2.1. Comparison Method
The comparison method deals with the determination of the energy efficiency and luminous efficacy of CL. Before describing the comparison method we shall briefly recall the definitions of energy efficiency and luminous efficacy. The energy efficiency η
e of CL is defined as the ratio of the power density
Pr of the CL and the power density
Pe of the e-beam that hits the phosphor layer (expressed in %):
The power density of the e-beam is the product of beam voltage and current density at the sample. The adjustment of the current density for phosphor samples is the essence of the comparison method, to be discussed hereafter. For powder samples, the angular distribution of the emitted light intensity is assumed to be Lambertian, which means that the power density of the CL can be calculated according to
where
R is the radiance expressed in W/(sr·cm
2). The radiance of the emitted light is defined as:
where SR(λ) is the spectral radiance (in W/(sr·cm
2·nm)) as a function of the wavelength λ and the integration limits,
a and
b, are determined by the eye sensitivity curve for reasons of comparison with the luminance. The luminance
L (in cd/m
2) of the CL is defined as:
where
V(λ) is the eye sensitivity curve. The luminous efficacy η
l (in lm/W) for a Lambertian light distribution is given by
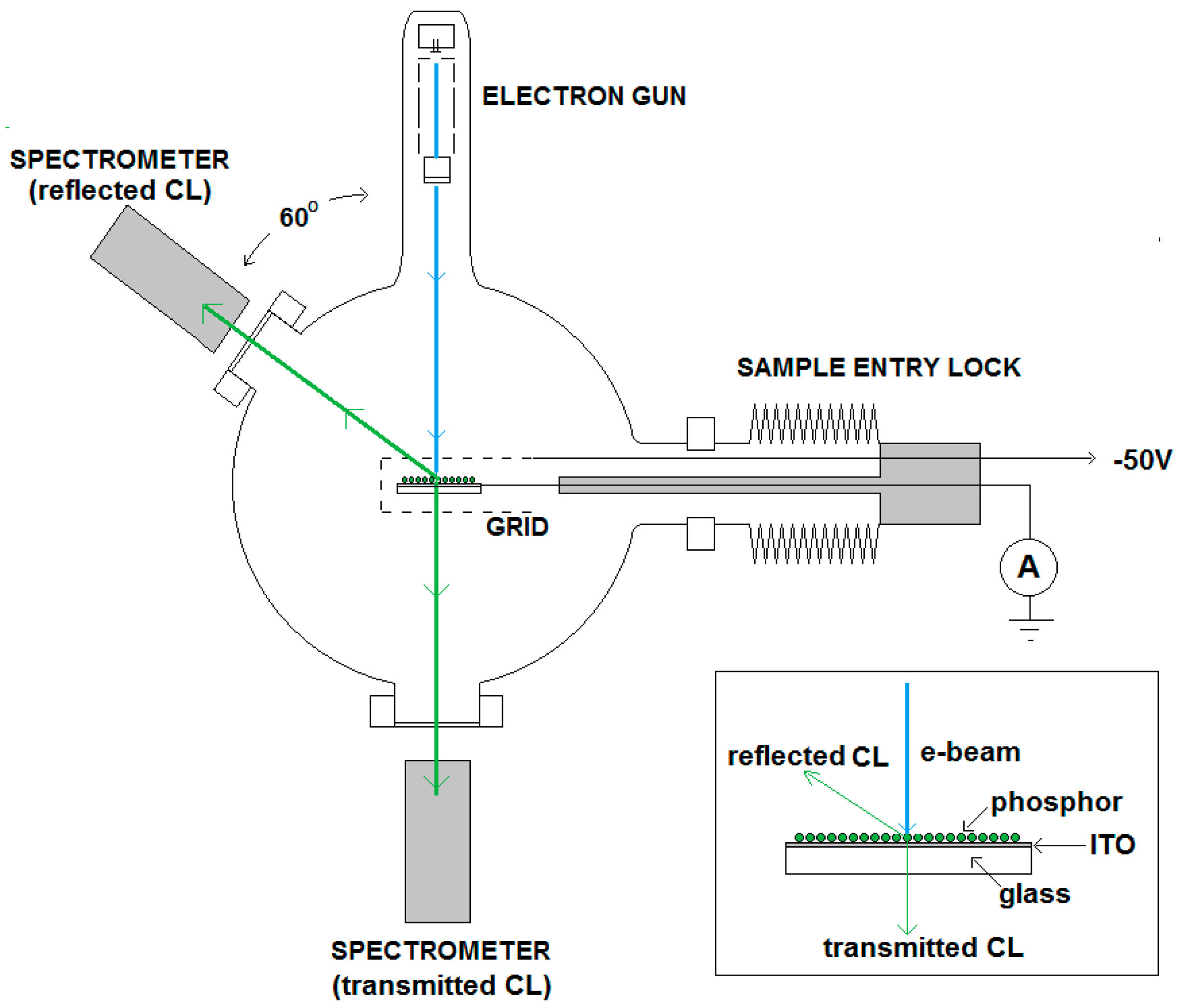
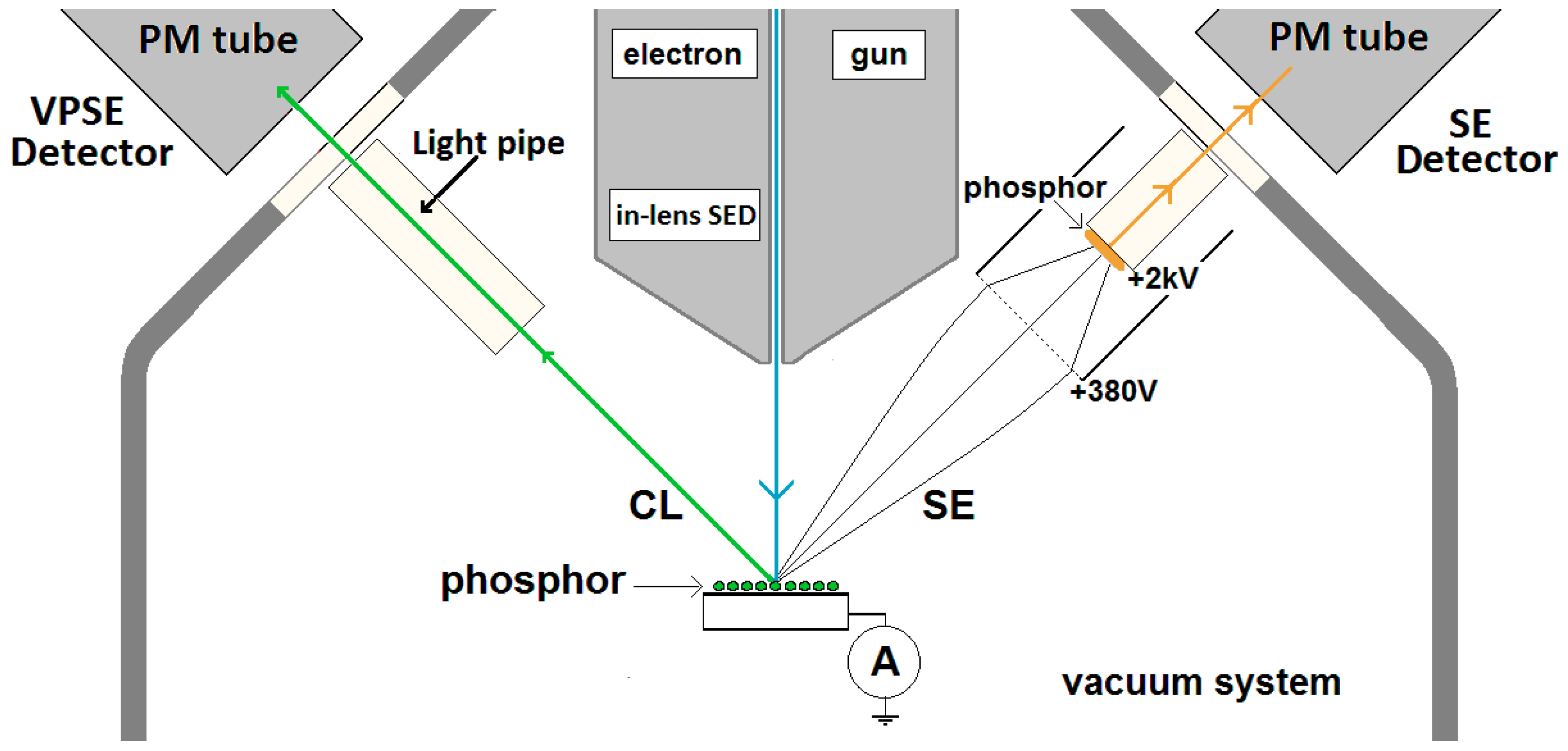
As mentioned above, the comparison method refers to an adjustment technique of the current density of an e-beam that is impinging on an insulating phosphor powder deposited as a thin film on a conductive substrate. The measuring set up for the samples has been depicted in
Figure 1, while a detail of a typical sample has been represented in the insert. The substrate is a glass plate coated with a thin conductive, transparent film of indium tin oxide (ITO). In
Figure 1, two principal directions for the emitted CL have been drawn: a reflection mode at the e-beam side and a transmission mode at the rear side of the glass plate. If the phosphor layer is very thick, the scattering events in the layer prevent transmission through the glass plate.
