Development of Coarse-Grained Liquid-Crystal Polymer Model with Efficient Electrostatic Interaction: Toward Molecular Dynamics Simulations of Electroactive Materials


{kind=link}
{kind=link}
{kind=link}
{kind=link}
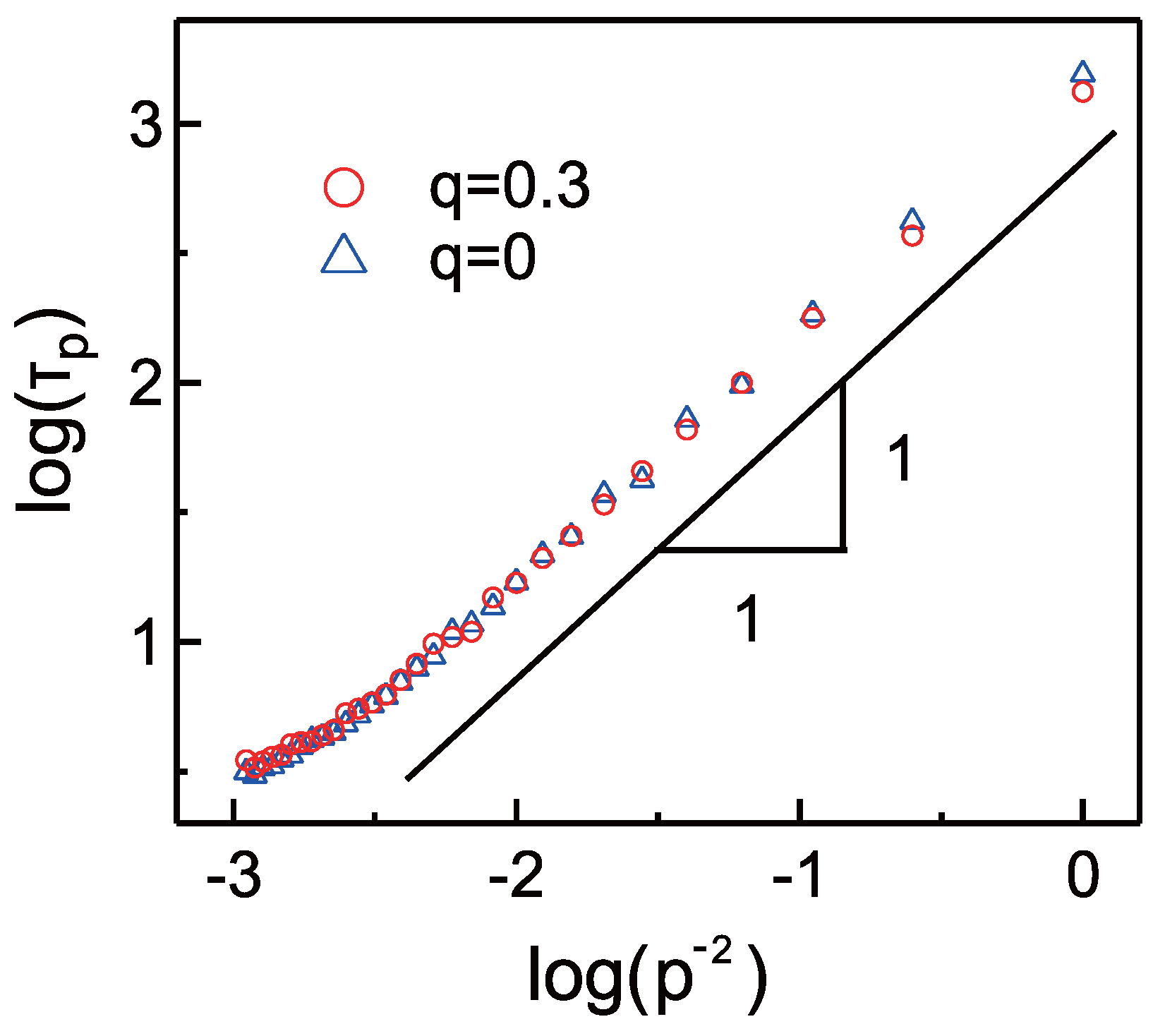{kind=link}
Abstract
1. Introduction
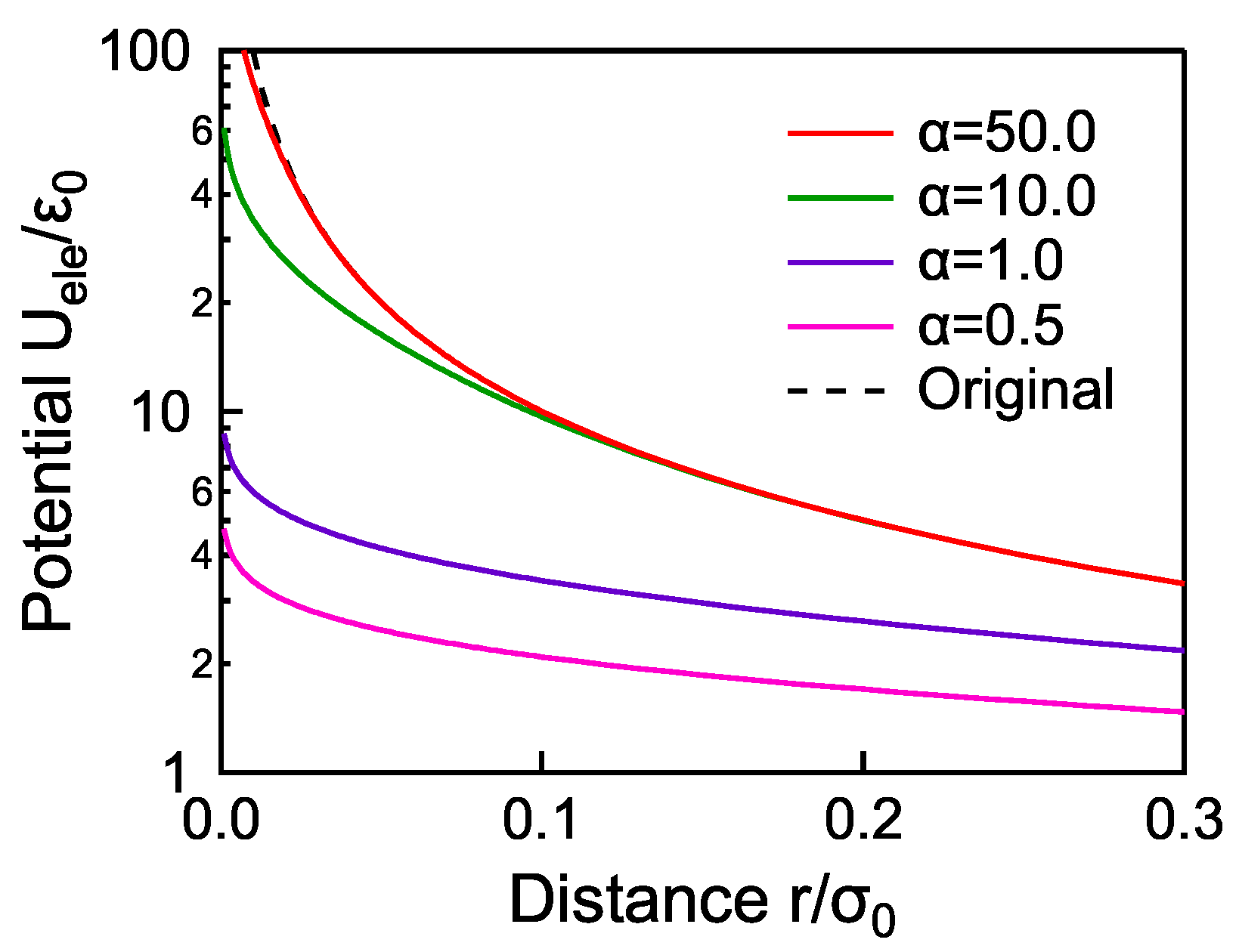
2. Methodology
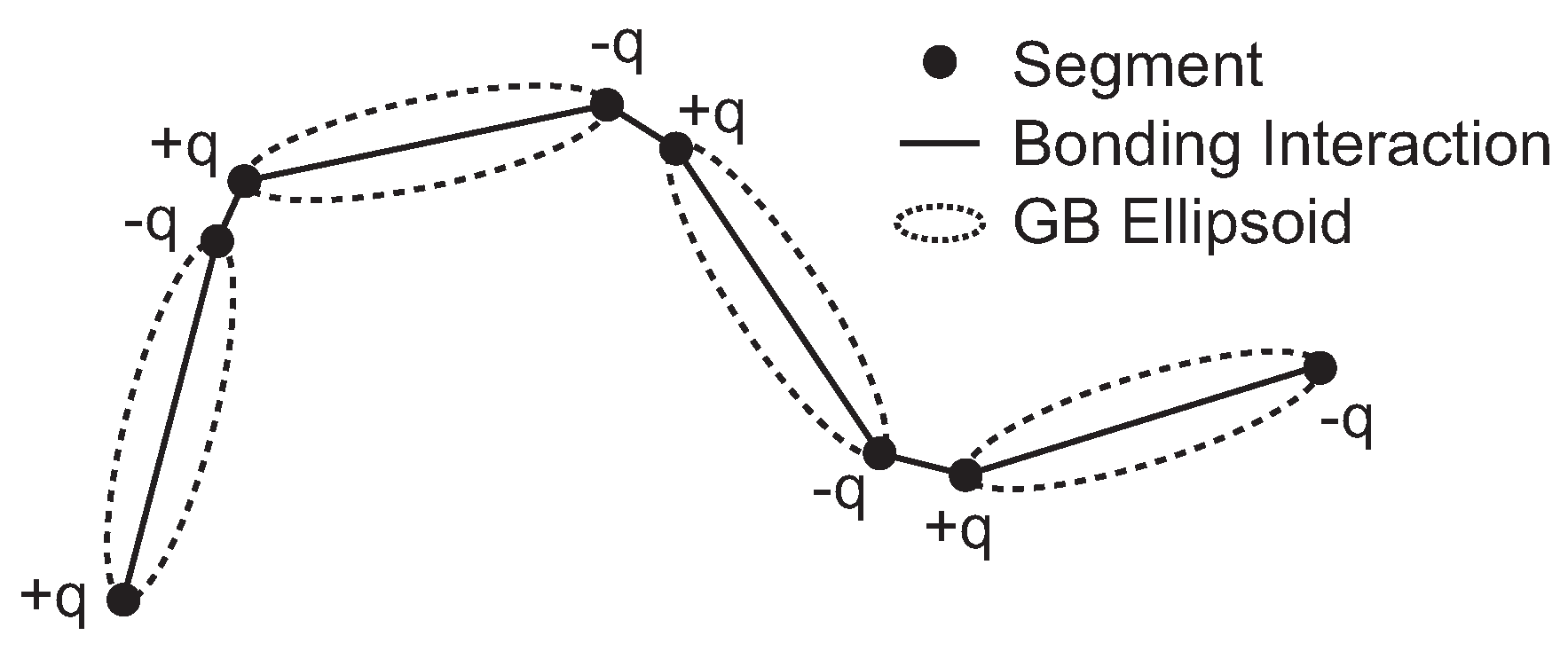
2.1. Coarse-Grained LCP Model
2.2. Simulation Detail
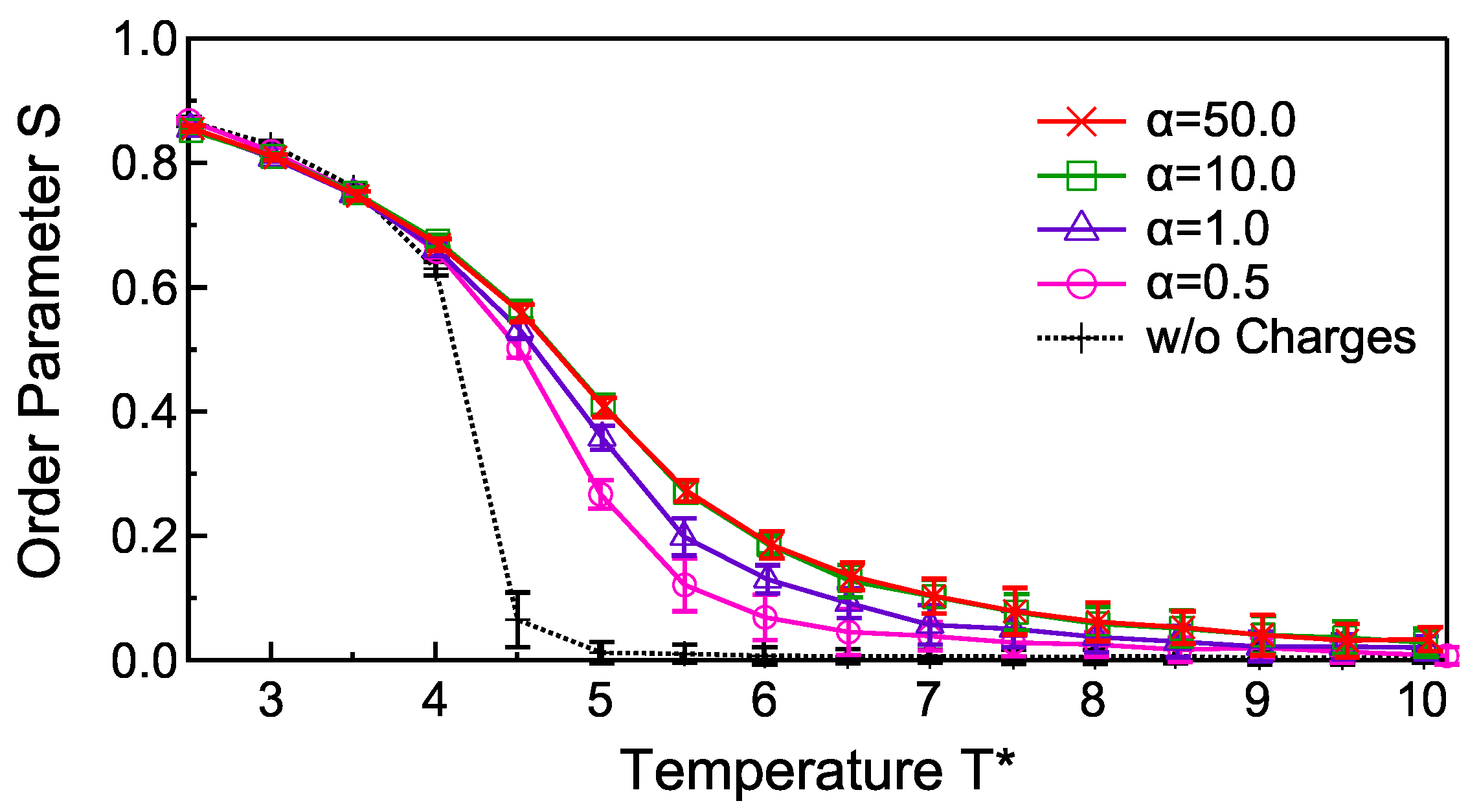
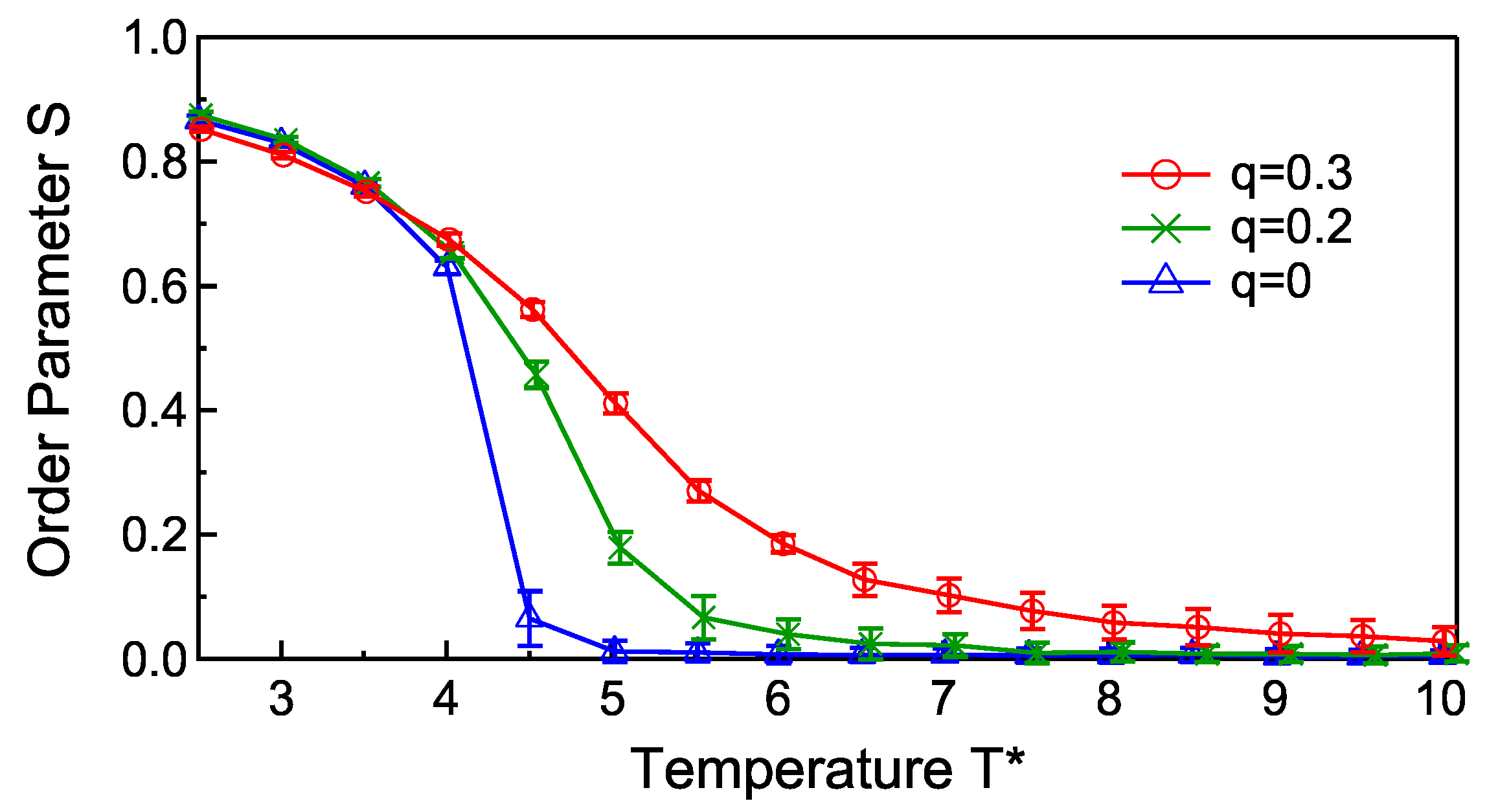
3. Results and Discussions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kato, T.; Tanabe, K. Electro- and photoactive molecular assemblies of liquid crystals and physical gels. Chem. Lett. 2009, 7, 634–639. [Google Scholar] [CrossRef]
- Bhargavi, R.; Nair, G.G.; Prasada, S.K.; Prabhu, R.; Yelamaggad, C.V. Anomalously large bend elastic constant and faster electro-optic response in anisotropic gels formed by a dipeptide. J. Appl. Phys. 2011, 109. [Google Scholar] [CrossRef]
