1. Introduction
The reciprocal influence of an electron-donating and an electron-accepting group is capable to give rise to structures with fascinating properties. Optically, the first manifestation of this mutual interaction is a color change when the two partners are mixed, resulting in the presence of an additional absorption band which is not detectable in the absorption spectra of the initial partners considered separately. This absorption band corresponds to the charge transfer (CT) interaction where part of the electronic density is transferred from the electron-rich to the electron-deficient partner. Depending on the fact that the two moieties are (or not) connected to each other, this CT transition can be intermolecular or intramolecular. Historically, intermolecular charge transfer complexes have been extensively studied, focused on the metallic conductivity resulting from the grinding of two insulating organic compounds, i.e., tetrathiafulvalene (TTF) and tetracyano-quinodimethane (TCNQ) [
1]. Following this pioneering work, the possibility to develop superconductors at low temperatures has driven extensive research efforts, and the emergence of the Bechgaard salts [
2]. If these structures are attractive for their electrical properties, the resulting charge transfer complex renders these structures insoluble, drastically limiting the scope of applicability. Conversely, materials exhibiting an intramolecular charge transfer (ICT) i.e., meaning that the electron donor is connected to the electron acceptor by mean of a spacer (aliphatic or conjugated) are not necessarily insoluble so that these structures found widespread applications ranging from dye-sensitized solar cells (DSSCs) [
3], nonlinear optical applications [
4,
5], solvatochromic probes [
6,
7,
8], photochromes [
9], single component semiconductors [
10,
11,
12], electrochromes [
13,
14], and piezochromes [
15]. Typically, these compounds are composed of a π-conjugated system with an electron-donating, and an electron-accepting part connected at both sides of the conjugated spacer [
16,
17,
18,
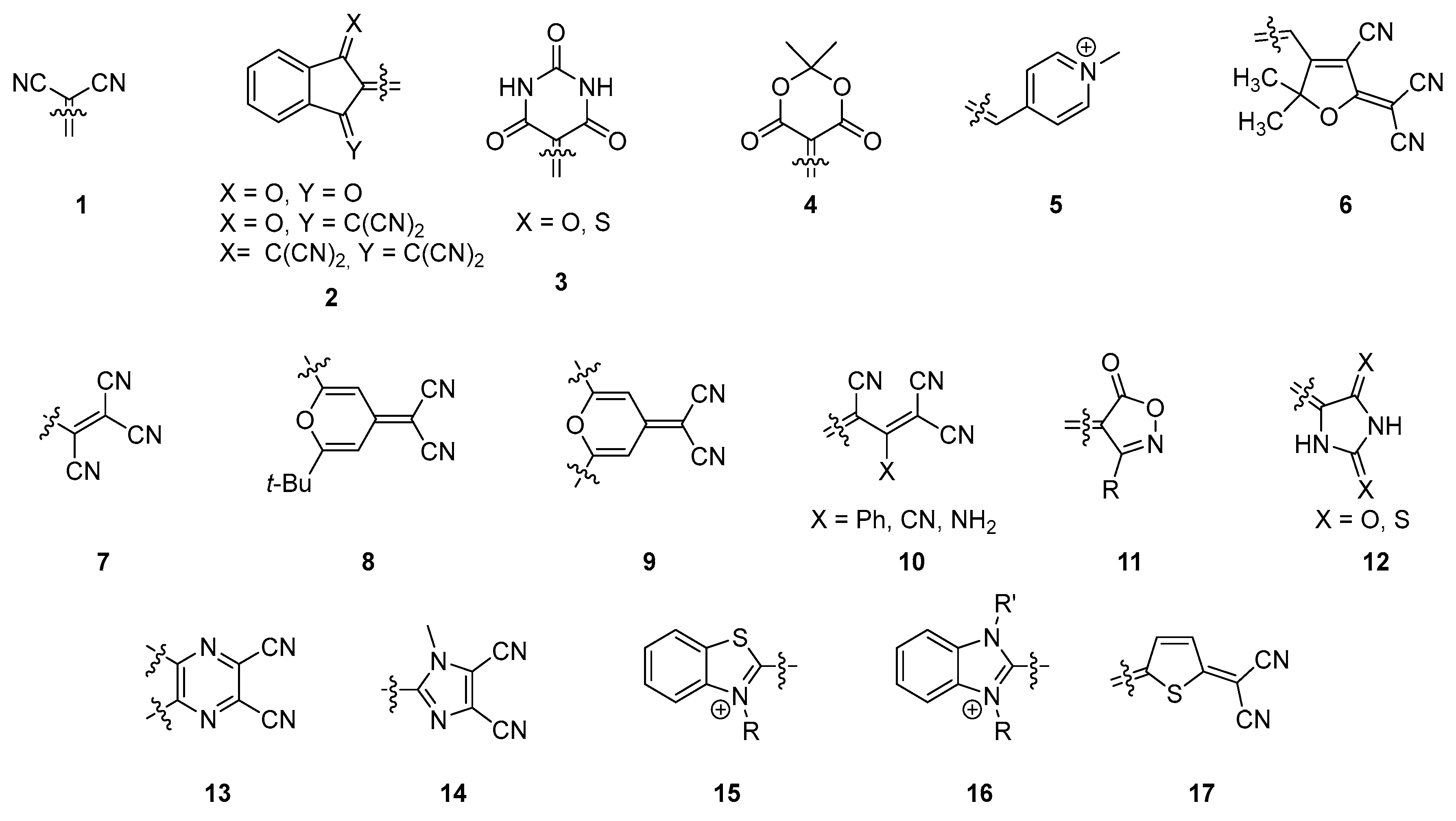
19]. If the number of groups used as electron donors cannot be calculated anymore in regards to the diversity of structures, the number of electron acceptors that can be covalently linked to electron donors is more limited, as exemplified by the list of molecules 1–17 presented in the
Figure 1. Notably, malononitrile
1 [
20], indanedione derivatives
2 [
21], (thio)barbituric derivatives
3 [
22], Meldrum derivatives
4 [
23], pyridinium
5 [
24], methyl-containing tricyanofurans
6 [
25], substituted tricyanopropenes
7 [
26], pyran derivatives
8 and
9 [
27,
28], 1,1,3-tricyano-2-substituted propenes
10 [
29], isoxazolones
11 [
30], hydantions and rhodanines
12 [
31], pyrazines
13 [
32], dicyanoimidazoles
14 [
33], benzo[
d]thiazoliums
15 [
34], benzo[
d]imidazoliums
16 [
35], and dicyanovinyl-thiophen-5-ylidenes
17 [
36] can be cited as the most common acceptors.
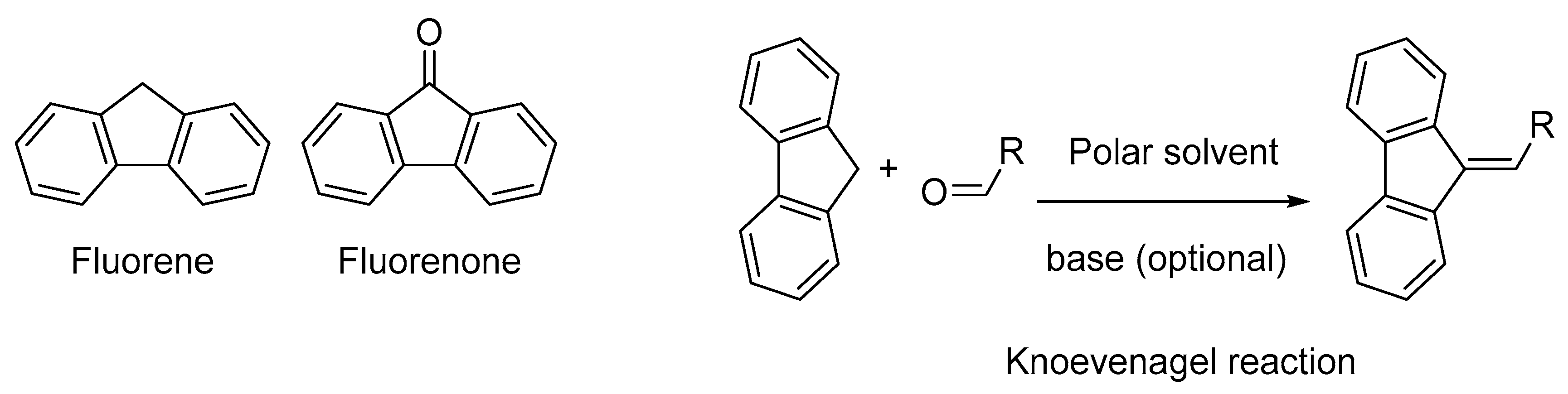
For all these acceptors and irrespective of the electron donors, the intramolecular charge transfer band is centered in the visible range and this latter can be displaced towards the near infrared region if the length of the π-conjugated spacer introduced between the two partners is extended as much as possible. However, the extension of the π-conjugated spacer is often a hard work from a synthetic point of view and alternatives are actively researched [
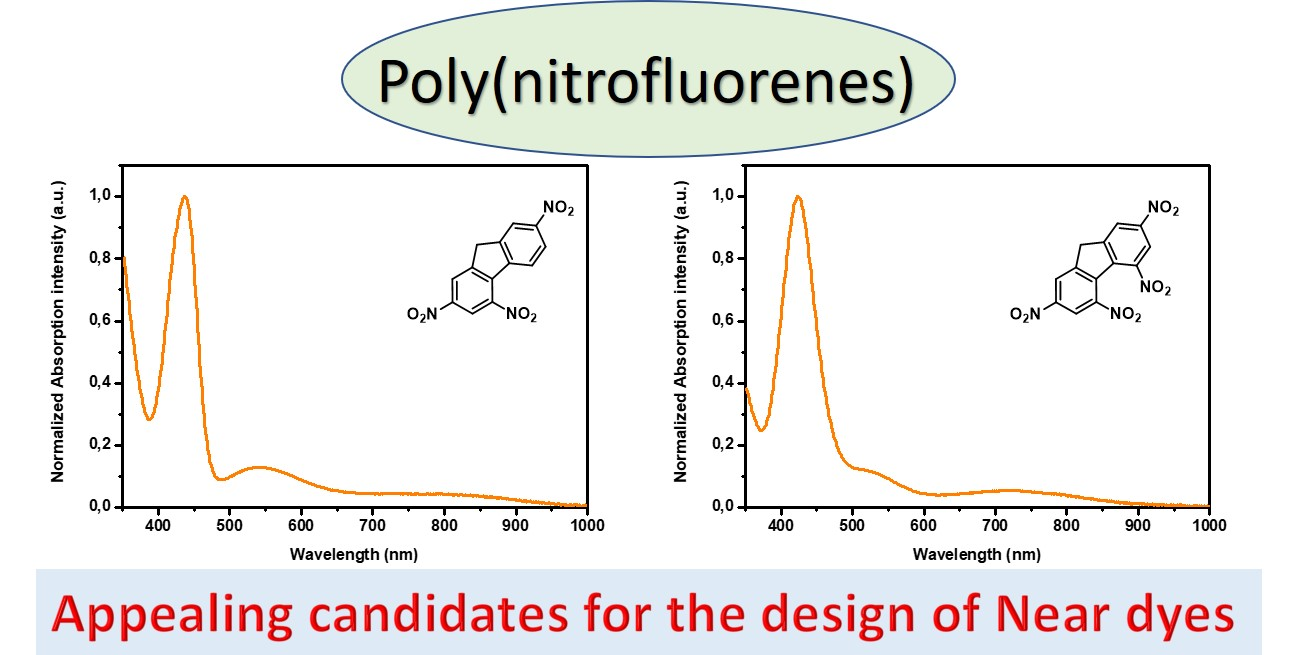
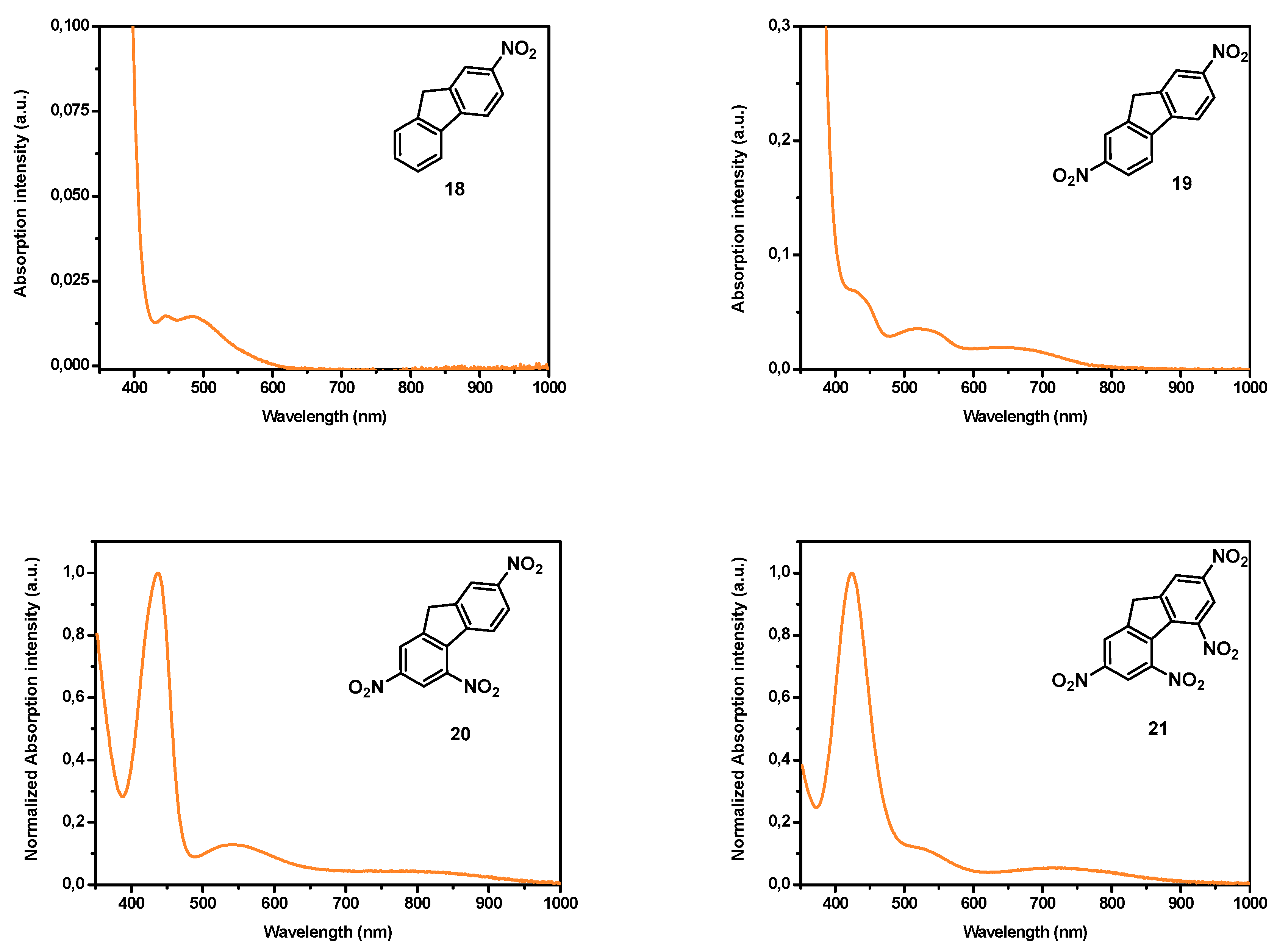
37]. Considering that for numerous applications, an absorption in the near-infrared region is required and face to the fact that all the above-mentioned electron acceptors are not sufficiently electro-deficient to inherently position the charge transfer band in the near infrared (NIR) region without taking recourse to the elongation of the π-conjugated spacer, electron acceptors already displaying an absorption band in the NIR region are actively researched. In this field, and to the best of our knowledge, only (poly)nitrofluorenes such as dinitrofluorene
