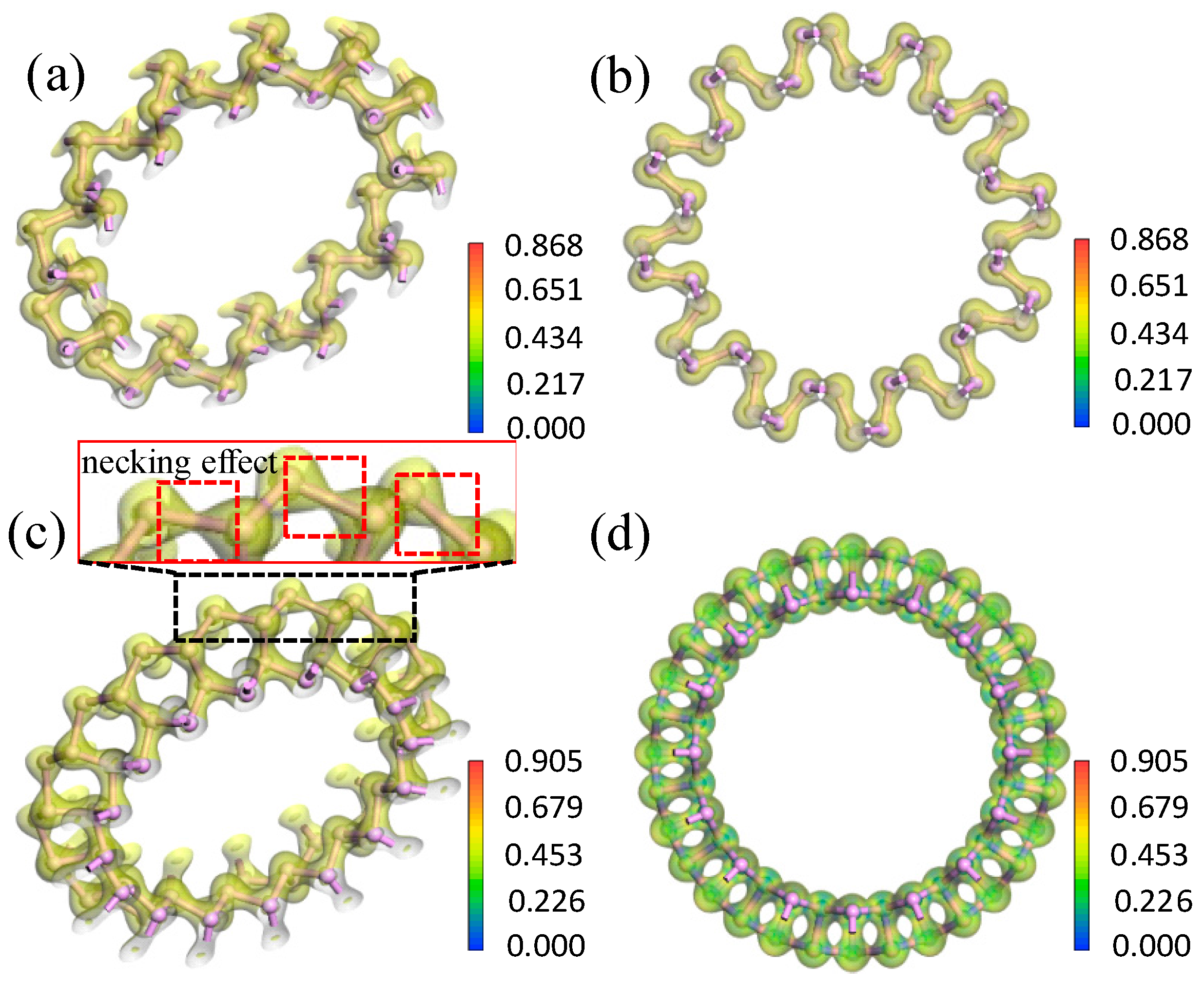
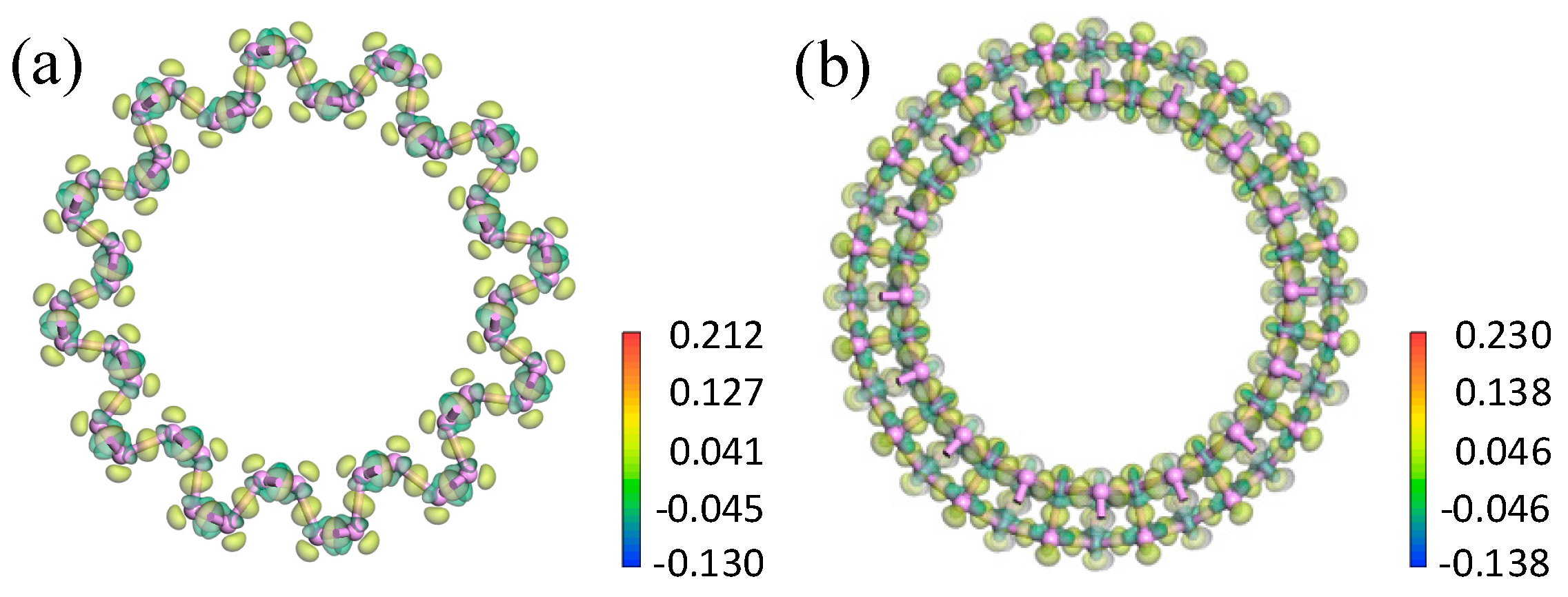
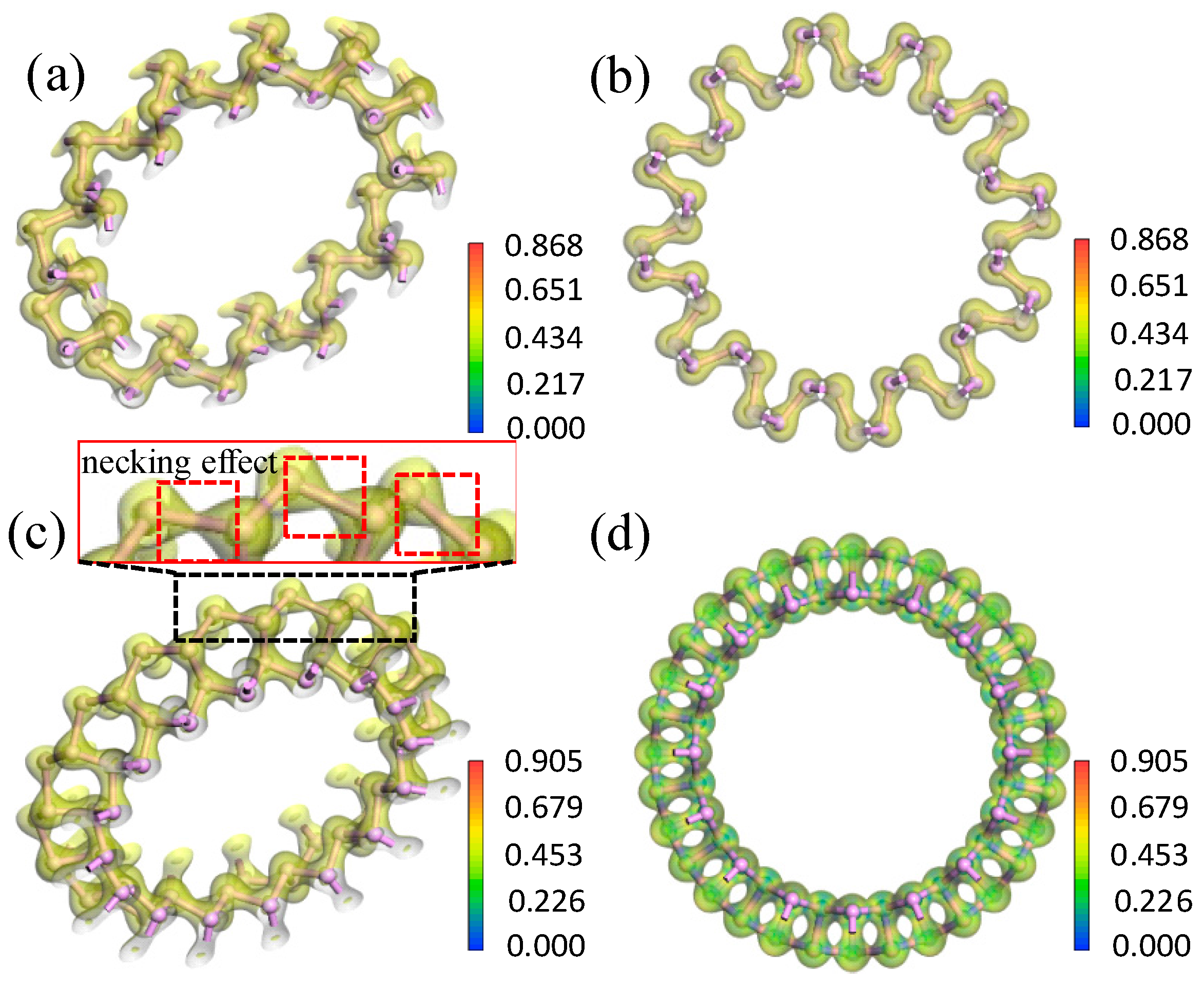
3.2.2. Electron Density and Difference in Electron Density
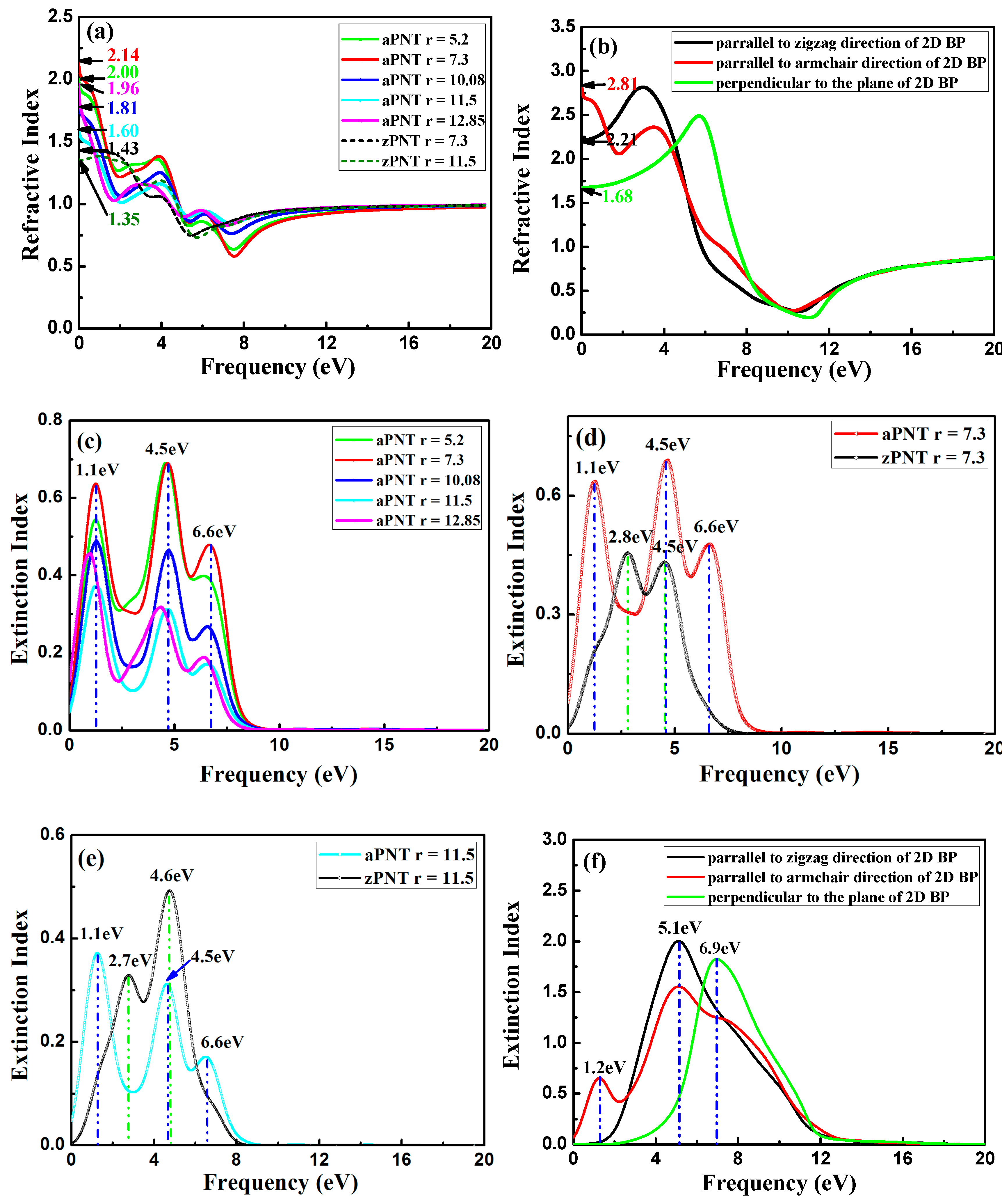
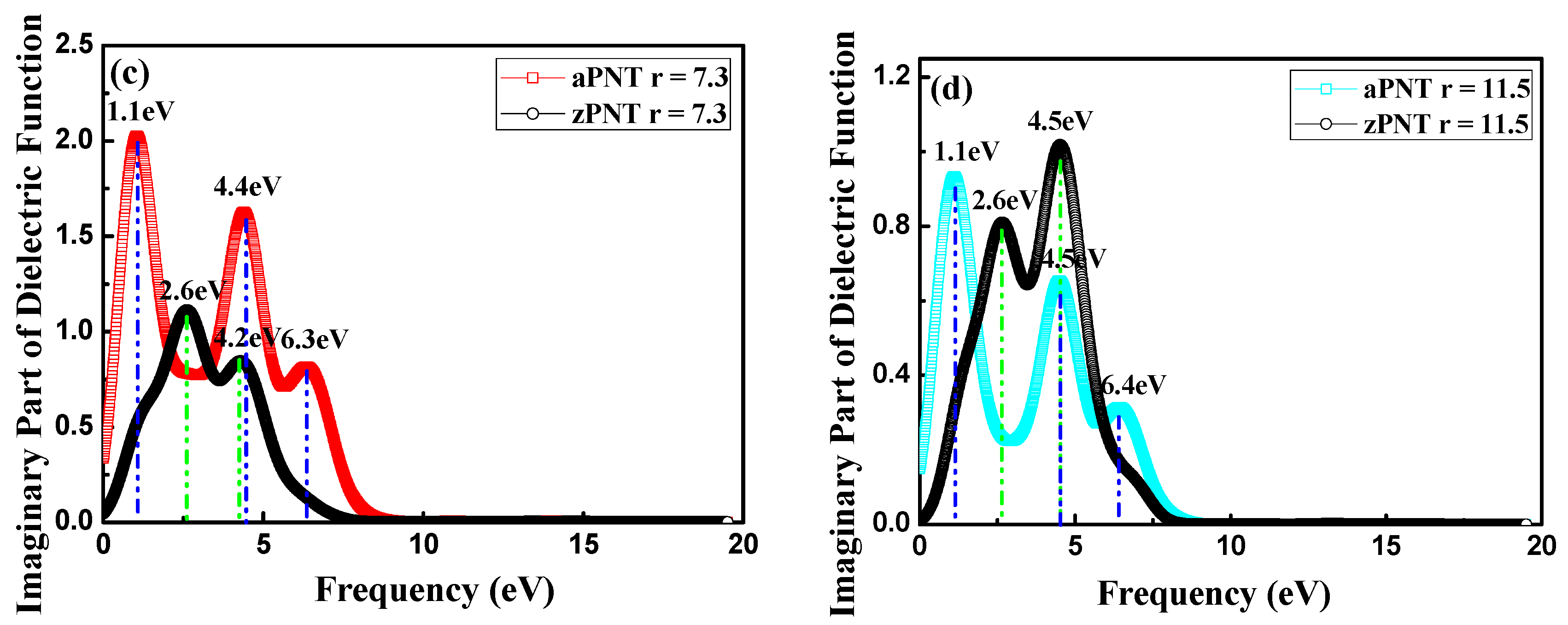
3.3. Optical Properties
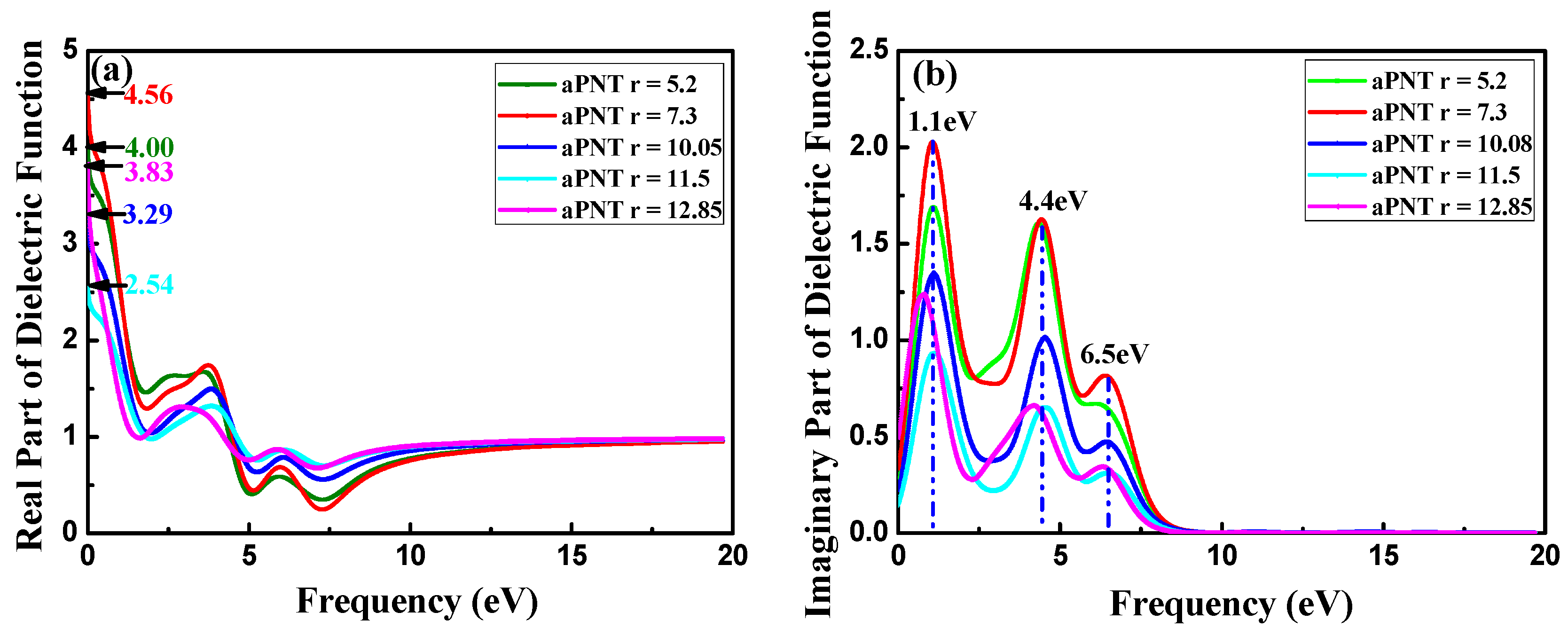
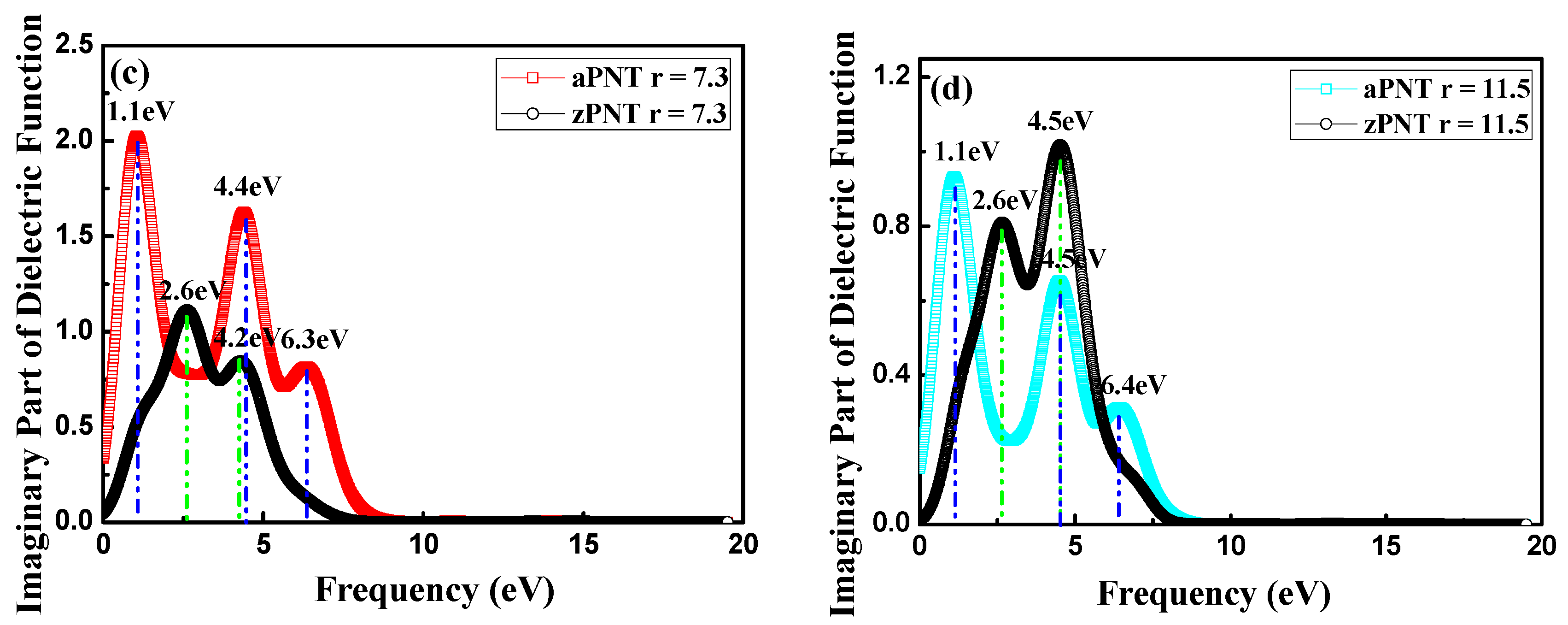
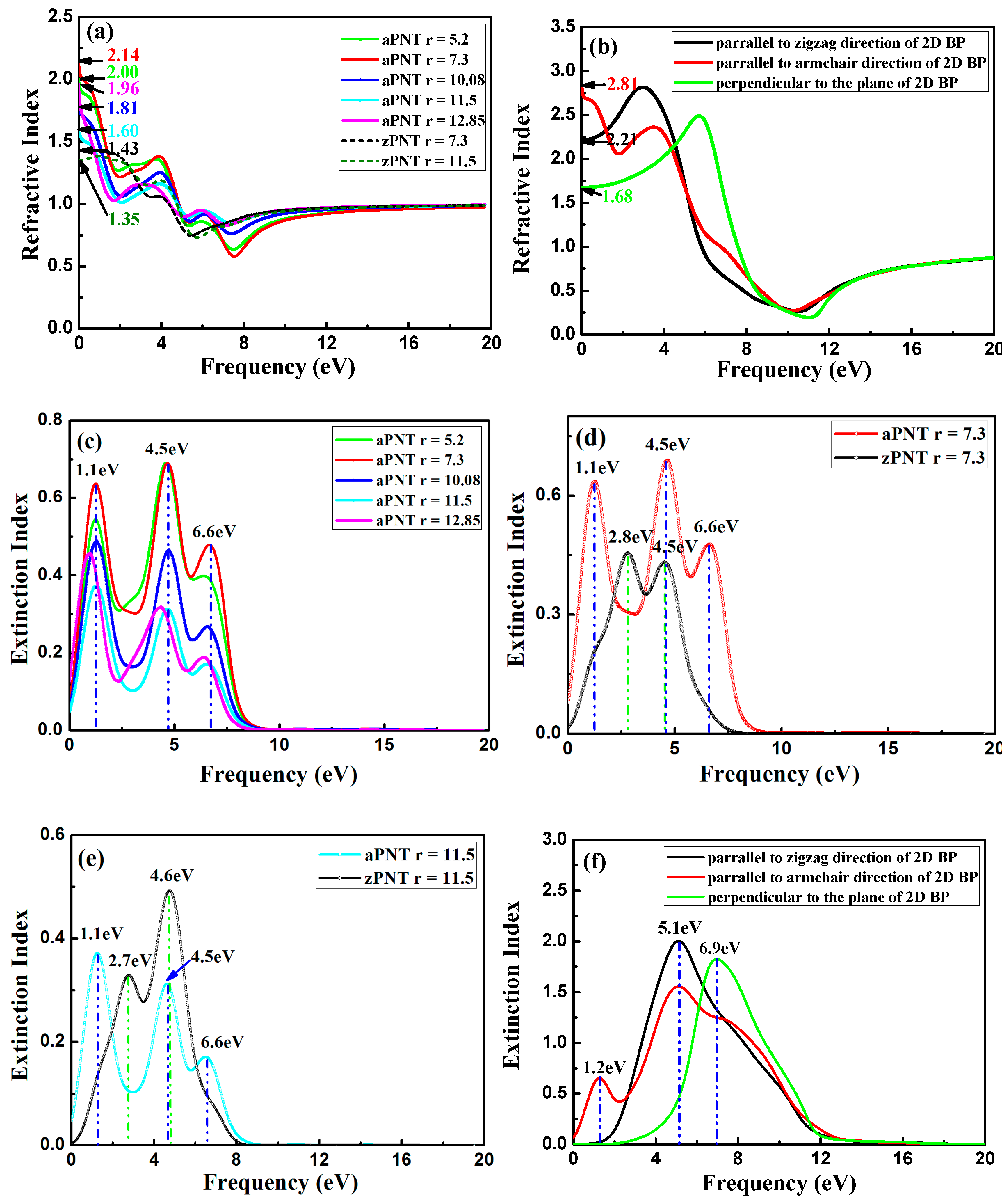
3.3.1. Complex Dielectric Constants
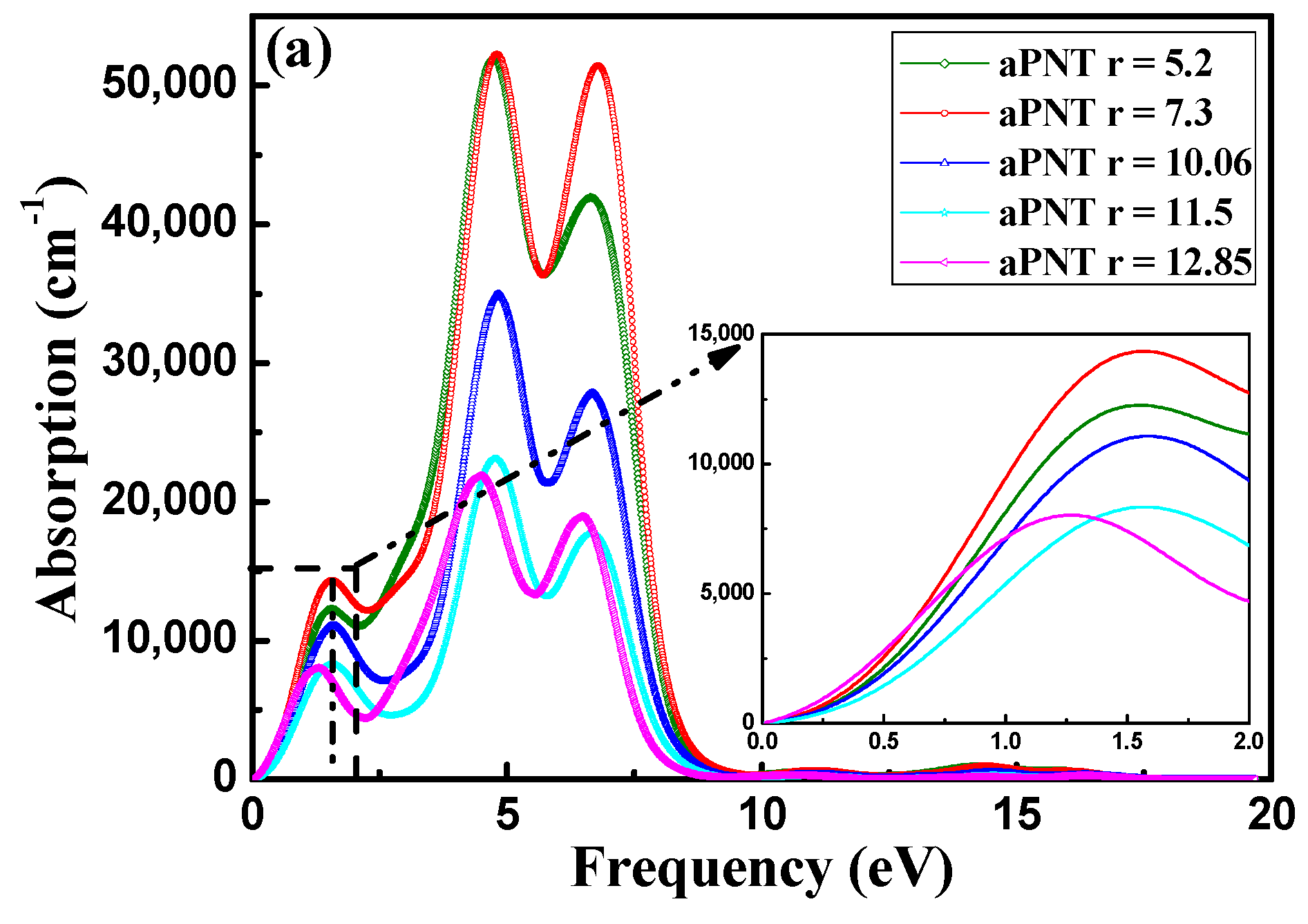
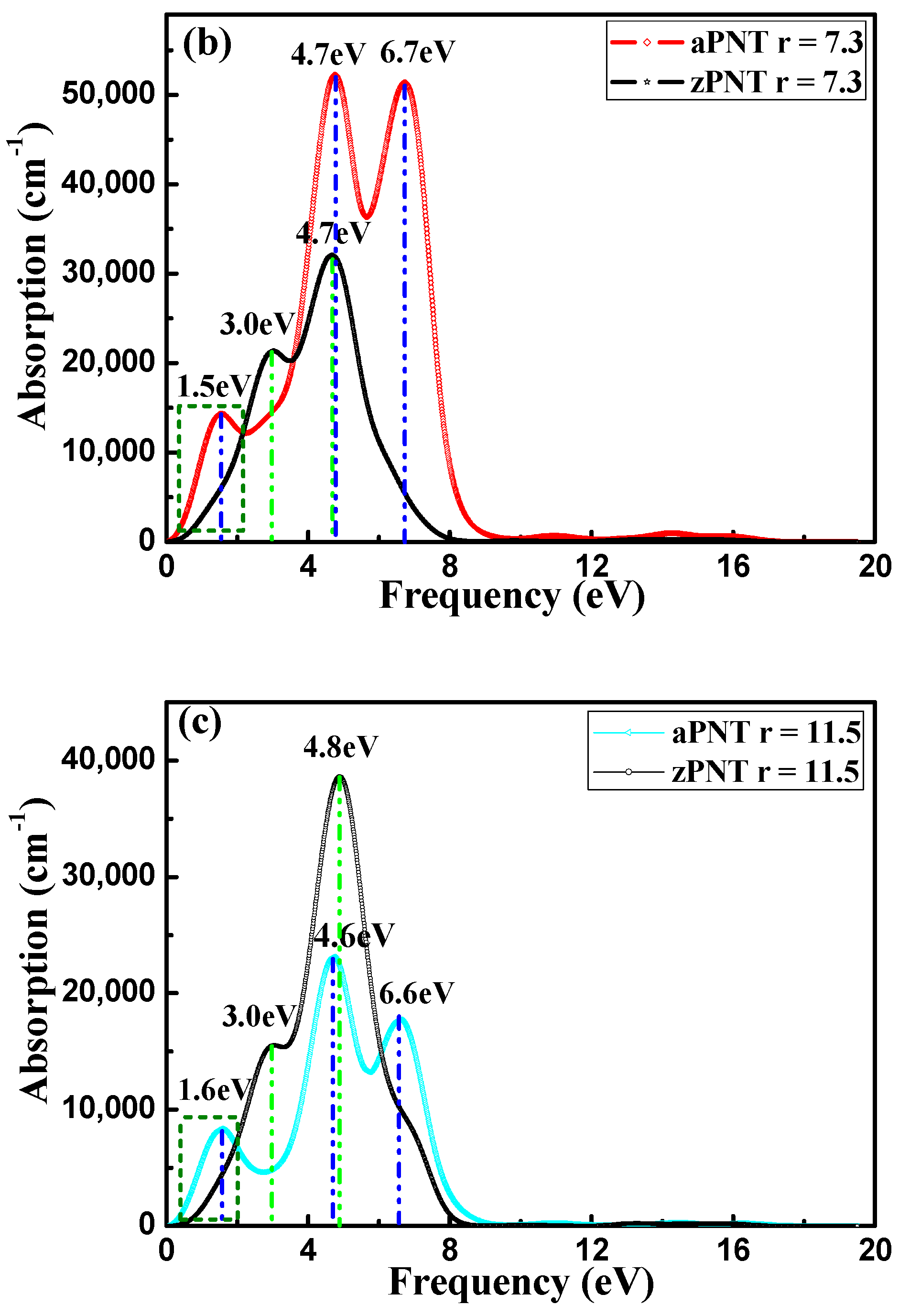
3.3.2. Absorption Coefficient
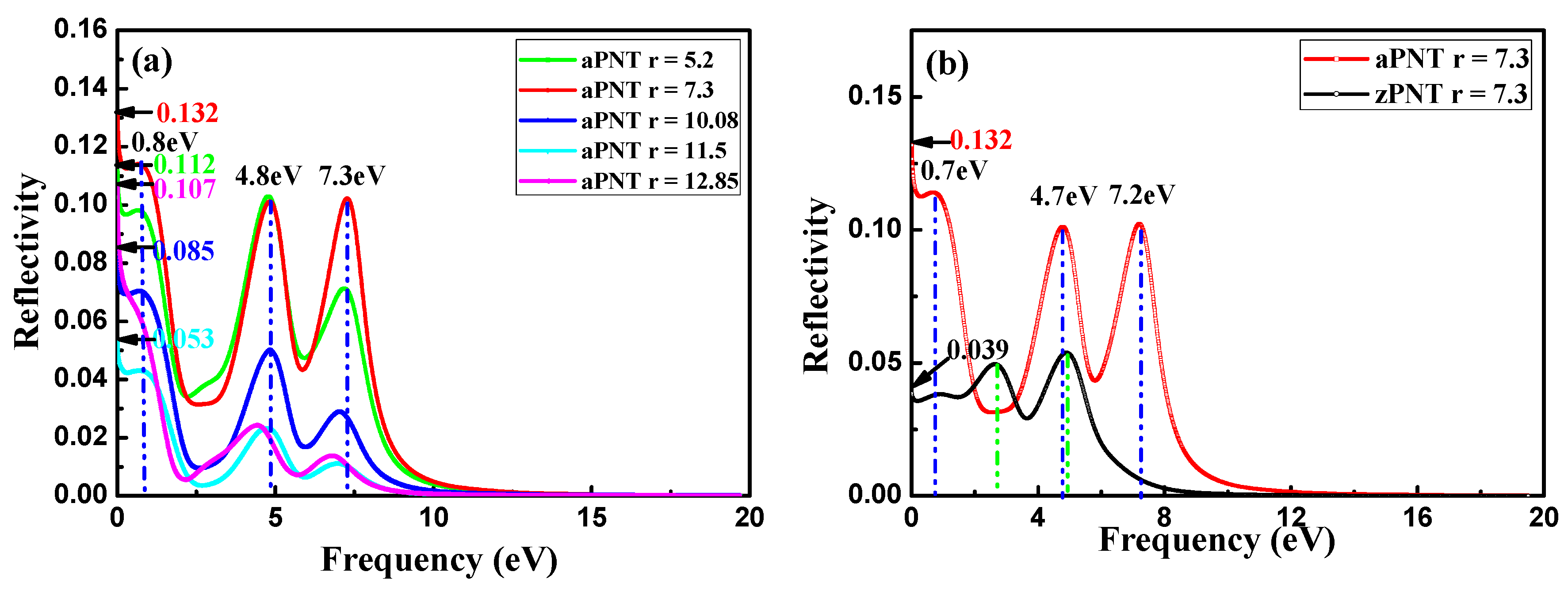
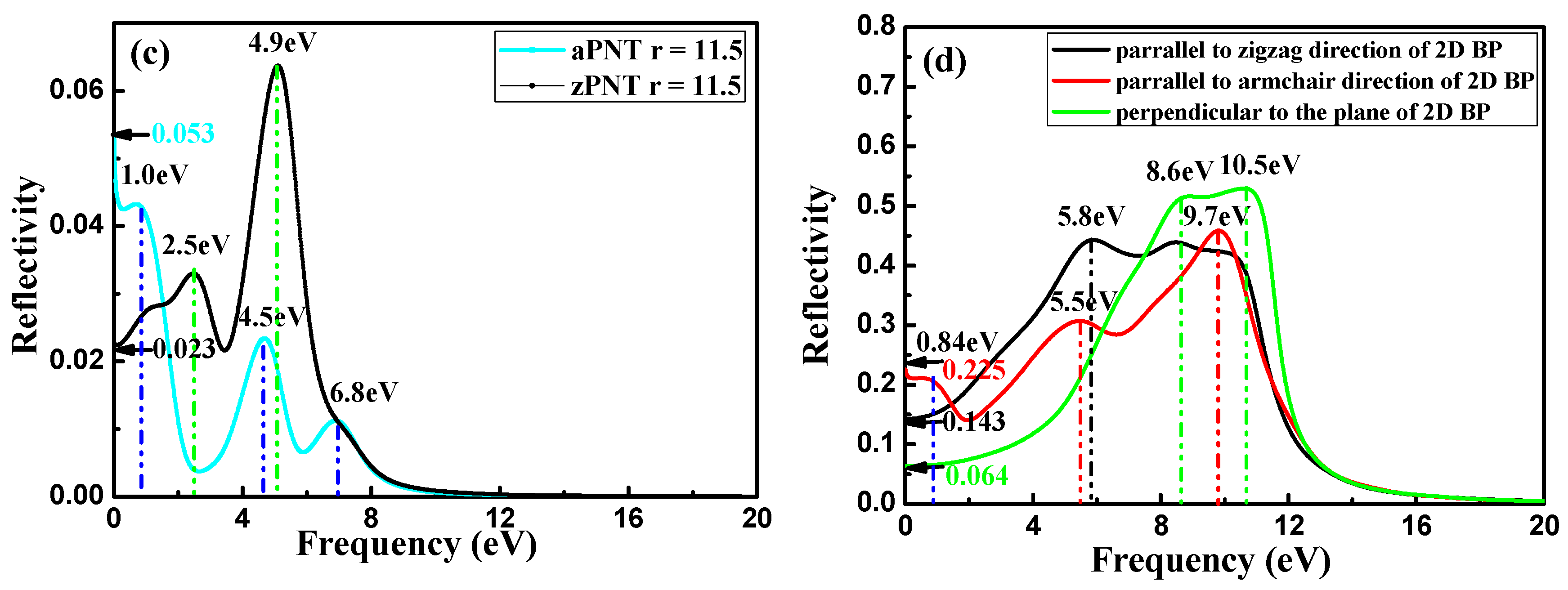
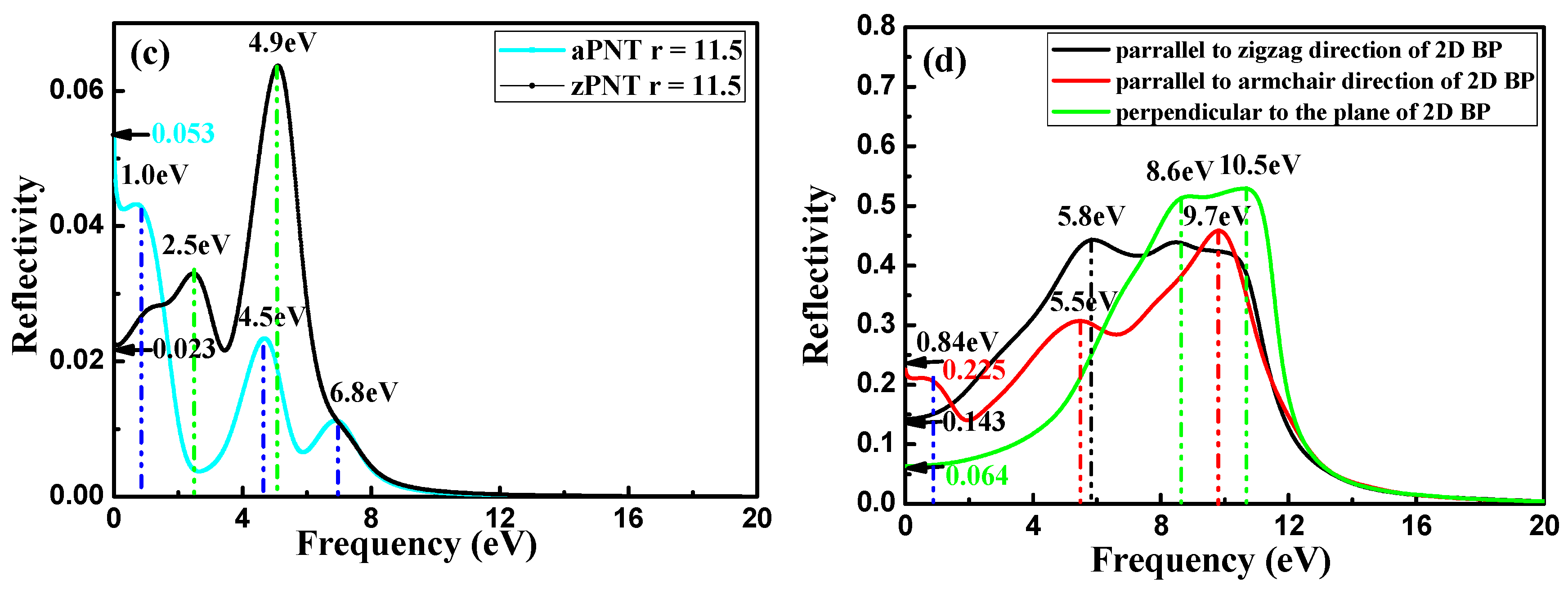
3.3.3. Reflection Coefficient
3.3.4. Refractive Index
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, K.S.; Zhao, Y.; Jang, H.; Lee, S.Y.; Kim, J.M.; Kim, K.S.; Ahn, J.H.; Kim, P.; Choi, J.; Hong, B.H. Large-scale pattern growth of graphene films for stretchable transparent electrodes. Nature 2009, 457, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Kim, S.; Jung, S.; Jang, S.; Kim, J. Suspended black phosphorus nanosheet gas sensors. Sens. Actuator B 2017, 250, 569–573. [Google Scholar] [CrossRef]