- Choi, Y.J.; Yoon, W.J.; Kim, D.Y.; Park, M.; Lee, Y.; Jung, D.; Kim, J.S.; Yu, Y.T.; Lee, C.R.; Jeong, K.Y. Stimuli-responsive liquid crystal physical gels based on the hierarchical superstructures of benzene-1,3,5-tricarboxamide macrogelators. Polym. Chem. 2017, 8, 1888–1894. [Google Scholar] [CrossRef]
- Li, C.; Guo, R.; Jiang, X.; Hu, S.; Li, L.; Cao, X.; Yang, H.; Song, Y.; Ma, Y.; Jiang, L. Reversible switching of water-droplet mobility on a superhydrophobic surface based on a phase transition of a side-chain liquid-crystal polymer. Adv. Mater. 2009, 21, 4254–4258. [Google Scholar] [CrossRef]
- White, T.J.; Broer, D.J. Programmable and adaptive mechanics with liquid crystal polymer networks and elastomers. Nat. Mater. 2015, 14, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Xiea, P.; Zhang, R. Liquid crystal elastomers, networks and gels: advanced smart materials. J. Mater. Chem. 2005, 15, 2529–2550. [Google Scholar] [CrossRef]
- Okamoto, T.; Urayama, K.; Takigawa, T. Large electromechanical effect of isotropic-genesis polydomain nematic elastomers. Soft Matter 2011, 7, 10585–10589. [Google Scholar] [CrossRef]