19 [
38], trinitrofluorene
20 and tetranitrofluorene (TNF)
21 exhibit such an absorption band (see
Figure 2). It has to be noticed that the absorption band detected in the NIR for
19–
21 is not detectable in
18. Based on this unique property, numerous push–pull dyes displaying an absorption peak in the NIR region have been designed. If (polynitro)fluorenone derivatives have been extensively studied for the design of intermolecular charge transfer complexes [
39], the presence of the carbonyl function in fluorenone totally impede this structure to be used for the design of push–pull molecules by connecting the electron withdrawing/releasing groups to the carbon inserted between the two aromatic rings. Fluorenone derivatives will not be discussed in this review (see
Figure 3). Indeed, by the presence of the activated CH
2 group standing between the two aromatic rings, poly(nitro)fluorene derivatives constitute candidates of choice for the synthesis in one step of push–pull molecules by a Knoevenagel reaction, when opposed to an electron donor comprising an aldehyde function (see
Figure 3).
As an interesting feature, when substituted with electron-withdrawing groups such as nitro groups, the methylene group standing between the two aromatic rings is sufficiently acid so that the Knoevenagel reaction on
20 and
21 can be carried out without any base, in non-toxic and polar solvents such as
N,
N-dimethylformamide (DMF). Principles of Green Chemistry can thus be applied to the synthesis of push–pull molecules (see
Figure 3). Conversely, the methylene group in
18 and
19 is often not sufficiently activated to allow the Knoevenagel reaction to be carried out without a base and piperidine or pyridine are classically used.
In this review, an overview of the different push–pull structures comprising (poly)nitrofluorenes as electron acceptors is presented. More precisely, four different aspects will be detailed. First, numerous push–push dyes have been obtained by functionalization of the 9-position of the fluorene acceptor. Second, a series of dyes has also been obtained by nucleophilic substitution of secondary amines on cyano-substituted fluorenes and this second approach constitutes the second most widely used synthetic procedure to access to poly(nitro)fluorene-based dyes. Third, the conceptual unimolecular rectifier proposed by Aviram and Ratner has been a source of inspiration for the design of numerous chromophores and a series of dyes has been designed based on this proposed structure. Finally, parallel to organic donors, organometallic donors have also been investigated, and it constitutes the last part of this review. To end, and parallel to the synthesis and the examination of the different photophysical properties of the dyes, the different applications justifying the design of these structures will be detailed.
2. Push–Pull Molecules Based on the Connection of Strong Electron Donors at the 9-Position of the Fluorene Acceptors
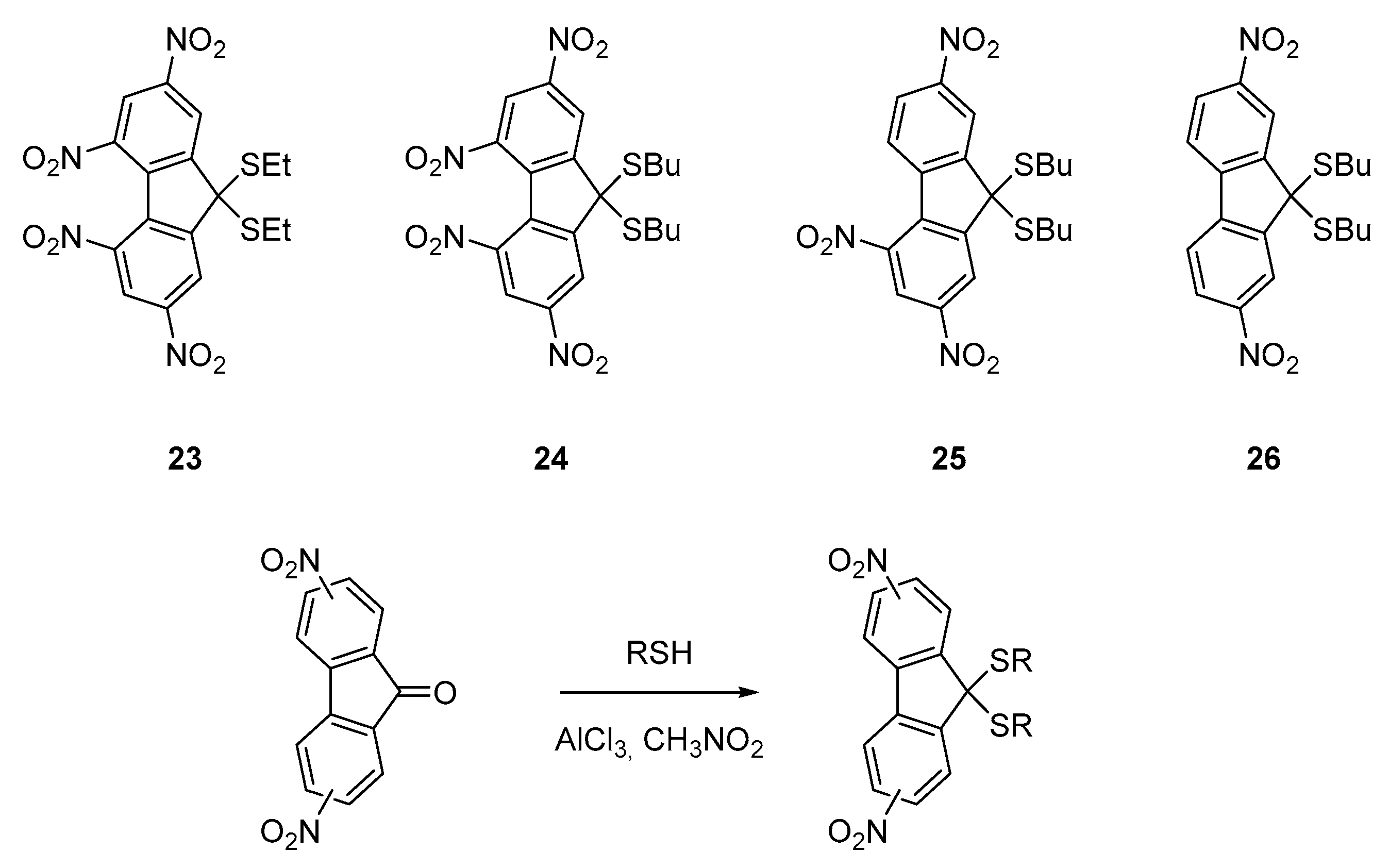
The first report mentioning the design of push–pull molecules with poly(nitrofluorenes)
23–
26 was published in 1984 [
40]. In this pioneering work, relatively weak electron donors were used since alkylthio groups were employed. By virtue of the electron-donating ability of the sulfur atom, an absorption extending until 480 nm could be obtained for
25 and
26. Despite the weak electron-donating ability of alkylthiols, the strong electron-accepting ability of
19–
21 could be evidenced, the standard conditions of thioacetalization of ketones being unable to provide the targeted molecules. Indeed, the charge transfer interactions existing between the alkylthiols and nitrofluoren-9-one derivatives hampered the classical condensation procedures to provide the push–pull structures
23–
26 (See
Figure 4).
