In Situ FT-IR Characterization of CuZnZr/Ferrierite Hybrid Catalysts for One-Pot CO2-to-DME Conversion

 ,
,  ,
,  ,
, 
 ,
, 

Abstract
1. Introduction
2. Experimental
3. Results and Discussion
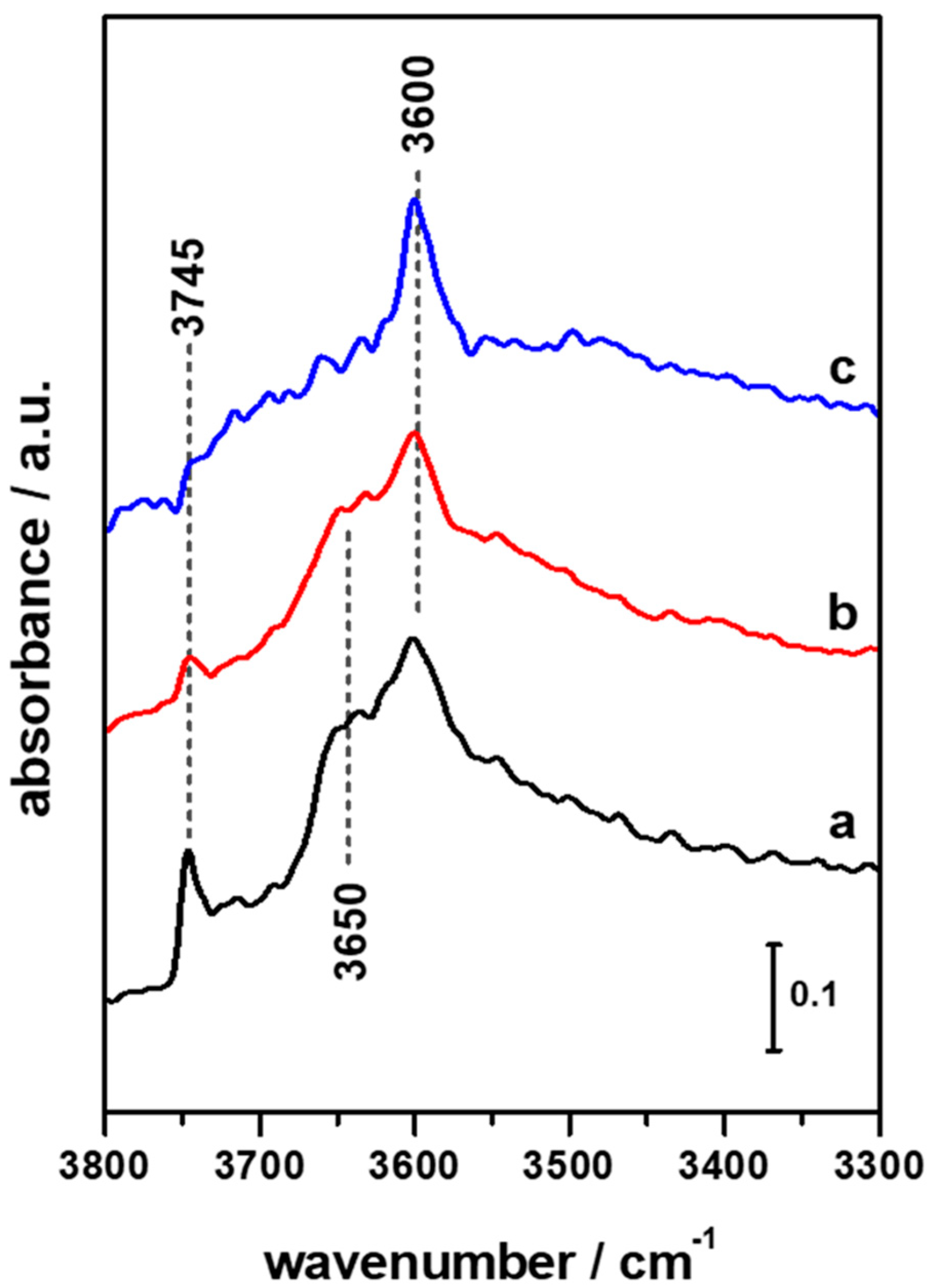
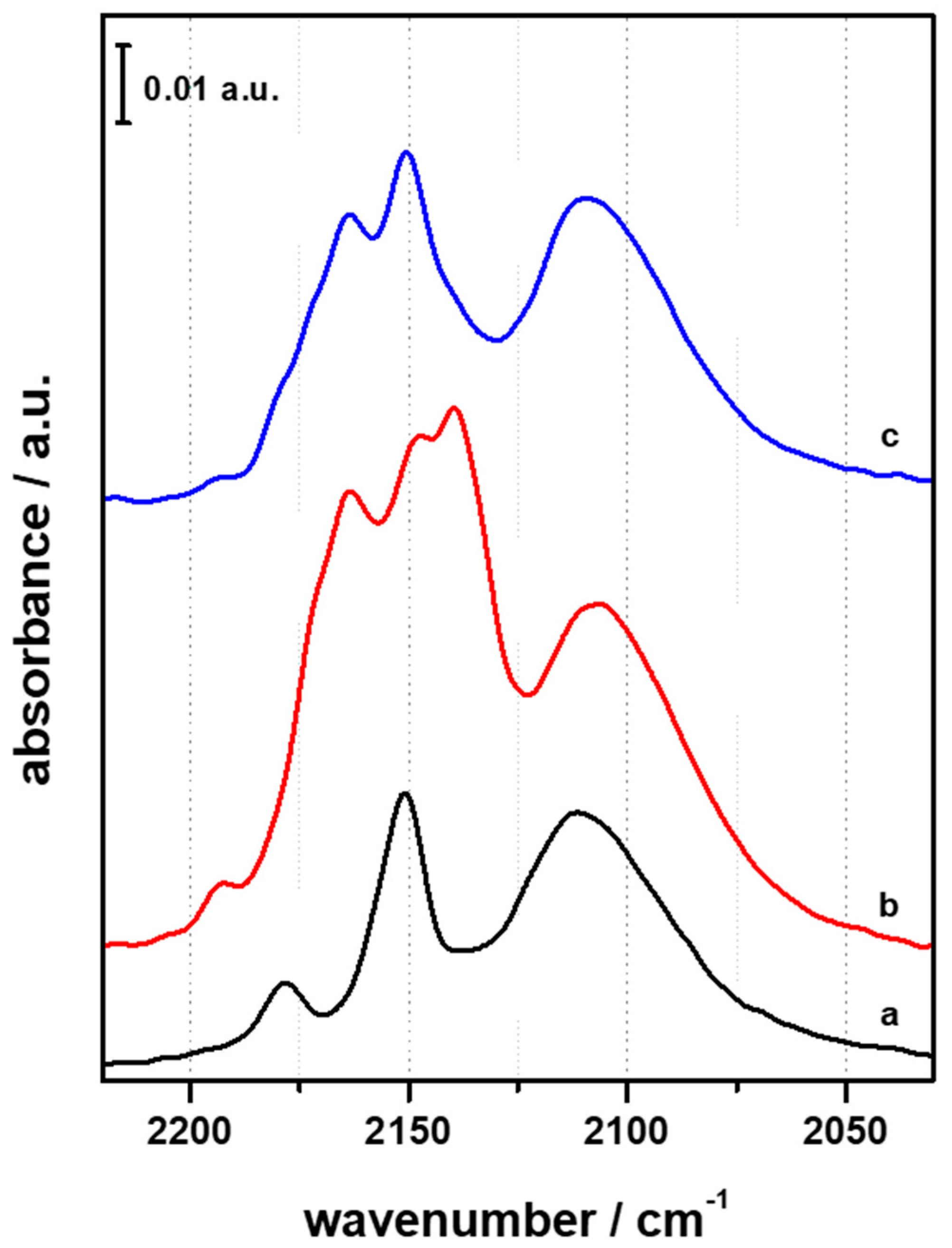
3.1. FTIR Investigation of the Pristine HFER Catalysts
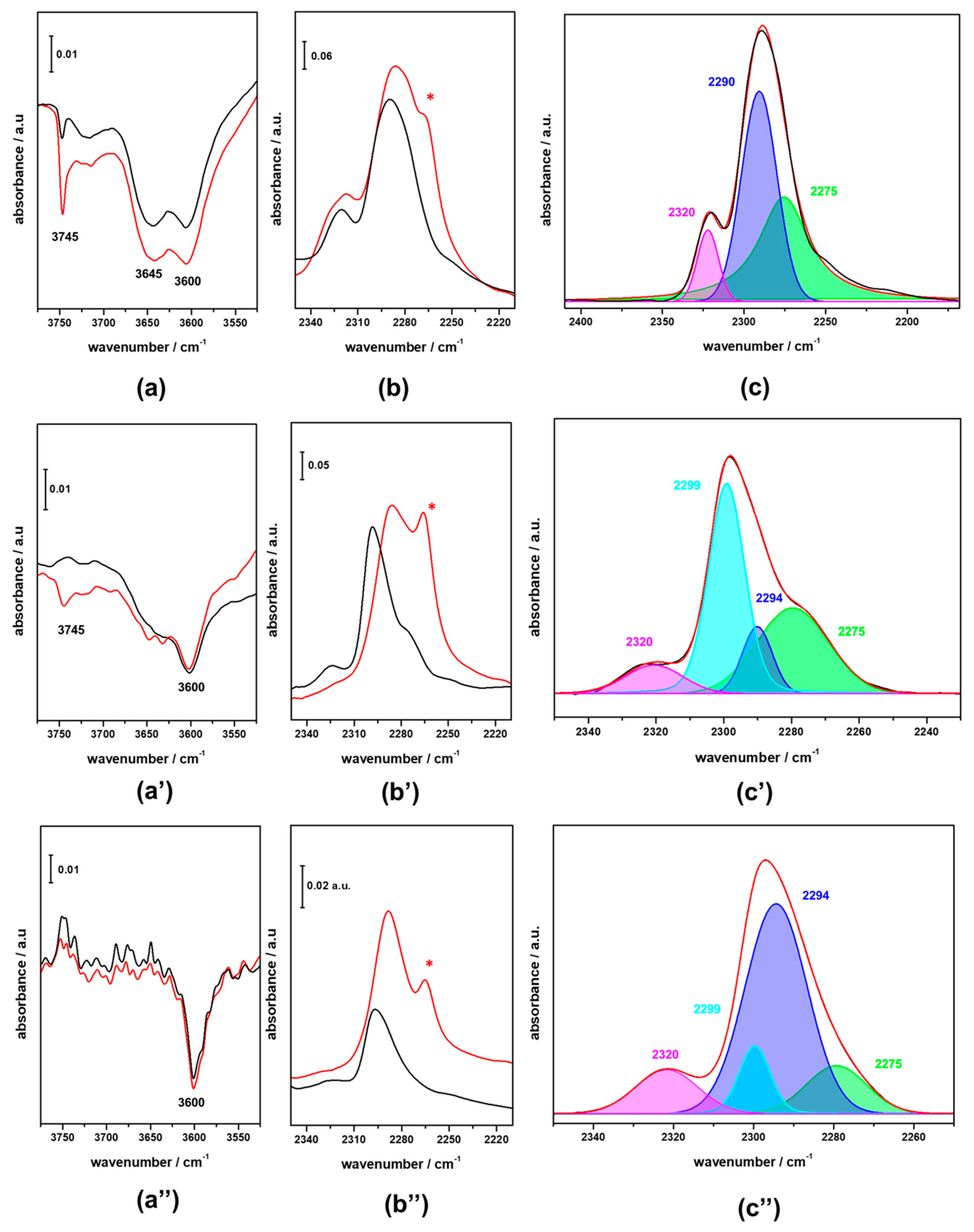
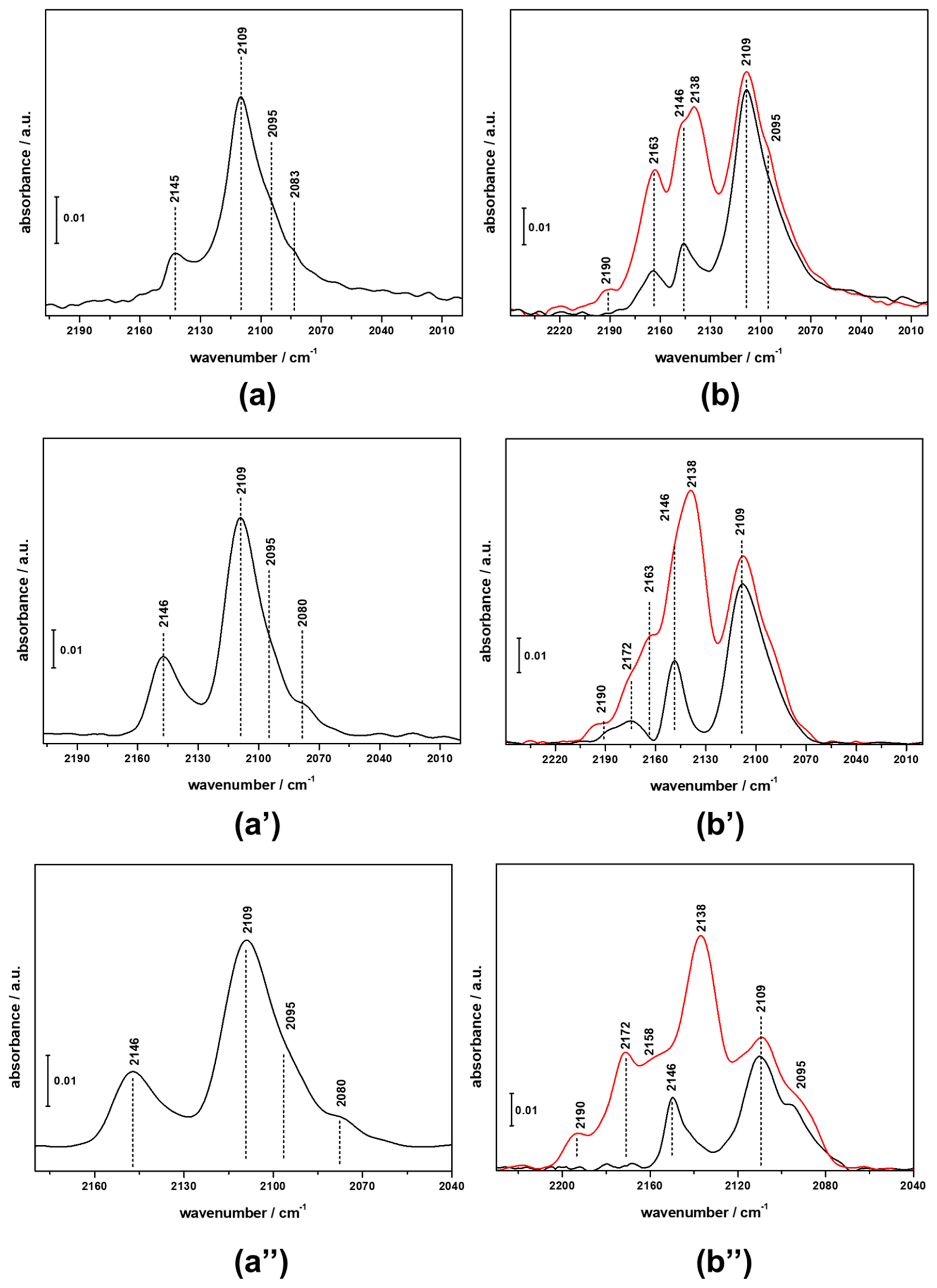
3.2. FTIR Investigation of the Fresh CZZ‒FER Catalysts
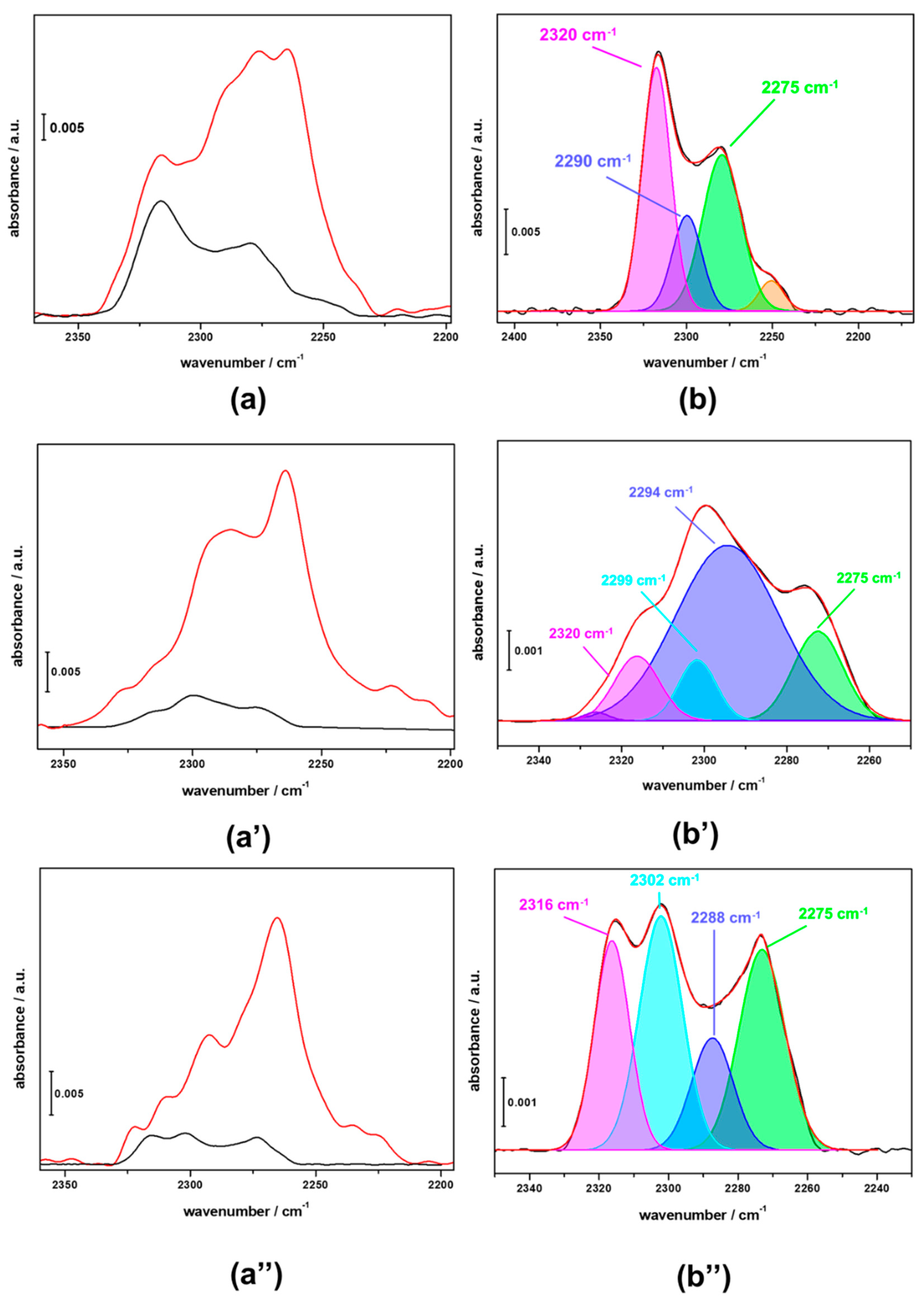
3.3. FTIR Investigation of the Used CZZ‒FER Catalysts and Insights about Deactivation during CO2-to-DME One-Pot Conversion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arcoumanis, C.; Bae, C.; Crookes, R.; Kinoshita, E. The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review. Fuel 2008, 87, 1014–1030. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, C. Applicability of dimethyl ether (DME) in a compression ignition engine as an alternative fuel. Energy Convers. Manag. 2014, 86, 848–863. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl ether (DME) as an alternative fuel. J. Pow. Source 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Haro, P.; Trippe, F.; Stahl, R.; Henrich, E. Bio-syngas to gasoline and olefins via DME—A comprehensive techno-economic assessment. Appl. Energy 2013, 108, 54–65. [Google Scholar] [CrossRef]
- Tian, P.; Wei, Y.; Ye, M.; Liu, Z. Methanol to Olefins (MTO): From fundamentals to commercialization. ACS Catal. 2015, 5, 1922–1938. [Google Scholar] [CrossRef]