- Ikeda, T.; Nakano, M.; Yu, Y.; Tsutsumi, O.; Kanazawa, A. Anisotropic bending and unbending behavior of azobenzene liquid-crystalline gels by light exposure. Adv. Mater. 2003, 15, 201–205. [Google Scholar] [CrossRef]
- Wu, W.; Yao, L.; Yang, T.; Yin, R.; Li, F.; Yu, Y. NIR-light-induced deformation of cross-linked liquid-crystal polymers using upconversion nanophosphors. J. Am. Chem. Soc. 2011, 133, 15810–15813. [Google Scholar] [CrossRef] [PubMed]
- Haan, L.T.; Verjans, J.M.N.; Broer, D.J.; Bastiaansen, C.W.M.; Schenning, A.P.H.J. Humidity-Responsive Liquid Crystalline Polymer Actuators with an Asymmetry in the Molecular Trigger That Bend, Fold, and Curl. J. Am. Chem. Soc. 2014, 136, 10585–10588. [Google Scholar] [CrossRef] [PubMed]
- Ohm, C.; Brehmer, M.; Zentel, R. Liquid crystalline elastomers as actuators and sensors. Adv. Mater. 2010, 22, 3366–3387. [Google Scholar] [CrossRef] [PubMed]
- McConney, M.E.; Martinez, A.; Tondiglia, V.P.; Lee, K.M.; Langley, D.; Smalyukh, I.I.; White, T.J. Topography from topology: photoinduced surface features generated in liquid crystal polymer networks. Adv. Mater. 2013, 25, 5880–5885. [Google Scholar] [CrossRef] [PubMed]