- Lopez-Sanchez, O.; Lembke, D.; Kayci, M.; Radenovic, A.; Kis, A. Ultrasensitive photodetectors based on monolayer MoS2. Nat. Nanotechnol. 2013, 8, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Schäffel, F.; Warner, J.H.; Bachmatiuk, A.; Rellinghaus, B.; Büchner, B.; Schultz, L.; Rümeli, M.H. On the catalytic hydrogenation of graphite for graphene nanoribbon fabrication. Phys. Status Solidi B 2009, 246, 2540–2544. [Google Scholar] [CrossRef]
- Guo, Y.; Robertson, J. Band offsets and metal contacts in monolayer black phosphorus. Microelectron. Eng. 2017, 178, 108–111. [Google Scholar] [CrossRef]
- Kim, J.; Huh, M.; Jung, S.W.; Ryu, S.H.; Sohn, Y.; Kim, K.S. Electronic band structure of surface-doped black phosphorus. J. Electron Spectrosc. 2017, 219, 86–91. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, W.; Luo, Z.; Zheng, Y.; Yu, Z.; Zhang, D.; Yang, J.; Tan, H.; Zhu, J.; Wang, X.; et al. Dou Functionalized few-layer black phosphorus with super-wettability towards enhanced reaction kinetics for rechargeable batteries. Nano Energy 2017, 40, 576–586. [Google Scholar] [CrossRef]