The coating thickness in our investigations was usually between 1 and 4 mg/cm
2; this implied that for small phosphor particles with diameters <1 μm the light distribution of the CL in both modes is assumed to be Lambertian [
18]. This assumption was explicitly made by Shea [
12] and Shea and Walko [
24], and implicitly by Chubun et al. [
13]. For layers with more than three particles on top of each other, this assumption has been well established; for monolayers of particles. the intensity of the CL deviates from a pure cosine distribution. Since we measured the radiance (and luminance) of the CL in the phosphor layer at an angle of 30°, there could be a slight underestimation of the measured radiance (and luminance) for samples with a very thin coating thickness. The coating thickness of layers deposited by either settling or electrophoresis in our experiments was measured by weighing [
18].
Figure 1 indicates that spectra are measured in both reflection and transmission mode. The advantage of this measuring method is that the sum of the radiances in the reflection and transmission modes is independent of the layer thickness for non-absorbing phosphor layers [
18] as long as the energy of the e-beam has completely been transferred to the phosphor particles. The latter condition is not satisfied in very thin phosphor layers, which contain pinholes. The second characteristic of our measuring technique is the adjustment of the beam current onto a conductive sample, which does not charge upon electron bombardment. For conductive materials it is straightforward to determine the effective current impinging on the surface. The actual current refers to the primary electrons hitting the surface minus the SEs leaving from that surface. Thus, in order to determine the effective current for CL, the SEs must be sent back to the phosphor to be collected. This can be done by biasing the target surface positively, or using a shield or grid that is biased negatively. The latter method was chosen, because in this way the collection of SEs emitted from the wall of the vacuum chamber could be avoided. In most of our experiments the shield was at −50V. In the case of a charging sample, it is impossible to guarantee that all SEs are collected by biasing the shield negatively. In that case a spurious current will be measured. In order to deal with this problem, a comparison method has been developed: CL from a charging sample is measured by applying the same current settings as used for a non-charging reference surface such as Cu, ITO or ZnO:Zn. Copper and ITO are chosen because they have almost the same backscattering yields as ZnO:Zn. The additional advantage of using ZnO:Zn as non-charging reference is that the CL of ZnO:Zn can be used to optimally align the spectrometers 1 and 2.
The reference and sample are positioned in the e-beam by a vertical translation (1–5 keV rig) or horizontal translation (2–15 keV rig) as indicated in
Figure 1. When making measurements, the reference is first positioned in the e-beam, the current is usually adjusted to 1 μA (as indicated by the high impedance ammeter), yielding a current density of 1 μA/cm
2, since the surface areas of references and samples are typically 1 cm
2. When the shield is biased to −50 V, low energy SEs from the reference sample are collected together with the primary electrons and thus a true measurement of the current striking the sample is made. The shield structure is designed to have as high a transparency as possible (about 98%) so that high energy backscattered electrons (BSEs) are able to escape unhindered: i.e., these are not detected in this way. BSEs do not contribute to the luminescence and must be discarded. It is worth noting that measurements of beam current made using enclosed Faraday cups normally collect these backscattered electrons, and so substantially over-estimate the energy absorbed by the phosphor. The conductive reference is ideally chosen to have a backscattering coefficient as close as possible to that of the sample, hence similar mean atomic number and morphology, so that the fraction of electrons backscattered from both is the same.