- Herzer, N.; Guneysu, H.; Davies, D.J.D.; Yildirim, D.; Vaccaro, A.R.; Broer, D.J.; Bastiaansen, C.W.M.; Schenning, A.P.H.J. Printable optical sensors based on H-bonded supramolecular cholesteric liquid crystal networks. J. Am. Chem. Soc. 2012, 134, 7608–7611. [Google Scholar] [CrossRef] [PubMed]
- Pasini, P.; Skačej, G.; Zannoni, C. A microscopic lattice model for liquid crystal elastomers. Chem. Phys. Lett. 2005, 413, 463–467. [Google Scholar] [CrossRef]
- Stimson, L.M.; Wilson, M.R. Molecular dynamics simulations of side chain liquid crystal polymer molecules in isotropic and liquid-crystalline melts. J. Chem. Phys. 2005, 123, 34908. [Google Scholar] [CrossRef] [PubMed]
- Skačej, G.; Zannoni, C. Molecular simulations elucidate electric field actuation in swollen liquid crystal elastomers. Proc. Natl. Acad. Sci. USA 2012, 109, 10193–10198. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, J.K.; Roberts, T.F.; Shekhar, R.; Abbott, N.L.; de Pablo, J.J. Modeling the polydomain-monodomain transition of liquid crystal elastomers. Phys. Rev. E 2013, 87, 020502. [Google Scholar] [CrossRef] [PubMed]