The four molecules could only be obtained using an unusual procedure making use of aluminium chloride as a mediator for the reaction [
41]. Using this strategy,
23–
26 could be obtained with reaction yields ranging from 85 to 93%. Interestingly, a weak charge transfer interaction could be evidenced for
23–
26, the lowest-energy transition exhibiting an onset at 470 nm for
25 and
26 and 410 nm for
23 and
24 respectively. Weakness of the interaction between the sulfur donor and the nitro acceptors and thus the low electronic delocalization in the ground state was confirmed by cyclic voltammetry, all molecules exhibiting redox potentials comparable to that obtained for alkyl 2,4,5,7-tetranitrofluorene-9,9-dipropionates that does not possess any electron-donating groups. These molecules possessing good electron acceptors connected to electron donors can efficiently sensitize the photoconductivity of semiconducting polymers by forming charge transfer complexes with these latter, especially with carbazole-containing polymers [
42,
43,
44], and this ability was examined with poly(
N-vinylcarbazole) (PVK) which is a standard semiconducting polymer. This interaction was demonstrated by using two different techniques i.e., the space-charge-limited xerographic discharge and by an electrochemical time-of-flight (ETOF) method. It has to be noticed that the first commercially available organic photoconductor was based on a charge-transfer complex between tetranitrofluorene (TNF) and PVK, justifying the different studies devoted to this topic. As interesting feature, the spectral zone of photoconductivity of the carbazole-containing polymers can be selected by the appropriate choice of the push–pull molecule. By comparing the electron mobilities of
23–
26 with that of the reference 2,4,7-trinitrofluoren-9-one, comparable transport properties could be determined [
45,
46,
47,
48], demonstrating the pertinence of the strategy for the design of photoconductive materials with poly(nitro)fluorene derivatives.
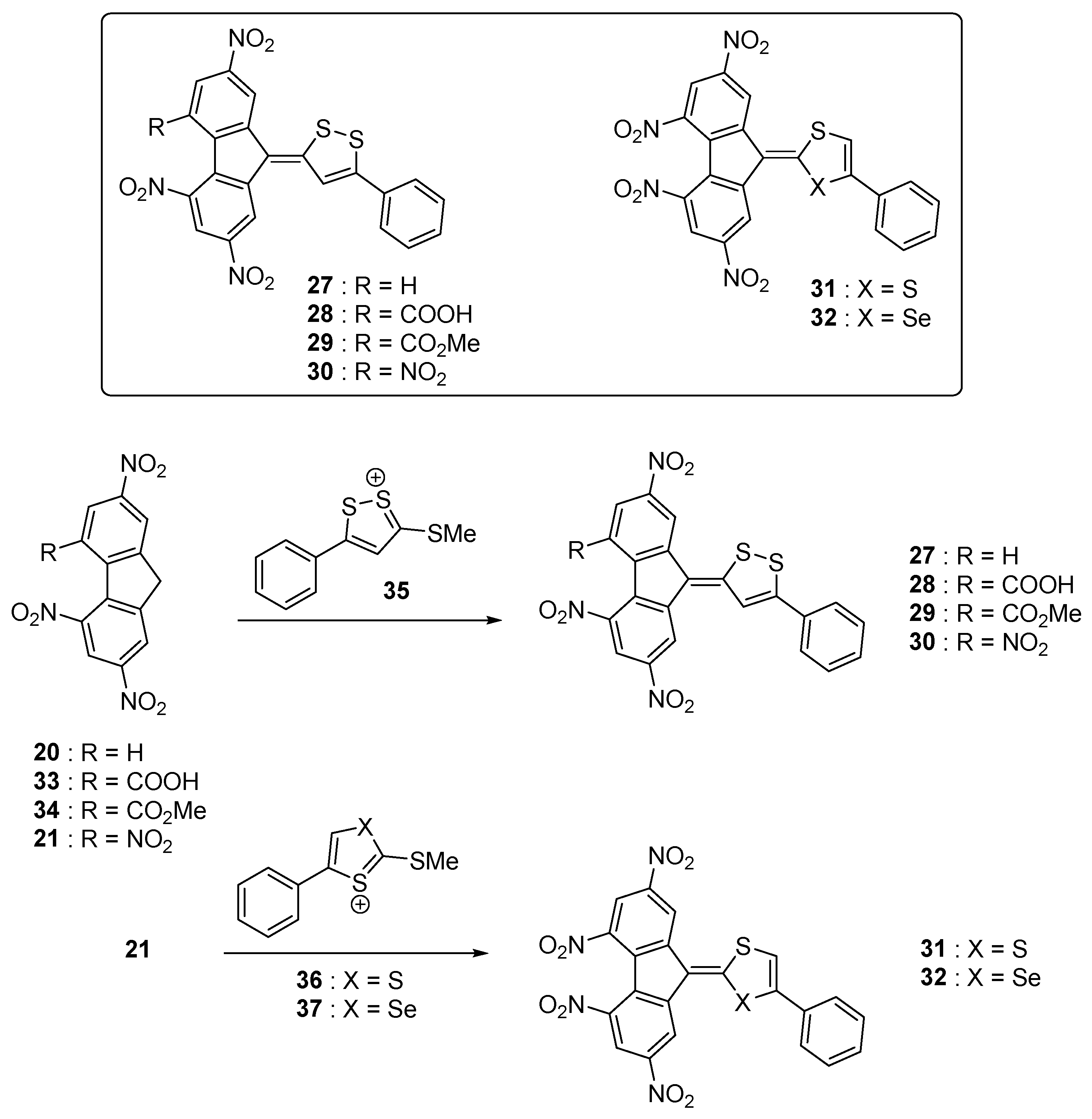
In contrast, by improving the electron donating ability of the donors, a significant red-shift of the absorption combined with the appearance of a second and new long-wavelength absorption band can be detected at lower energy than the intramolecular charge transfer. This specific band could be evidenced with the series of push–pull molecules
27–
32 (see
Figure 5) [
49]. This additional absorption band is really a characteristic from the poly(nitro)fluorene acceptors. As electron donors, dithiolylidene and selenathiolylidene groups were used, these groups being among the best electron donors [
50,
51].
27–
32 were obtained by condensation of the corresponding dithiolylium
35,
36 and selenathiolylium salts
37 with the electron acceptors
20,
21,
33 and
34 [
52,
53]. More precisely, due to the presence of multiple electron-accepting nitro groups on
20 and
21, these latter are strong C-H acids [
54] allowing the condensation in a highly polar solvent such as DMF to be carried out without any base. From a photophysical point of view, two charge transfer bands could be detected for the whole series
27–
32, the first one being detected between 446 and 500 nm and the second one between 587 and 633 nm respectively. Almost no influence of the heteroatom on the electronic transitions of
31 and
32 could be evidenced (see
Table 1).
In sharp contrast, positions of the sulfur atoms clearly influence the ICT bands and a blue-shift of 60 nm for
31 relative to that of
30 was determined. Considering that the same electron acceptor is used for
30 and
31 (i.e.,
21), the enhanced electron donating ability of 1,2-dithiol-3-ylidene compared to that of 1,3-dithiol-2-ylidene was demonstrated. As attended, improvement of the electron accepting ability of the substituents in
27–
30 resulted in a bathochromic shift of the ICT bands (see
Table 1) and a bathochromic shift of the absorption maximum was also determined by increasing the solvent polarity. It thus evidences the higher polarity of the excited state for these compounds relative to that of their ground states. Intramolecular nature of the charge transfer was proved by protonation of the different molecules in sulfuric acid, resulting in the disappearance of the two ICT bands and the formation of colorless acidic solutions.