- Chen, J.H.; Jang, C.; Xiao, S.; Ishigami, M.; Fuhrer, M.S. Fuhrer Intrinsic and extrinsic performance limits of graphene devices on SiO2. Nat. Nanotechnol. 2008, 3, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Zhu, Y.F.; Jiang, Q. Bandgap Opening of Bilayer Graphene by Dual Doping from Organic Molecule and Substrate. J. Phys. Chem. C 2013, 117, 12873–12881. [Google Scholar] [CrossRef]
- Yong, K.S.; Otalvaro, D.M.; Duchemin, I.; Saeys, M.; Joachim, C. Calculation of the conductance of a finite atomic line of sulfur vacancies created on a molybdenum disulfide surface. Phys. Rev. B Condens. Matter 2008, 77, 998–1002. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nano 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Fuhrer, M.S.; James, H. Measurement of mobility in dual-gated MoS2 transistors. Nat. Nanotechnol. 2013, 8, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Allec, S.I.; Wong, B.M. Inconsistences in the electronic properties of phosphorene nanotubes: New insights from large-scale DFT calculations. J. Phys. Chem. Lett. 2016, 7, 4340–4345. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, V.; Cai, Y.; Ong, Z.; Zhang, G.; Zhang, Y.W. Recent advances in the study of phosphorene and its nanostructures. Crit. Rev. Solid State Mater. Sci. 2017, 42, 1–82. [Google Scholar] [CrossRef]