- Benjeddou, A. Advances in piezoelectric finite element modeling of adaptive structural elements: A survey. Comput. Struct. 2000, 76, 347–363. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Chertovich, A.V.; Kramarenko, E.Y. Dissipative particle dynamics for systems with high density of charges: Implementation of electrostatic interactions. J. Chem. Phys. 2016, 145. [Google Scholar] [CrossRef] [PubMed]
- Groot, R.D. Electrostatic interactions in dissipative particle dynamics–simulation of polyelectrolytes and anionic surfactants. J. Chem. Phys. 2003, 118, 11265–11277. [Google Scholar] [CrossRef]
- Berardi, R.; Zannoni, C.; Lintuvuori, J.S.; Wilson, M.R. A soft-core Gay-Berne model for the simulation of liquid crystals by Hamiltonian replica exchange. J. Chem. Phys. 2009, 131. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Sawa, F.; Shoji, T.; Fukunaga, H.; Takimoto, J.; Doi, M. A general-purpose coarse-grained molecular dynamics program. Comput. Phys. Commun. 2002, 145, 267–279. [Google Scholar] [CrossRef]
- Fukunaga, H.; Takimoto, J.; Doi, M. Molecular dynamics simulation study on the phase behavior of the Gay-Berne model with a terminal dipole and a flexible tail. J. Chem. Phys. 2004, 120, 7792–7800. [Google Scholar] [CrossRef] [PubMed]