The sample is maintained at earth potential and so the kinetic energy of the electrons is proportional to the applied cathode voltage. This is not always the case in other studies; for example, in studies by Shea [
12,
24] and Wakefield et al. [
25], the samples were biased positively, and therefore a correction must be made to calculate the kinetic energy of the primary electrons. In making CL measurements from charging and non-charging phosphor samples, the current adjustment as determined for the reference is not altered. The ITO-layer is connected to earth during the CL-measurements, but the current that may have changed in the case of a charging sample is not recorded. The assumption is that the same quantity of primary electrons hit the charging phosphor layer as determined for the non-charging reference. Obviously, this is only correct if the voltage of the charged phosphor surface does not deviate much from the voltage of the conductive reference. The work of Seager et al. [
16] provides evidence for this hypothesis and by recording the so-called SE-yield curves we found that we could usually comply with this condition. This will be discussed hereafter.
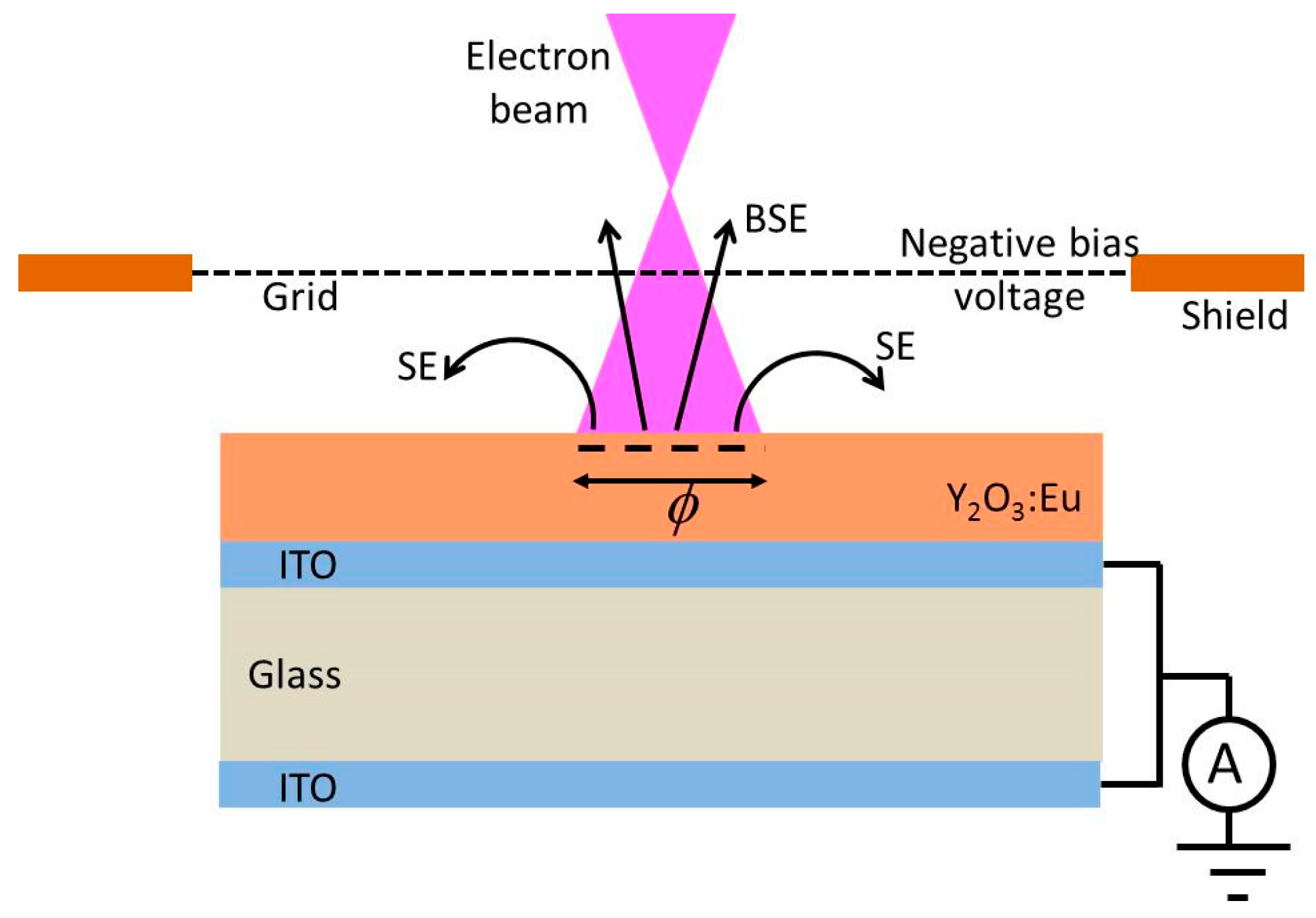
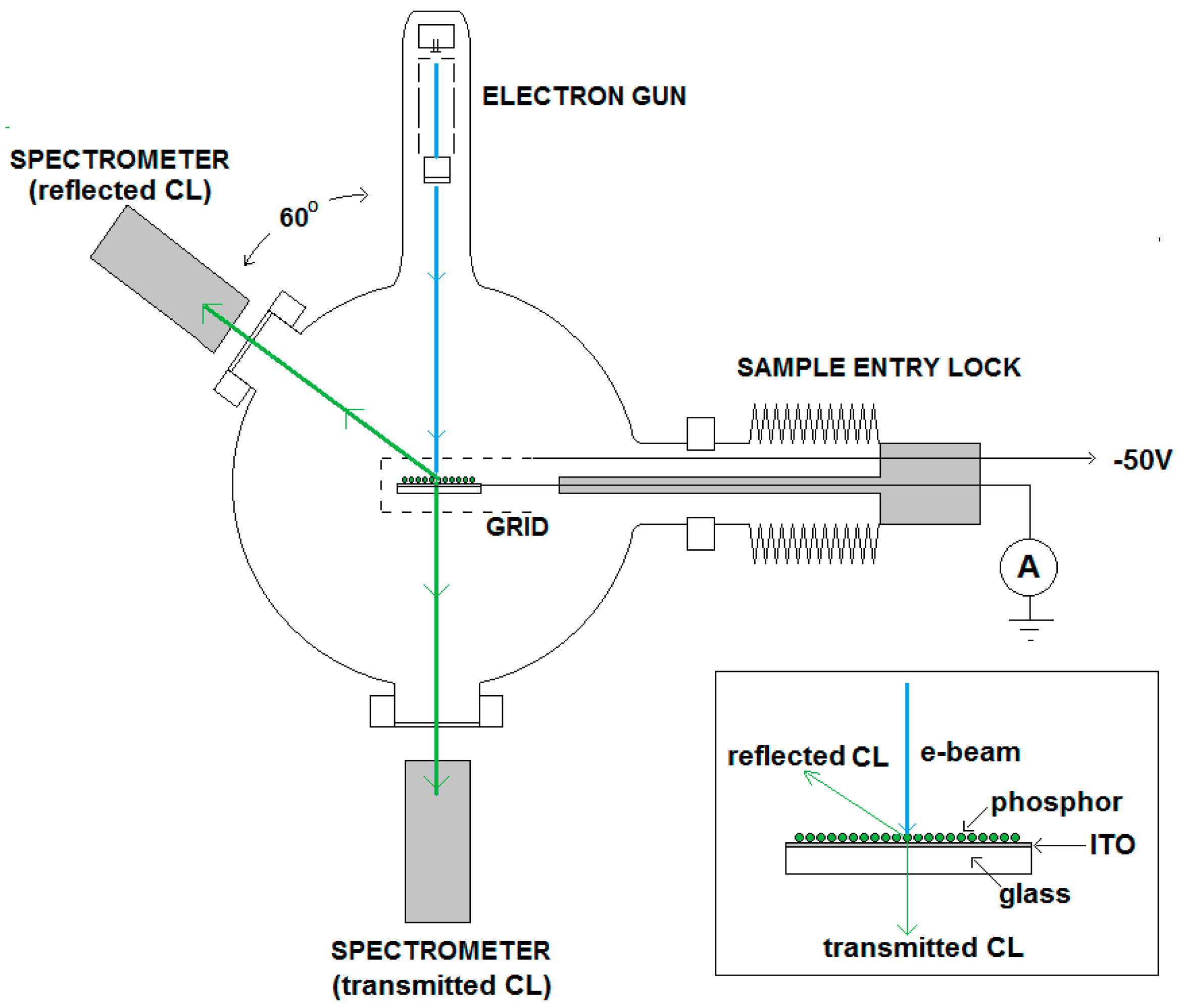
The size and luminance uniformity of the spot on the reference is visually optimized by adjusting the focus voltage of the electron gun. The focus voltage is about ~45% of the beam voltage in measuring the CL. At this focus voltage, the focus point of the electron gun is in front of the sample (over focus condition), as shown in
Figure 2.