- Li, L.; Yu, Y.; Ye, G.J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nano 2014, 9, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yin, J.R.; Zhang, P.; Ding, Y.H. Strain/stress engineering on the mechanical and electronic properties of phosphorene nanosheets and nanotubes. RSC Adv. 2017, 7, 51466–54174. [Google Scholar] [CrossRef]
- Chen, W.H.; Yu, C.F.; Chen, I.C.; Cheng, H.C. Mechanical property assessment of black phosphorene nanotube using molecular dynamics simulation. Comput. Mater. Sci. 2017, 133, 35–44. [Google Scholar] [CrossRef]
- Deng, Y.X.; Luo, Z.; Conrad, N.J.; Liu, H.; Gong, Y.J.; Najmaei, S.; Ajayan, P.M.; Lou, J.; Xu, X.F.; Ye, P.D. Black Phosphorus-Monolayer MoS2 van der Waals Heterojunction p-n Diode. ACS Nano 2014, 8, 8292–8299. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Maassen, J.; Deng, Y.X.; Du, Y.C.; Garrelts, R.P.; Lundstrom, M.S.; Ye, P.D.; Xu, X.F. Anisotropic in-plane thermal conductivity observed in few-layer black phosphorus. Nat. Commun. 2015, 8572–8580. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Gomez, A.; Vicarelli, L.; Prada, E.; Island, J.O.; Narasimha-Acharya, K.L.; Blanter, S.I.; Groenendijk, D.J.; Buscema, M.; Steele, G.A.; Alvarez, J.V.; et al. Isolation and characterization of few-layer black phosphorus. 2D Mater. 2014, 1, 025001. [Google Scholar] [CrossRef]
- Liu, H.; Du, Y.; Deng, Y.; Ye, P.D. Semiconducting black phosphorus: Synthesis, transport properties and electronic applications. Chem. Soc. Rev. 2014, 44, 2732–2743. [Google Scholar] [CrossRef] [PubMed]
- Yasaei, P.; Kumar, B.; Foroozan, T.; Wang, C.H.; Asadi, M.; Tuschel, D.; Indacochea, E.J.; Klie, F.R.; Salehi-Khojin, A. High-Quality Black Phosphorus Atomic Layers by Liquid-Phase Exfoliation. Adv. Mater. 2015, 27, 1887. [Google Scholar]
- Yu, S.; Zhu, H.; Eshun, K.; Arab, A.; Badwan, A.; Li, Q.L. A computational study of the electronic properties of one-dimensional armchair phosphorene nanotubes. J. Appl. Phys. 2015, 118, 164306. [Google Scholar] [CrossRef]
- Hu, T.; Hashmi, A.; Hong, J.S. Geometry, electronic structures and optical properties of phosphorus nanotubes. Nanotechnology 2015, 26, 415702. [Google Scholar] [CrossRef] [PubMed]