- Gay, G.B.; Berne, B.J. Modification of the overlap potential to mimic a linear site–site potential. J. Chem. Phys. 1981, 74, 3316–3319. [Google Scholar] [CrossRef]
- Swope, W.C.; Andersen, H.C.; Berens, P.H.; Wilson, K.R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters. J. Chem. Phys. 1982, 76, 637–649. [Google Scholar] [CrossRef]
- Satoh, K.; Mita, S.; Kondo, S. Monte Carlo simulations using the dipolar Gay-Berne model: effect of terminal dipole moment on mesophase formation. Chem. Phys. Lett. 1996, 255, 99–104. [Google Scholar] [CrossRef]
- Bose, T.K.; Saha, J. Ferroelectric order in liquid crystal phases of polar disk-shaped ellipsoids. Phys. Rev. E 2014, 89, 052509. [Google Scholar] [CrossRef] [PubMed]
- Gramsbergen, E.F.; Longa, L.; Jeu, W.H. Landau theory of the nematic-isotropic phase transition. Phys. Rep. 1986, 135, 195–257. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1990; pp. 91–96. [Google Scholar]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tagashira, K.; Takahashi, K.Z.; Fukuda, J.-i.; Aoyagi, T. Development of Coarse-Grained Liquid-Crystal Polymer Model with Efficient Electrostatic Interaction: Toward Molecular Dynamics Simulations of Electroactive Materials. Materials 2018, 11, 83. https://doi.org/10.3390/ma11010083
Tagashira K, Takahashi KZ, Fukuda J-i, Aoyagi T. Development of Coarse-Grained Liquid-Crystal Polymer Model with Efficient Electrostatic Interaction: Toward Molecular Dynamics Simulations of Electroactive Materials. Materials. 2018; 11(1):83. https://doi.org/10.3390/ma11010083
Chicago/Turabian StyleTagashira, Kenji, Kazuaki Z. Takahashi, Jun-ichi Fukuda, and Takeshi Aoyagi. 2018. "Development of Coarse-Grained Liquid-Crystal Polymer Model with Efficient Electrostatic Interaction: Toward Molecular Dynamics Simulations of Electroactive Materials" Materials 11, no. 1: 83. https://doi.org/10.3390/ma11010083
APA StyleTagashira, K., Takahashi, K. Z., Fukuda, J.-i., & Aoyagi, T. (2018). Development of Coarse-Grained Liquid-Crystal Polymer Model with Efficient Electrostatic Interaction: Toward Molecular Dynamics Simulations of Electroactive Materials. Materials, 11(1), 83. https://doi.org/10.3390/ma11010083