- Raoof, F.; Taghizadeh, M.; Eliassi, A.; Yaripour, F. Effects of temperature and feed composition on catalytic dehydration of methanol to dimethyl ether over γ-alumina. Fuel 2008, 87, 2967–2971. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Xu, M.; Lunsford, J.H.; Goodman, D.W.; Bhattacharyya, A. Synthesis of dimethyl ether (DME) from methanol over solid-acid catalysts. Appl. Catal. A Gen. 1997, 149, 289–301. [Google Scholar] [CrossRef]
- Catizzone, E.; Bonura, G.; Migliori, M.; Frusteri, F.; Giordano, G. CO2 recycling to dimethyl ether: State-of-the-art and perspectives. Molecules 2018, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Tan, Y.; Han, Y. A comparative study on the thermodynamics of dimethyl ether synthesis from CO hydrogenation and CO2 hydrogenation. Ind. Eng. Chem. Res. 2006, 45, 1152–1159. [Google Scholar] [CrossRef]
- Pan, S.-Y.; Chiang, P.-C.; Pan, W.; Kim, H. Advances in state-of-art valorization of technologies for captured CO2 towards sustainable carbon cycle. Crit. Rev. Environ. Sci. Technol. 2018, 48, 471–534. [Google Scholar] [CrossRef]
- Perathoner, S.; Centi, G. CO2 recycling: A key strategy to introduce green energy in the chemical production chain. Chem. Sus. Chem. 2014, 7, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. From 1-D to 3-D zeolite structures: Performance assessment in catalysis of vapour-phase methanol dehydration to DME. Microp. Mesop. Mater. 2017, 243, 102–111. [Google Scholar] [CrossRef]