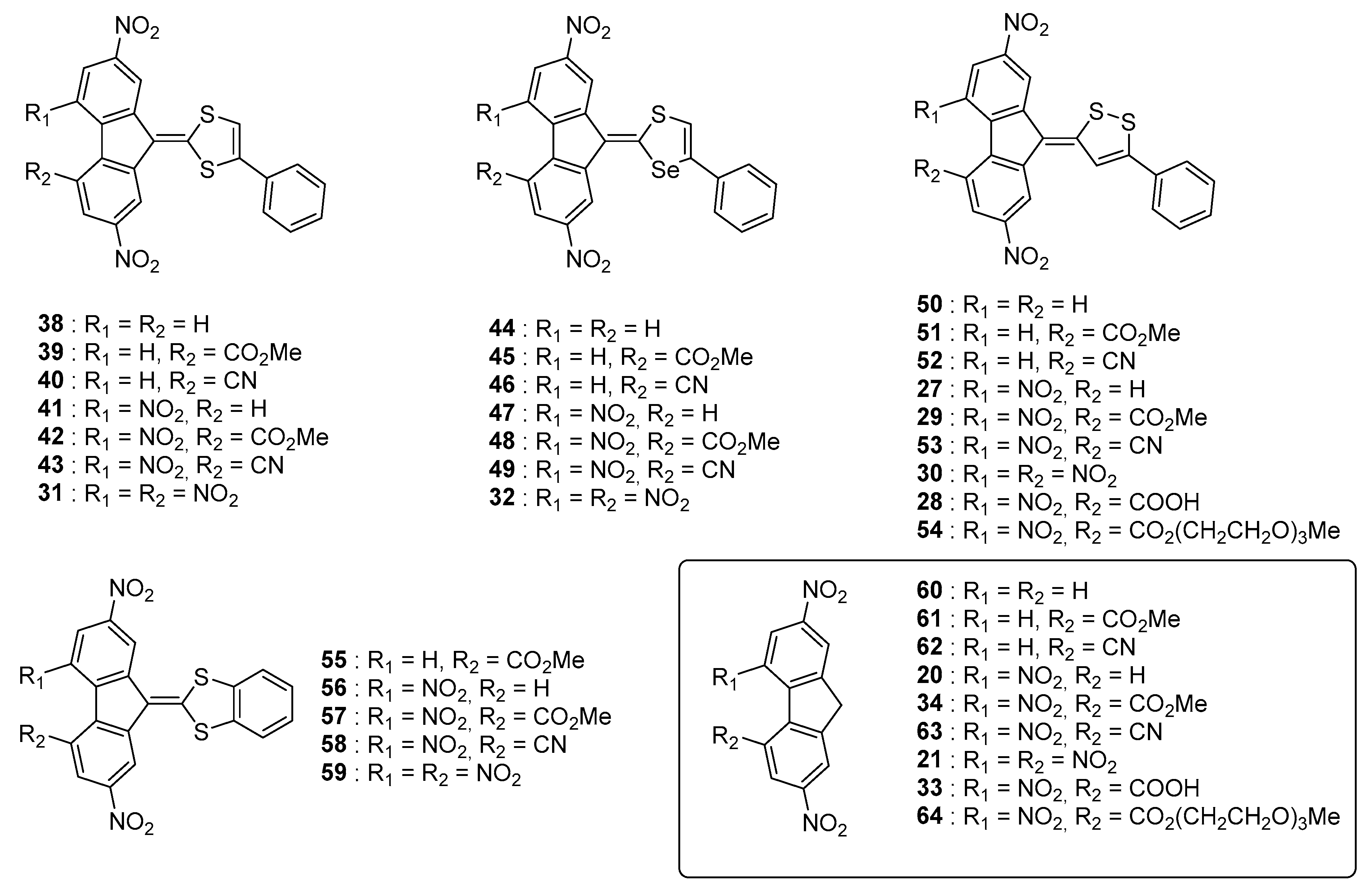
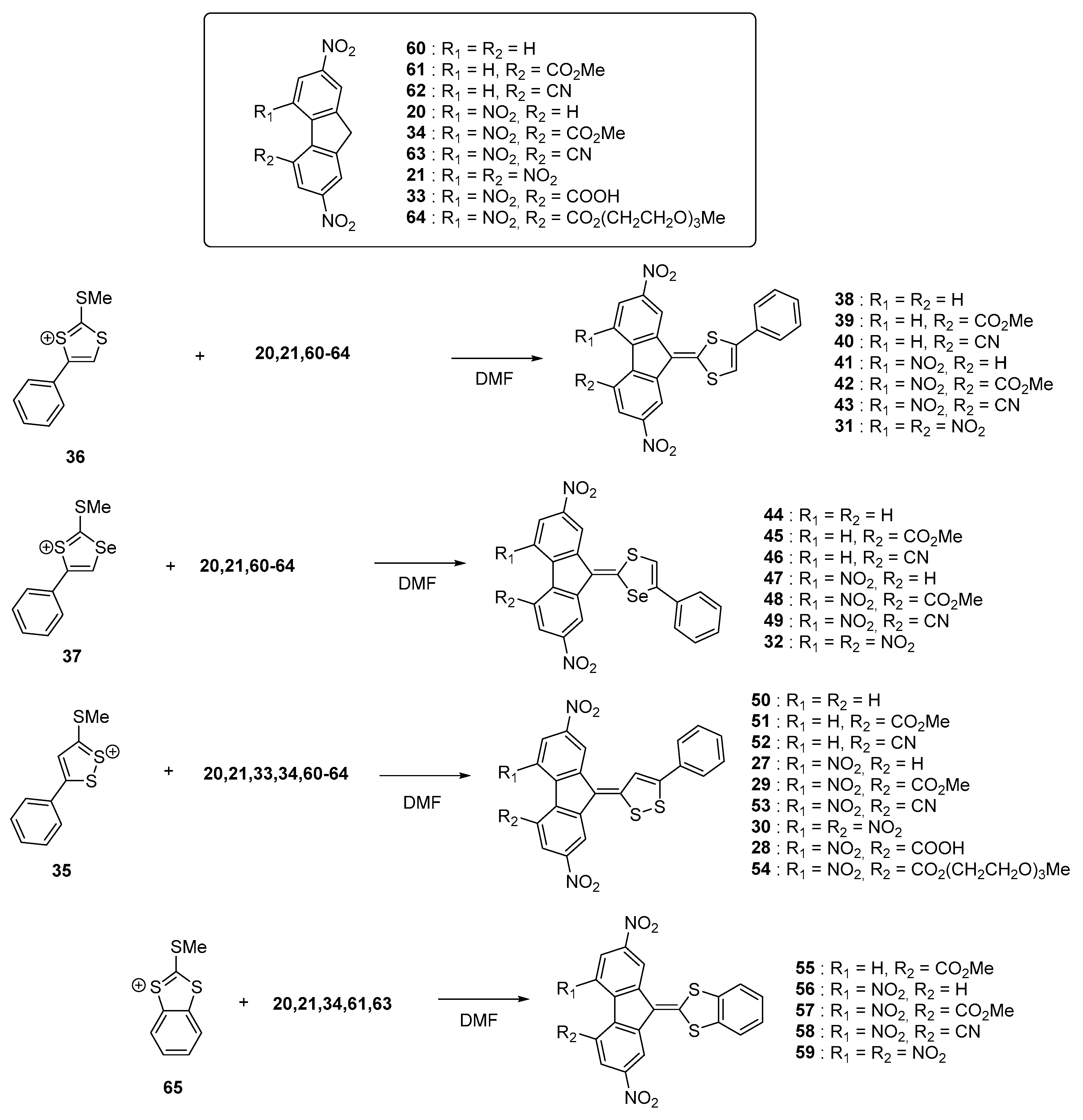
These different trends (lack of influence of the heteroatoms on the ICT maxima, positive solvatochromism) were confirmed in a subsequent study devoted to a series of 28 molecules (
27–
32,
38–
59) (See
Figure 6) [
55] Several molecules of the former study (namely
27–
32) were revisited in the context of this new work with aim at establishing a structure-photophysical properties relationship.
Nine acceptors
20,
21,
33,
34,
60–
64 were examined and the synthesis of the different dyes were carried out using procedures similar to that previously reported, using
35–
37 and
65 as the electron donors. New conclusions could be determined from this work examining a wide range of structures. First, the reaction yields significantly decreased by reducing the electron-accepting ability of R
1 and R
2 so that use of a base was required while using
60–
62 as acceptors (see
Figure 7). As specificity,
60–
62 are not substituted with nitro groups, deactivating the methylene group of the fluorene acceptor. The intramolecular nature of the charge transfer complex was proved by dissolving the different compounds in sulfuric acid, resulting in the formation of colorless solutions by protonation of the electron-donating part. Reversibility of the process was also evidenced upon addition of water, regenerating the initial compounds.
Only acidic sensitive compounds comprising nitrile or ester groups could not be entirely recovered, these groups being partially hydrolysed in strongly acidic conditions. Intramolecular nature of the charge transfer band was also proved by UV-visible spectroscopy, with a perfect and linear concentration dependence of the absorbance of their absorption maxima.
As other finding, modification of the linkage of the benzene ring to the donor group drastically alter of the absorption properties, with a hypsochromic shift of the absorption peaks of about 70 nm when the aromatic ring is fused to the donor (see series of compounds
55–
59 vs. the series of molecules
27–
30,
50–
54). Examination of the solvatochromism proved to be quite complex, all the dyes exhibiting two intramolecular charge transfer bands originating from different transitions. (see
Table 2).
Considering that the electronic transitions involved in these different absorption bands are not the same, the solvatochromic behaviors of the two bands varied separately and opposite behaviors were sometimes found for the two bands of a same dye. While using correlations based on the basicity and/or the polarity of the solvent such as the Koppel–Palm correlation, no linear relationships could be obtained [
56,
57]. A similar behavior was observed while using more traditional multi-parameter polarity scales such as the normalized Reichardt’s parameter (E
NT) or the Dimroth–Reichardt E
T(30) index [
58]. Parallel to the solvatochromism, a thermochromism could be detected for all compounds, evidencing a modification of the solute-solvent interactions with the temperature [
59,
60]. Thus, a hypsochromic shift while increasing the temperature was determined for all compounds, with a more pronounced shift for the low energy ICT band. To illustrate this, a blue shift of 72 nm was observed in chlorobenzene for the second ICT band of
31 while increasing the temperature from 3 to 92°C whereas a blue shift of only 7 nm was detected for the high-energy transition.
To quantitatively examine the temperature effect, a linear dependence with inverse temperature could be evidenced. As attended, a negative halochromic behavior was detected, resulting from the protonation of the electron-donating dithiolylium. Interestingly, at certain concentration, an additional band in the 800–1000 nm region was detected for
31 and
32, assigned to the formation of radical ion species. Precisely, the generation of radical species was assigned to bimolecular interactions between the neutral form regenerated upon dilution and the protonated form, resulting to an intermolecular electron transfer between the neutral and the cationic form, producing a radical and a radical cation following the Equation (1).