- Ansari, R.; Shahnazari, A.; Rouhi, S. A density-functional-theory-based finite element model to study the mechanical properties of zigzag phosphorene nanotubes. Physica E 2017, 88, 272–278. [Google Scholar] [CrossRef]
- Appalakondaiah, S.; Vaitheeswaran, G.; Lebegue, S. Effect of van der Waals interactions on the structural and elastic properties of black phosphorus. Phys. Rev. B 2012, 86, 41–48. [Google Scholar] [CrossRef]
- Elahi, M.; Khaliji, K.; Tabatabaei, S.M. Modulation of electronic and mechanical properties of phosphorene through strain. Phys. Rev. B 2015, 91, 115412. [Google Scholar] [CrossRef]
- Cartz, L.; Srinivasa, S.R.; Riedner, R.J. Effect of pressure on bonding in black phosphorus. J. Chem. Phys. 1979, 71, 1718–1721. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Pederson, M.; Perdew, J. Fully self-consistent Fermi-orbital self-interaction correction in density-functional theory. Bull. Am. Phys. Soc. 2017, 62, 052505–052514. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Crystal System | Space Group | Lattice Parameter (Å) | Bond Length (Å) | Interlayer Spacing (Å) | |||
|---|---|---|---|---|---|---|---|---|
| Armchair Direction | Zigzag Direction | P1–P2 | P2–P3 | Nearest Atomic | Layer-to-Layer Distance | |||