- Migliori, M.; Catizzone, E.; Aloise, A.; Bonura, G.; Gòmez-Hortiguela, L.; Frusteri, L.; Cannilla, C.; Frusteri, F.; Giordano, G. New insights about coke deposition in methanol-to-DME reaction over MOR-, MFI- and FER-type zeolites. J. Ind. Eng. Chem. 2018. [Google Scholar] [CrossRef]
- Prasad, P.S.S.; Bae, J.W.; Kang, S.-H.; Lee, Y.-J.; Jun, K.-W. Single-step synthesis of DME from syngas on Cu–ZnO–Al2O3/zeolite bifunctional catalysts: The superiority of ferrierite over the other zeolites. Fuel Process. Technol. 2008, 89, 1281–1286. [Google Scholar] [CrossRef]
- Montesano, R.; Narvaez, A.; Chadwick, D. Shape-selectivity effects in syngas-to-dimethyl ether conversion over Cu/ZnO/Al2O3 and zeolite mixtures: Carbon deposition and by-product formation. Appl. Catal. A Gen. 2014, 448, 69–77. [Google Scholar] [CrossRef]
- Garcia-Trenco, A.; Martinez, A. Direct synthesis of DME from syngas on hybrid CuZnAl/ZSM-5 catalysts: New insights into the role of zeolite acidity. Appl. Catal. A Gen. 2012, 411–412, 170–179. [Google Scholar] [CrossRef]
- Cai, M.; Palcic, A.; Subramanian, V.; Moldovan, S.; Ersen, O.; Valtchev, V.; Ordomsky, V.V.; Khodakov, A.Y. Direct dimethyl ether synthesis from syngas on copper-zeolite hybrid catalysts with a wide range of zeolite particle size. J. Catal. 2016, 338, 227–238. [Google Scholar] [CrossRef]
- Frusteri, F.; Migliori, M.; Cannilla, C.; Frusteri, L.; Catizzone, E.; Aloise, A.; Giordano, G.; Bonura, G. Direct CO2-to-DME hydrogenation reaction: New evidences of a superior behavior of FER-based hyb rid systems to obtain high DME yield. J. CO2 Util. 2017, 18, 353–361. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. Dimethyl ether synthesis via methanol dehydration: Effect of zeolite structure. Appl. Catal. A Gen. 2015, 502, 215–220. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Spadaro, L.; Cannilla, C.; Arena, F.; Frusteri, F. Hbrid Cu–ZnO–ZrO2/H-ZSM5 system for the direct synthesis of DME by CO2 hydrogenation. Appl. Catal. B Environ. 2013, 140, 16–24. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B Environ. 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Ge, Q.; Huang, Y.; Qiu, F.; Li, S. Bifunctional catalysts for conversion of synthesis gas to dimethyl ether. Appl. Catal. A Gen. 1998, 167, 23–30. [Google Scholar] [CrossRef]
- Frusteri, F.; Bonura, G.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Stepwise tuning of metal-oxide and acid sites of CuZnZr-MFI hybrid catalysts for the direct DME synthesis by CO2 hydrogenation. Appl. Catal. B Environ. 2015, 176–177, 522–531. [Google Scholar] [CrossRef]
- Bonura, G.; Frusteri, F.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Catalytic features of CuZnZr-zeolite hybrid systems for the direct CO2-to-DME hydrogenation reaction. Catal. Today 2016, 277, 48–54. [Google Scholar] [CrossRef]
- Bonura, G.; Migliori, M.; Frusteri, L.; Cannilla, C.; Catizzone, E.; Giordano, G.; Frusteri, F. Acidity control of zeolite functionality on activity and stability of hybrid catalysts during DME production via CO2 hydrogenation. J. CO2 Util. 2018, 24, 398–406. [Google Scholar] [CrossRef]