Electrochemical investigations revealed the different molecules to exhibit two redox processes, with two closely spaced reduction waves centered on the fluorene moieties, corresponding to the formation of the radical anion and dianion. For the series of compounds
32,
44–
49, an additional reversible reduction process was detected at about −2.0 V (vs. Fc
0/Fc
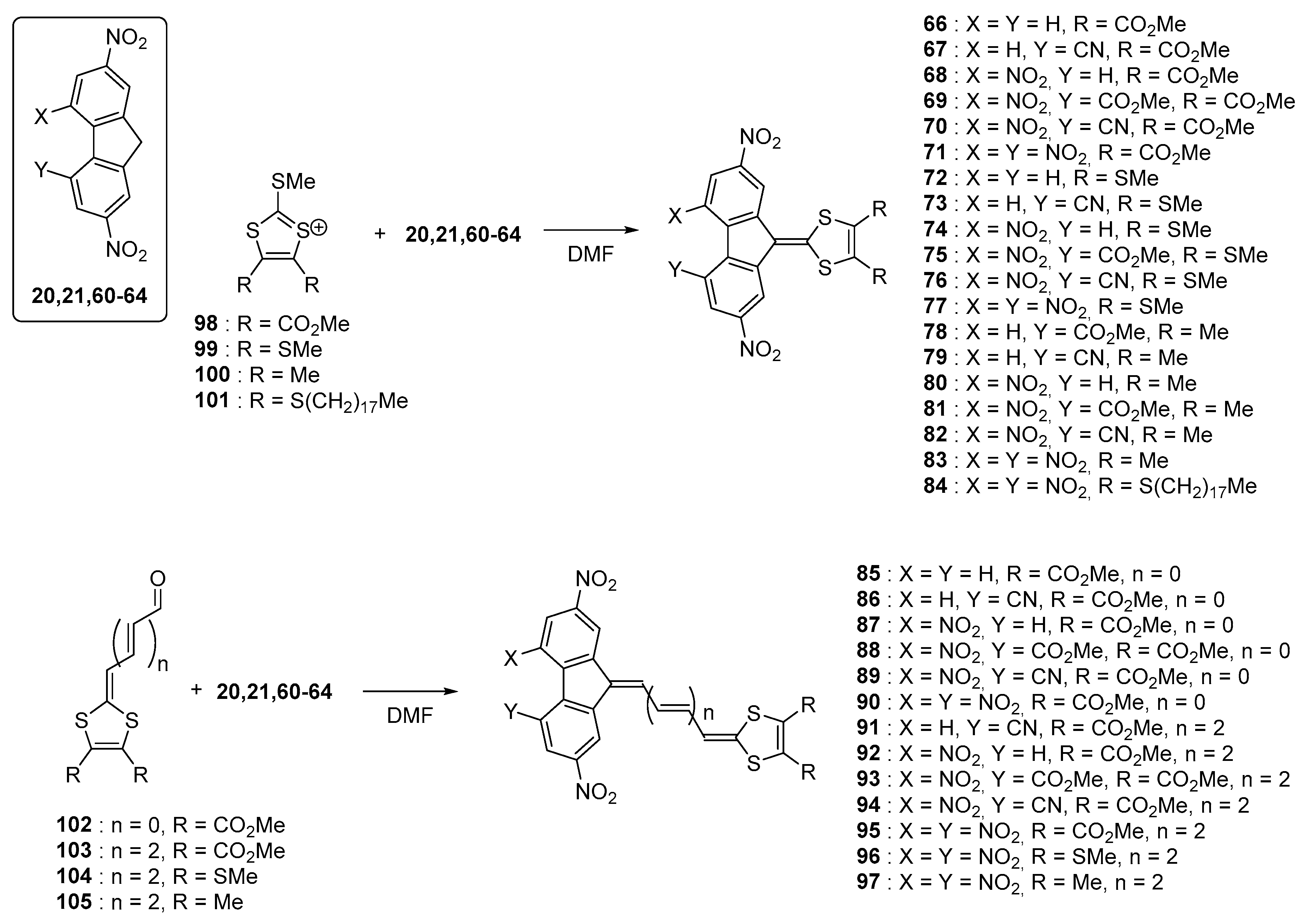
+ couple) and this process was assigned to the formation of the radical trianion species. Investigation of the substitution effects on the fluorene acceptors revealed the difference between the two first reduction processes to be almost constant, whatever the substitution of the fluorene core was. Position of the ICT band is highly sensitive to the strength of the electron donors and acceptors that are selected for designing push–pull dyes but position of this transition can also be modified with lengthening the conjugated chain between the two partners. Influence of this parameter was notably examined with a series of chromophores bearing 1,3-dithiolylium donors [
61]. The different dyes
66–
97 were synthesized using a strategy comparable to that used for the former series (see
Figure 8).
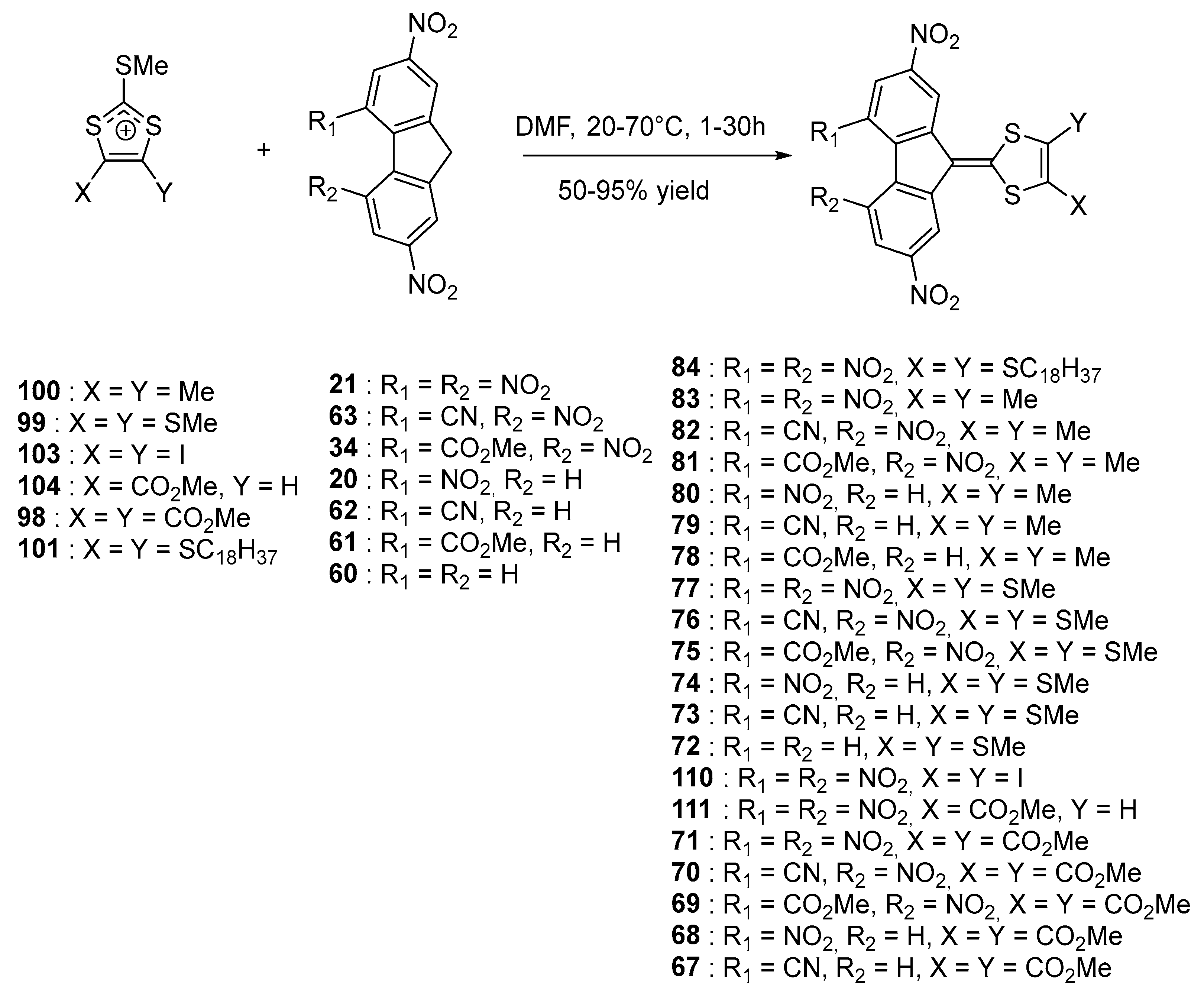
98–
105 were used as the electron donors and
20,
21,
60–
64 as the electron acceptors.
Upon elongation of the conjugated spacer, an extension of the highest occupied molecular orbital (HOMO) level over the donor part and the π-conjugated system is logically observed, resulting in a decrease of the HOMO-LUMO gap (where LUMO stands for lowest unoccupied molecular orbitals). Thus, comparison between
71,
83, and
90 revealed the second ICT band to shift from 544 to 588 and 636 nm respectively in 1,2-dichloroethane (see
Table 3). Parallel to this, the molar extinction coefficient drastically increased from 9800 to 21000 and 39000 dm
3·mol
−1·cm
−1 respectively upon elongation of the conjugated spacer. By electrochemistry, a cathodic shift could be detected (E
red (71) = −0.80 V, −1.02 V and E
red (83) = −0.83 V, −1.05 V vs. Ferrocene/Ferrocenium (Fc
0/Fc
+) couple), consistent with an improvement of the electron-donating ability in
83 and thus a lower ability for the fluorene moiety to accept electrons.
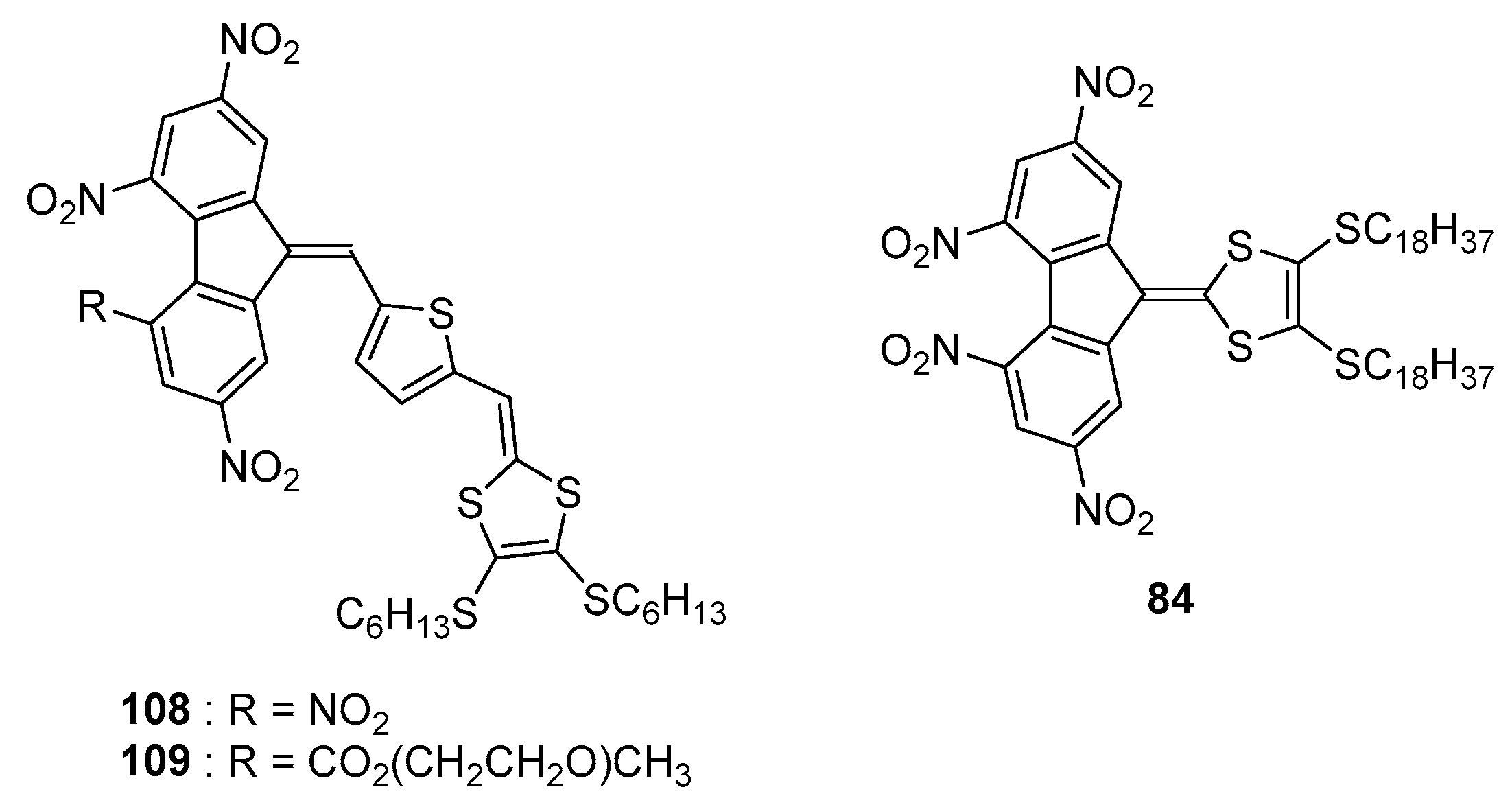
As potential application for these structures, the most soluble derivative
84 was examined as a sensitizer in photothermoplastic storage media (PTSM) based on poly[
N-(2,3-epoxypropyl)carbazole] (PEPC) [
44]. Notably, photothermoplastic materials can find numerous applications in aerospace and astrophysics [
62]. In the present case, high values of holographic sensitivity in the ICT region were determined, outperforming the results obtained with tetranitrofluorene
21, when used as the sensitizer for the same polymer. Especially
84 is a promising candidate for this application as it possesses a significant electron affinity (2.1 eV deduced from its reduction potentials (E
red1 = −0.81 V, E
red2 = −1.04 V vs. Fc
0/Fc
+ couple), facilitating the formation of charge transfer complexes with electron donors such as poly(carbazoles).