| This work | Orthorhombic | CMCA | 4.55 | 3.28 | 2.20 | 2.24 | 3.10 | 5.24 |
| Other work | - | - | 4.58 [14] 4.54 [26] 4.54 [27] | 3.32 [14] 3.28 [26] 3.31 [27] | - | - | 3.20 [14] | - |
| Experimental value | - | - | 4.37 [28] | 3.31 [28] | - | - | - | - |
| Modes | r (Å) | Strain | Atom Number | E/Atom (eV) | Eb (eV/atom) | Eb (KJ/mol) | Ef (eV/atom) | Ef (KJ/mol) | Band Gap (eV) |
|---|---|---|---|---|---|---|---|---|---|
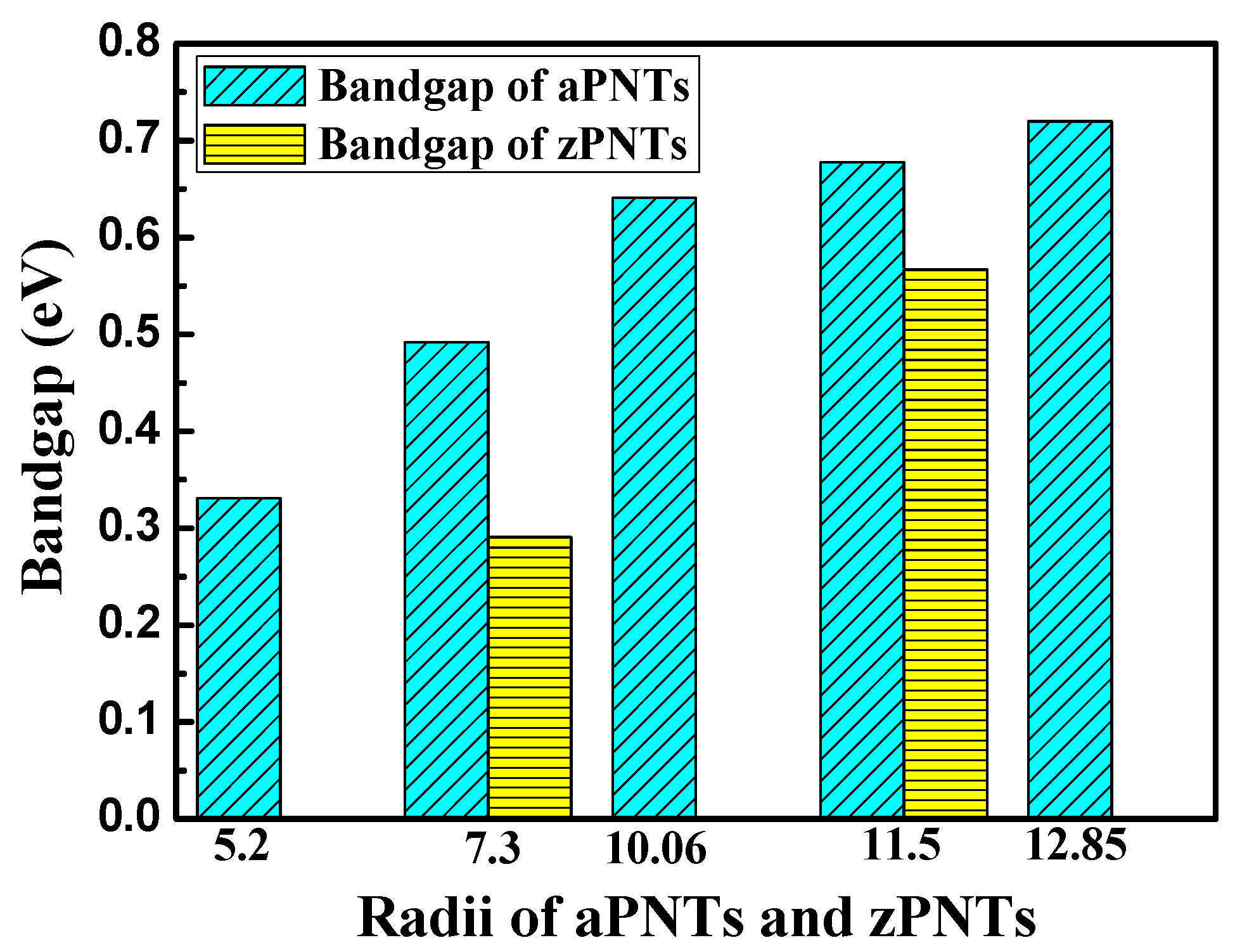
| aPNT | 5.2 | 20.2% | 36 | −179.60 | 0.060 | 5.73 | 5.22 | 502.13 | 0.331 |
| aPNT | 7.3 | 14.4% | 48 | −179.62 | 0.034 | 3.30 | 5.24 | 504.05 | 0.492 |
| aPNT | 10.06 | 10.5% | 64 | −179.64 | 0.020 | 1.94 | 5.26 | 505.97 | 0.641 |
| aPNT | 11.5 | 9.1% | 72 | −179.65 | 0.016 | 1.55 | 5.27 | 506.93 | 0.678 |
| aPNT | 12.85 | 8.2% | 80 | −179.66 | 0.014 | 1.29 | 5.28 | 507.89 | 0.720 |
| zPNT | 7.3 | 14.4% | 64 | −179.49 | 0.163 | 15.66 | 5.11 | 491.55 | 0.291 |
| zPNT | 11.5 | 9.1% | 96 | −179.56 | 0.101 | 9.69 | 5.18 | 498.28 | 0.567 |
| Bonds | Planar | Bond Population | aPNT | Deviation (%) | Bond Population | zPNT | Deviation (%) | Bond Population |
|---|---|---|---|---|---|---|---|---|