- Bordiga, S.; Turnes Palomino, G.; Pazè, C.; Zecchina, A. Vibrational spectroscopy of H2, N2, CO and NO adsorbed on H, Li, Na, K-exchanged ferrierite. Microp. Mesop. Mater. 2000, 34, 67–80. [Google Scholar] [CrossRef]
- Zholobenko, V.L.; Lukyanov, D.B.; Dwyer, J. Ferrierite and SUZ-4 zeolite: Characterization of acid sites. J. Phys. Chem. B 1998, 102, 2715–2721. [Google Scholar] [CrossRef]
- Szanyi, J.; Paffett, M.T. FTIR investigation of the co-adsorption of acetonitrile and carbon monoxide on Cu-ZSM-5. J. Chem. Soc. Faraday Trans. 1996, 92, 5165–5175. [Google Scholar] [CrossRef]
- Garcia-Trenco, A.; Vidal-Moyam, A.; Martinez, A. Study of the interaction between components in hybrid CuZnAl/HZSM-5 catalysts and its impact in the syngas-to-DME reaction. Catal. Today. 2012, 179, 43–51. [Google Scholar] [CrossRef]
- Garcia-Trenco, A.; Valencia, S.; Martinez, A. The impact of zeolite pore structure on the catalytic behaviour of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis. Appl. Catal. A Gen. 2013, 468, 102–111. [Google Scholar] [CrossRef]
- Arena, F.; Italiano, G.; Barbera, K.; Bordiga, S.; Bonura, G.; Spadaro, L.; Frusteri, F. Solid-state interaction, adsorption sites and functionality of Cu-ZnO/ZrO2 catalysts in the CO2 hydorgenation to CH3OH. Appl. Catal. A Gen. 2008, 350, 16–23. [Google Scholar] [CrossRef]
- Scarano, D.; Bordiga, S.; Lamberti, C.; Spoto, G.; Ricchiardi, G.; Zecchina, A.; Otero Areàn, C. FTIR study of the interaction of CO with pure and silica-supported copper(I) oxide. Surf. Sci. 1998, 411, 272–285. [Google Scholar] [CrossRef]