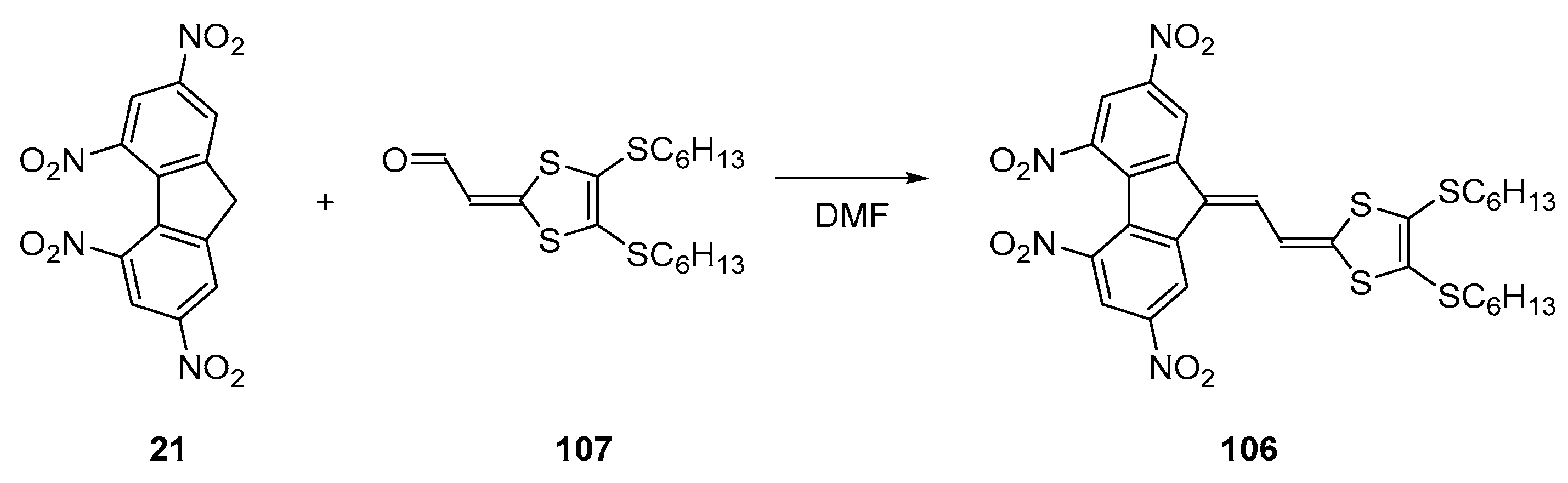
Finally, the best comprise between electron affinity and intramolecular charge transfer energy was obtained with the molecule 106 [
63]. This molecule was obtained by condensation of the aldehyde
107 with
21 in DMF (See
Figure 9). Comparison of the electrochemical properties of this molecule with those of
21 evidenced the two molecules to exhibit similar redox potentials (−0.89 V, −1.04 V and −1.78 V for 106 vs. −0.88 V, −1.15 V and −1.79 V for 21 vs. Fc
0/Fc
+ couple respectively), providing similar electron affinities and thus electron accepting abilities to the two molecules. However, compared to its analogue
84, a red-shift of the two ICT bands was obtained for
106, with a red-shift of 95 and 33 nm for the two ICT bands and an increase of the peak intensity of 1.5 and 3 for the two ICT bands respectively (See
Table 4). This enhancement of the molar extinction coefficient is consistent with the elongation of the π-conjugated system, increasing the oscillator strength and thus the electronic delocalisation upon excitation.
While comparing 84 and 106, the ability of 106 to sensitize the photoconductivity of the carbazole-based polymer PEPK-1 was greatly enhanced, by the red-shift of its absorption bands but also by the similar intensity of the two ICT bands, widening the photoresponse over the 500–700 nm region. Indeed, the photoresponse of 84 was limited to the 550–650 nm region due to the presence of a unique ICT band.
As already mentioned, elongation of the π-conjugated spacer is favorable to increase the molar extinction coefficient of the intramolecular charge transfer and to redshift the absorption maximum. An improvement of the photoconductivity sensitization is also attended if the electron affinity of the chromophore is enhanced, favouring the formation of a charge transfer complex with the sensitized polymer. With aim at optimizing these different parameters (length of the π-conjugated spacer, electron donating/accepting ability of the two partners), a thiophene was introduced between the electron-donating 1,3-dithiole group and the fluorene acceptor (see
Figure 10) [
64].
From a photophysical point of view, only few details were provided in this work, excepted that the two ICT bands are detected in the 400–900 nm region, with a significant red-shift of the absorption maximum accompanied by a substantial increase of the molar extinction coefficient for 108/109 relative to that of 84. By electrochemistry, unexpectedly, an irreversible one-electron reduction process was observed for 108 and 109, identified as resulting from the presence of the hydrogen on the double bond close to the acceptor moiety. As a consequence of this electrochemical irreversibility, when tested as sensitizers for the carbazole-based polymer PEPK, only a poor photoresponse in the ICT band of 108/109 was observed and this result was assigned to the poor photochemical stability of the radical anion previously demonstrated by the irreversibility of the reduction process. Solubility of the sensitizer is another major issue, especially for fluorene derivatives that are prone to aggregate, and this issue was addressed by elongating the chain (109 vs. 108). The higher solubility of 109 compared to that of 108 was evidenced.
Recently, the design used for the synthesis of 84 was revisited in a more ambitious study where
21 structures were examined [
65]. Their chemical structures are depicted in the
Figure 11. In fact, compared to the previous study [
61], only two new structures were added, namely
110 and
111. However, a more detailed study was carried out in this new work.
All molecules
67–
84 and
110,
111 exhibited a strong color ranging from red to black and the different dyes were also characterized by a low solubility in most of the common organic solvents. This is the reason
84 was synthesized: in order to greatly improve the solubility of this chromophore. As the main characteristic of this series, the different molecules are characterized by multiple redox states, ranging from four to five redox states what is rarely observed for purely organic molecules [
66,
67,
68]. Precisely, until four reduction processes could be detected for
71,
70, and
68, what is also observed for fullerene derivatives [
69]. Considering that upon reduction, the radical anion, the dianion, the radical trianion, and the tetraanion are centered on the fluorene moiety, a major influence of the substitution pattern was evidenced on these different reduction processes. A summary of the redox potentials is provided in the
Table 5. Precisely, a linear correlation for the first and second reduction processes could be determined while using the Hammett constants which quantify the impact of the substitution pattern on the redox potentials [
70]. No influence of the substitution pattern of the dithiole moiety on the reduction potentials was detected, evidencing the negative charge to be exclusively localized on the fluorene fragment. These results were confirmed by the theoretical calculations done on the different molecules, evidencing the LUMO level to be mostly centered on the fluorene moiety.
While examining the position of the ICT bands, classical trends were determined such as a clear bathochromic shift of the ICT band while improving the accepting ability of the fluorene moiety or by improving the electron donating ability of the 1,3-dithiole moiety. Here again, a linear correlation with the sum of the Hammett parameters was demonstrated, evidencing this series of molecules to exhibit a classical modification of the ICT band with the strength of the donors and the acceptors. The most redshifted absorption was detected for 83, with an ICT band peaking at 611 nm. Only a weak positive solvatochromism was determined for all molecules by increasing the solvent polarity, the variation of the ICT band being lower than 20 nm. This result is indicative of a highly polar ground state. Conversely, a pronounced thermochromic behavior was evidenced with a hypsochromic shift of the ICT band while increasing the temperature. This phenomenon was notably demonstrated in high-boiling point solvents such as chlorobenzene and 1,2-dichlorobenzene. Such a behavior is not so unusual since it was previously reported for polythiophenes [
71,
72]. Finally, among the series of 27 molecules, only
84 was examined as an electron acceptor for the preparation of CTC with carbazole-containing polymers. Precisely, 84 was used for recording holograms on photothermoplastic storage media (PTSM) materials in combination with poly(
N-epoxypropylcarbazole) (PEPK). To reach the maximum sensitivity, a concentration of only 2 wt.% was required for
84 compared to 10 wt.% for benchmark sensitizers such as 2,4,7-trinitrofluorenone and 2,4,7-trinitro-9-dicyanomethylene-fluorene, as a result of a broader UV-visible absorption spectrum.