| P1–P2 | 2.20 | 0.84 | 2.23 | 1.3 | 0.87 | 2.41 | 9.2 | 0.32 |
| P2–P3 | 2.24 | 0.40 | 2.25 | 0.4 | 0.34 | 2.20 | −2.1 | 0.43 |
| P3–P4 | 2.20 | 0.84 | 2.19 | −0.7 | 1.05 | 2.15 | −2.2 | 0.36 |
| P1–P2–P3 | 103.86 | - | 100.09 | −3.6 | - | 96.93 | −6.7 | - |
| P3–P4–P5 | 103.86 | - | 109.13 | 5.1 | - | 107.57 | 3.6 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Xie, Z.; Chen, Z.; Cheng, N.; Wang, J.; Zhu, G. Tunable Bandgap and Optical Properties of Black Phosphorene Nanotubes. Materials 2018, 11, 304. https://doi.org/10.3390/ma11020304
Li C, Xie Z, Chen Z, Cheng N, Wang J, Zhu G. Tunable Bandgap and Optical Properties of Black Phosphorene Nanotubes. Materials. 2018; 11(2):304. https://doi.org/10.3390/ma11020304
Chicago/Turabian StyleLi, Chunmei, Zhongjing Xie, Zhiqian Chen, Nanpu Cheng, Jinghui Wang, and Guoan Zhu. 2018. "Tunable Bandgap and Optical Properties of Black Phosphorene Nanotubes" Materials 11, no. 2: 304. https://doi.org/10.3390/ma11020304
APA StyleLi, C., Xie, Z., Chen, Z., Cheng, N., Wang, J., & Zhu, G. (2018). Tunable Bandgap and Optical Properties of Black Phosphorene Nanotubes. Materials, 11(2), 304. https://doi.org/10.3390/ma11020304