- Pritchard, J.; Catterick, T.; Gupta, R.K. Infrared spectroscopy of chemisorbed carbon monoxide on copper. Surf. Sci. 1975, 53, 1–20. [Google Scholar] [CrossRef]
- Kohler, M.A.; Cant, N.W.; Wainwright, M.S.; Trimm, D.L. Infrared spectroscopy studies of carbon monoxide adsorbed on a series of silica-supported copper catalysts in different oxidation states. J. Catal. 1989, 117, 188–201. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Coluccia, S.; Martra, G.; Ravasio, N. Cu/SiO2 and Cu/SiO2-TiO2 catalysts: I. TEM, DR UV-Vis-NIR, and FTIR characterization. J. Catal. 1999, 184, 316–326. [Google Scholar] [CrossRef]
- Hadjiivanov, K. Chapter Two—Identification and Characterization of Surface Hydroxyl Groups by Infrared Spectroscopy. In Advances in Catalysis; Jentoft, F.C., Ed.; Academic Press: London, UK, 2014; pp. 99–318. [Google Scholar]
- Penkova, A.; Dzwigaj, S.; Kefirov, R.; Hadjiivanov, K.; Che, M. Effect of preparation method on the state ions in BEA zeolites. A study by Fourier Transform Infrared Spectroscopy of adsorbed CO and NO, Temperature-Programmed Reduction, and X-Ray diffraction. J. Phys. Chem. C 2007, 111, 8623–8631. [Google Scholar] [CrossRef]
- Mihaylov, M.; Hadjiivanov, K. FTIR study of CO and NO adsorption and coadsorption on Ni-ZSM-5 and Ni/SiO2. Langmuir 2002, 18, 4376–4383. [Google Scholar] [CrossRef]