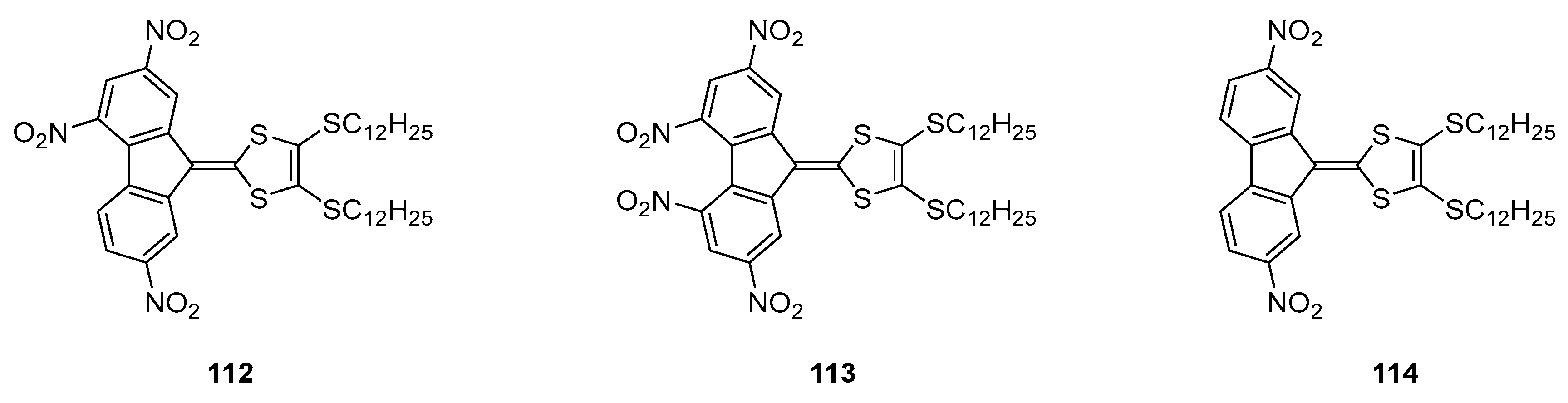
Until 2008, the unique application reported for poly(nitro)fluorene dyes concerned the electrophotographic processes with the sensitization of carbazole-containing polymers [
73]. In 2009, the scope of applicability of poly(nitro)fluorene dyes was extended to aggregation-induced emission (AIE) and the design of red-emitting materials [
74]. For this purpose, three molecules (
112–
114) were examined and the slight differences in chemical structures drastically impacted the optical properties and the supramolecular structures (See
Figure 12). To examine this last point, symmetrically and asymmetrically substituted push–pull molecules were investigated.
While refluxing 112 and 113 in hexanes and cooling the solutions at 0 °C afforded in the two cases precipitates that were examined by optical microscopy. If the formation of spheres of 6–8 μm diameter and a nanostructured surface was observed for 112, conversely, microtubes of 2.5–5 μm diameter were obtained for 113. The size and morphology of the microstructures could be easily tuned by varying the cooling temperature. Thus, “flowers with petals” could be obtained while cooling the solution at 15 °C for 112, replacing the microspheres previously obtained when the solution was cooled at 0 °C. By optically characterizing the microstructures, the two charge transfer bands detected in hexanes at 417 and 520 nm for 112 gradually evolved to a band at 480 nm with a shoulder at 520 nm upon aggregation. This new band was assigned to an intermolecular charge transfer between adjacent molecules in the solid state. Based on this finding, the cohesion between molecules in the solid-state results from intermolecular interactions between molecules, outperforming the intramolecular interactions. A similar behavior was evidenced for 113, the intramolecular charge transfer band at 552 nm splitting into two new bands at 385 and 460 nm. While examining the photoluminescence properties, a 50-fold enhancement of the photoluminescence quantum yield (4.5%) was determined for 112 upon aggregation. An AIE process was thus evidenced. Conversely, the symmetrical substituted 113 did not show any enhancement of the photoluminescence properties upon aggregation. In fact, authors determined from the crystallographic investigations that the structure of 113 was more twisted than that of 112 in the solid state, quenching the fluorescence. Parallel to this, intermolecular interactions are responsible of the aggregation of 112 in the solid state whereas dipole interactions govern the packing of 113 in the solid state. It was thus concluded the molecular dipole moments in chromophores to be a crucial parameter governing the packing mode and the ability to design AIE emitters.
Parallel to 1,3-dithiole electron donors, thiophenes are also extensively used for the design of semiconducting polymers [
75] and materials for NLO applications due to their exceptional electron donating ability and their oxidation potentials [
76]. In this context, a series of poly(nitrofluorenes) where the thiophene group was not used as a spacer but as an electron donor were prepared [
77]. More precisely, the 1,3-dithiole moiety was fused with the thiophene ring, furnishing an extended donor (see
Figure 13).
For this series of dyes 114–131, the synthesis of the acceptors and especially the selectivity during the mono- and dinitration reactions of the fluorene esters proved to be challenging and the reaction conditions had to be carefully optimized for each of them. To illustrate this, 138–142 could only be obtained by using a HNO3: AcOH 3:20 ratio starting from 133–137 whereas 143 and 144 could be more easily prepared while using a HNO3: AcOH 1:1 ratio. Conversely, 148–150 could only be obtained while reacting 133–135 in fuming nitric acid. In the case on long-chain esters (undecanyl and triethyleneglycol monomethyl ether and triethyleneglycol monoethyl ether), the purification of 145 and 146 was almost impossible due to the presence of a mixture of di- and trinitro derivatives that could not be separated due to similar polarities on the column chromatography. As an alternative strategy to access to 145 and 146, the ester 144 was hydrolyzed in acidic conditions providing 153 and esterified with the appropriate alcohol furnishing the two expected esters 145, 146 and even 154.
As other surprising result, the synthesis of
151 could not be realized in the conditions used for
150, the Nuclear Magnetic Resonance (NMR) analysis evidencing that the terminal ethyl group was lost (see
Figure 14). Loss of the ethyl group and formation of a nitrate was confirmed by Infrared spectroscopy analyses. This unexpected behavior was not observed for
150.
From a synthetic point of view,
114–
126 were obtained in conditions adapted for each acceptor. Thus, due to the high reactivity of the tri- and tetranitrofluorene acceptors, condensation with salt
155 could be realized simply in DMF whereas pyridine was used as the solvent for the less reactive dinitro acceptors. Long reaction times were required for the reaction with mononitro derivatives, yielding the targeted molecules in low yields (40 to 76% yield) due to the degradation of the salt
155 over time. Compounds
114–
116 could not be isolated pure. Face to this result, reactivity of the dihydrothiophene salt
157 towards condensation with fluorene acceptors was examined. Due to a greater delocalisation of the positive charge over the whole salt, reaction temperature for the synthesis of
127–
131 could be considerably reduced (room temperature for the synthesis of
127–
131 versus 100 °C for
114–
125, 50 °C for
126) (see
Figure 15). Reaction yields ranging from 47 to 64% were determined for the synthesis of
127–
131.
While examining the optical properties of the series of dyes
114–
131, a bathochromic shift of the two ICT bands was unexpectedly observed for
127–
131 compared to that of their analogues
114–
126. This counter-intuitive behavior can be assigned to the fact that when the thiophene ring is fused to 1,3-dithiole, this latter doesn’t behave as a donor but as an acceptor, reducing the electron donating ability of the 1,3-dithiole moiety. A similar behavior was previously reported for TTF derivatives [
78,
79]. The most red-shifted ICT was detected for
131, bearing
21 as the acceptor and the ICT was detected at 596 nm (see
Table 6).
The electrochemical behavior of
114–
131 revealed an irreversible oxidation process to take place upon oxidation. As deduced by UV-visible spectroscopy, the negative impact of the electron-withdrawing ability of the fused thiophene on the redox properties was confirmed, the oxidation potentials being detected at more cathodic potentials for the
114–
126 vs.
127–
131. An oxidation process centered on the 1,3-dithiole fragment could be confirmed. Two closely-spaced reduction processes could be detected for all molecules with the presence of an additional reduction peak detected around −1.8 V (vs. Fc
0/Fc
+ couple), that was reversible, quasi-reversible or irreversible depending of the electron-withdrawing acceptor. No regular dependence with the substitution pattern of the fluorene moiety was evidenced for this third and additional peak. Finally, attempts to electropolymerize the series
114–
126 failed, as a result of the formation of the radical cation onto the 1,3-dithiole part which is thus inappropriate for dimerization. The steric hindrance of the monomer was also suggested as impeding the monomeric units to connect to each other. This drawback was overcome with the terthiophene derivative
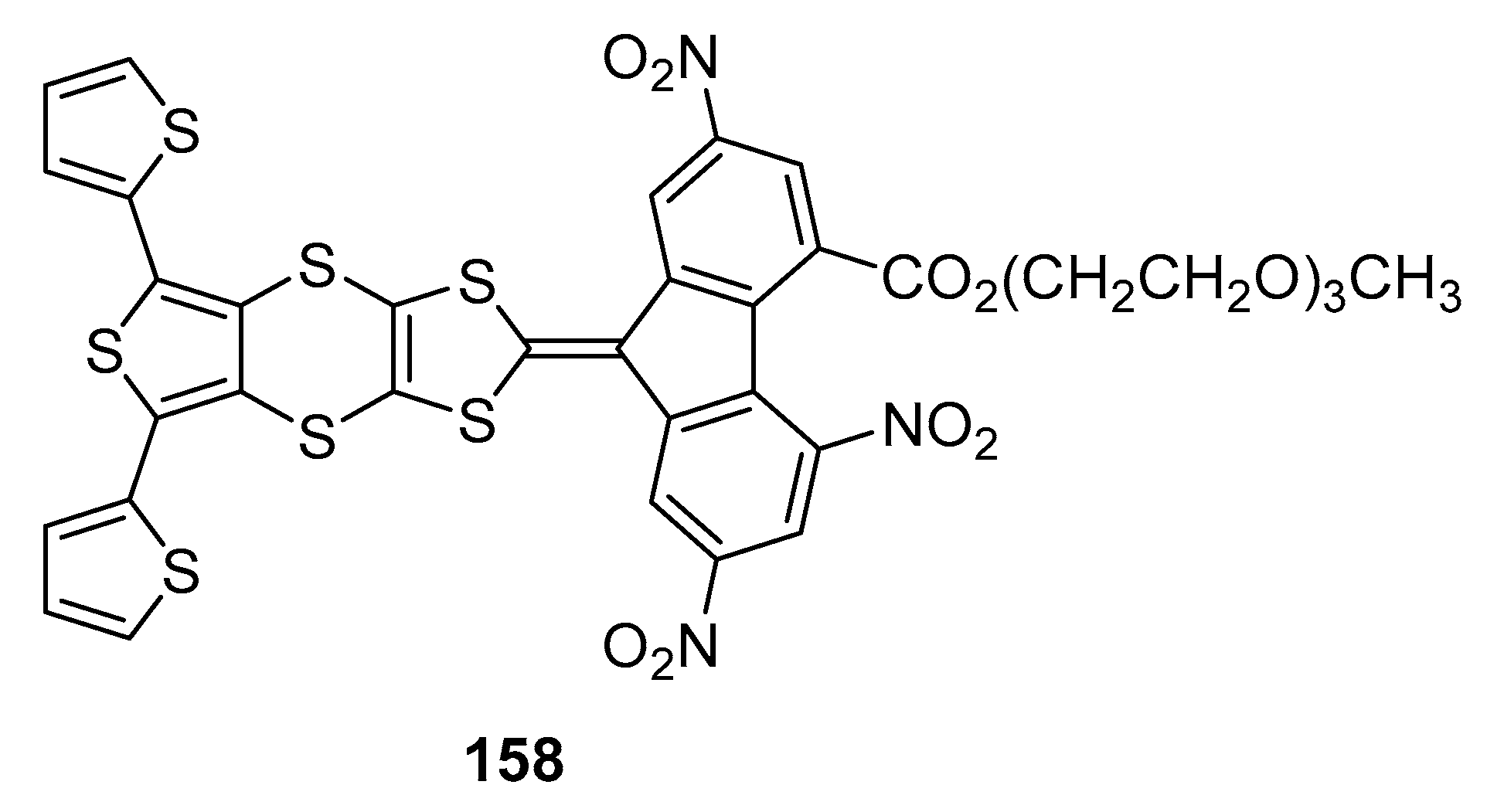
158 where two electroactive thiophene groups were introduced on both side of the thieno-1,3-dithiol-2-ylidene moiety (See
Figure 16) [
80]. In these conditions, a sufficient distance was introduced between the bulky fluorene units so that a polythiophene polymer could be formed by cycling between 0 and 1.40 V.