- Ordomsky, V.V.; Cai, M.; Sushkevich, V.; Moldovan, S.; Ersen, O.; Lancelot, C.; Valtchev, V.; Khodakov, A.Y. The role of external acid sites of ZSM-5 in deactivation of hybrid CuZnAl/ZSM-5 catalyst for direct dimethyl ether synthesis from syngas. Appl. Catal. A Gen. 2014, 486, 266–275. [Google Scholar] [CrossRef]
- Catizzone, E.; Migliori, M.; Purita, A.; Giordano, G. Ferrierite vs. γ-Al2O3: The superiority of zeolites in terms of water-resistance in vapour-phase dehydration of methanol to dimethyl ether. J. Energy Chem. 2018. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Erena, J.; Sierra, I.; Olazar, M.; Bilbao, J. Deactivation and regeneration of hybrid catalysts in the single-step synthesis of dimethyl ether from syngas and CO2. Catal. Today 2005, 106, 265–270. [Google Scholar] [CrossRef]
- Sierra, I.; Erena, J.; Aguayo, A.T.; Arandes, J.M.; Olazar, M.; Bilbao, J. Co-feeding water to attenuate deactivation of the catalyst metallic function (CuO-ZnO-Al2O3) by coke in the direct synthesis of dimethyl ether. Appl. Catal. B Environ. 2011, 106, 167–173. [Google Scholar] [CrossRef]
- Fierro, G.; Ferraris, G.; Moretti, G. CuO nanoparticles entrapped in MFI framework: Investigation of textural, magnetic and catalytic properties of Cu-ZSM-5 and Cu-1 catalysts. Appl. Catal. B Environ. 2009, 91, 499–506. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Band Position and Assignment | Number of Acid Sites (mmol g−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HFER8 | CZZFER8 | CZZFER8 Used | HFER30 | CZZFER30 | CZZFER30 Used | HFER60 | CZZFER60 | CZZFER60 Used | |
| 2320 cm−1 Lewis sites | 0.150 | 0.120 | 0.08 | 0.030 | 0.005 | 0.004 | 0.018 | 0.003 | 0.003 |
| 2290 cm−1 Brønsted sites | 0.807 | 0.08 | 0.02 | 0.480 | 0.063 | 0.05 | 0.387 | 0.02 | 0.02 |
| 2275 cm−1 Silanols | 0.930 | 0.520 | 0.24 | 0.250 | 0.035 | 0.01 | 0.190 | 0.01 | 0.01 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miletto, I.; Catizzone, E.; Bonura, G.; Ivaldi, C.; Migliori, M.; Gianotti, E.; Marchese, L.; Frusteri, F.; Giordano, G. In Situ FT-IR Characterization of CuZnZr/Ferrierite Hybrid Catalysts for One-Pot CO2-to-DME Conversion. Materials 2018, 11, 2275. https://doi.org/10.3390/ma11112275
Miletto I, Catizzone E, Bonura G, Ivaldi C, Migliori M, Gianotti E, Marchese L, Frusteri F, Giordano G. In Situ FT-IR Characterization of CuZnZr/Ferrierite Hybrid Catalysts for One-Pot CO2-to-DME Conversion. Materials. 2018; 11(11):2275. https://doi.org/10.3390/ma11112275
Chicago/Turabian StyleMiletto, Ivana, Enrico Catizzone, Giuseppe Bonura, Chiara Ivaldi, Massimo Migliori, Enrica Gianotti, Leonardo Marchese, Francesco Frusteri, and Girolamo Giordano. 2018. "In Situ FT-IR Characterization of CuZnZr/Ferrierite Hybrid Catalysts for One-Pot CO2-to-DME Conversion" Materials 11, no. 11: 2275. https://doi.org/10.3390/ma11112275
APA StyleMiletto, I., Catizzone, E., Bonura, G., Ivaldi, C., Migliori, M., Gianotti, E., Marchese, L., Frusteri, F., & Giordano, G. (2018). In Situ FT-IR Characterization of CuZnZr/Ferrierite Hybrid Catalysts for One-Pot CO2-to-DME Conversion. Materials, 11(11), 2275. https://doi.org/10.3390/ma11112275