To reduce the negative impact of the 1,3-dithiole moiety on the electrochemical oxidation of the thiophene moiety and the localization of the radical cation onto the 1,3-dithiole moiety, a strategy consisting in isolating electronically the two groups from each other was examined and in this aim, the
160–
164 series was developed [
81]. By analogy with the previous work, a series of molecules
158,
166–
169 where additional and lateral thiophene groups have been introduced to reduce the steric hindrance was also prepared (See
Figure 17).
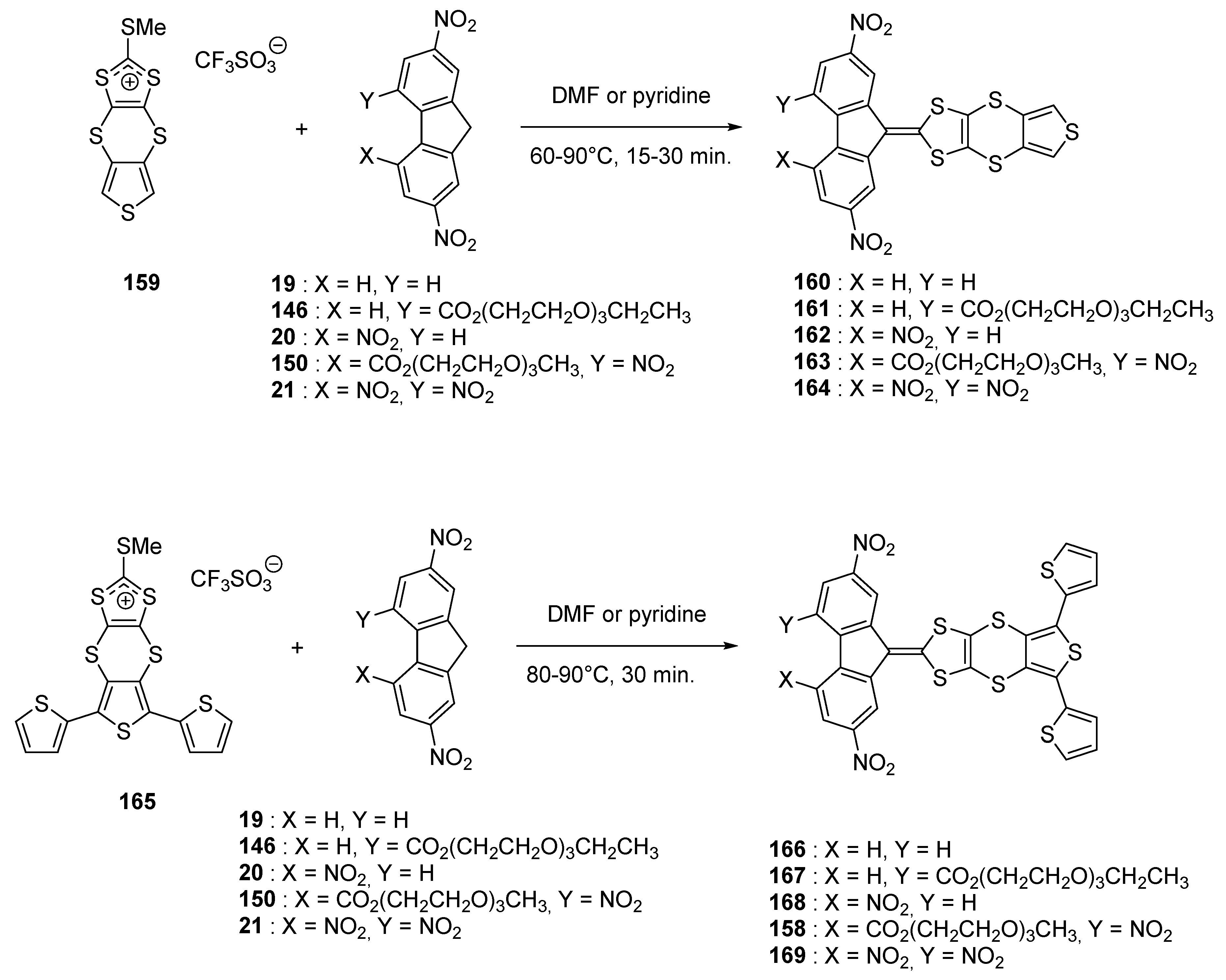
The different molecules were prepared using the standard procedure, by reaction consisting in the condensation of the substituted fluorenes with the appropriate dithiolium salt 159 or 165. It has to be noticed that 165 is unstable so that it has to be used immediately after synthesis, in the next step, without purification. If 166 and 168 could be synthesized, their low solubilities impeded their purifications so that, even if obtained, these two compounds were thus further investigated.
By UV-visible spectroscopy, the low influence of the thiophene moiety as well as the 1,4-dithiino spacer on the charge transfer interaction was evidenced, especially by comparing the positions of the ICT bands with those of unsubstituted analogues previously reported (See
Table 7) [
55,
61,
82] Similarly, comparison of the oxidation potentials of the
167–
169 series with those of the series
160–
164 previously studied [
77] evidenced the 1,4-dithiino spacer to decrease the electronic effects of the fluorene moiety on the oxidation ability of the thiophene unit, what is favorable for the oxidative electropolymerization of thiophene. Finally, for electropolymerization, only
158 was examined, this latter exhibiting the best compromise between solubility and electron-accepting ability of the fluorene fragment.
Poly(158) was obtained by electrochemical oxidation, by successive cycling between 0 and 1.4 V in dichloromethane. A dark blue polymer formed at the surface of the Gold electrode. By UV-visible spectroscopy, an ICT band located at 567 nm could be determined for the electrogenerated polymer. For comparison, poly(158) was also synthesized chemically, by oxidation with FeCl3, and an ICT band at the same position could be detected. By FTIR spectroscopy, analysis of the photoinduced IR spectrum of poly(158) revealed the presence of two representative infrared active vibration (IRAV) bands, one evidencing the presence of radical cations delocalized over the polythiophene backbone and a second one demonstrating the accepting part of the polymer to be localized over the fluorene units.


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
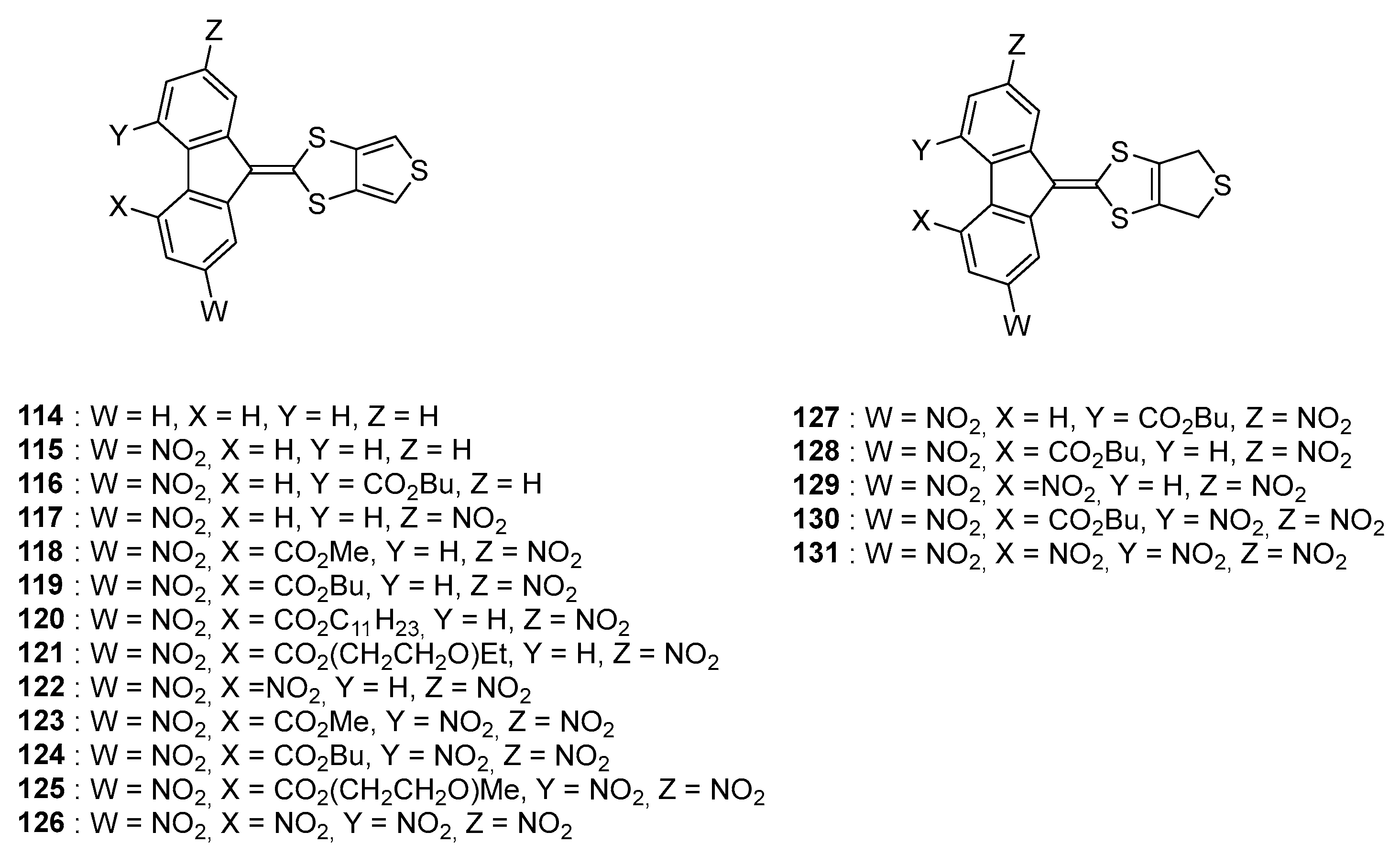{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}