In
Figure 2 the BSEs pass the negatively biased grid, whereas the slow SEs are deflected. However, in the case of charging, not all SEs will be collected, because some are repulsed by the negatively charged phosphor surface. By changing the focus voltage the size of the spot on the phosphor layer changed, implying that the the current density in the spot varies as well. The current density of the e-beam is, besides the voltage, the most important parameter to affect the charging of an insulating phosphor layer. This is shown in
Figure 3, where the sample current has been plotted as a function of V
shield. The curves in
Figure 3 are called SE-yield curves.
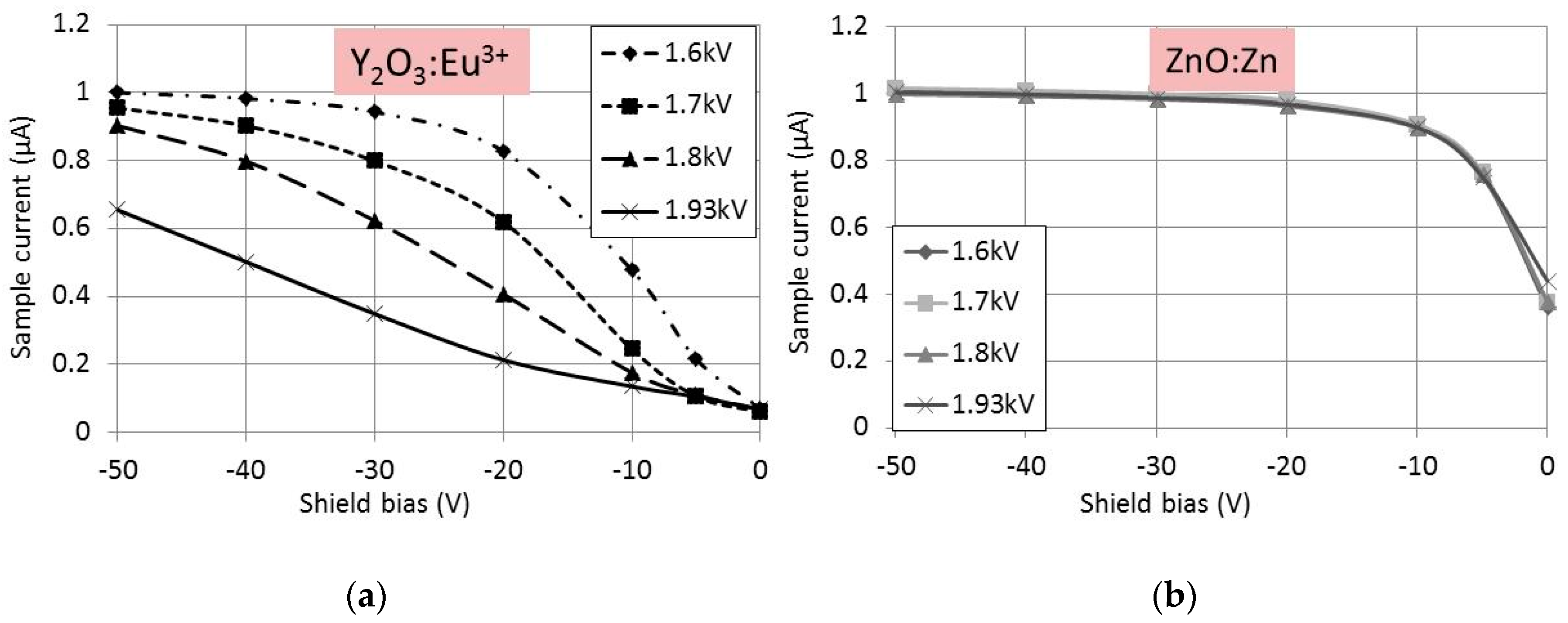
In
Figure 3 the SE-yield curves at various focus voltages for Y
2O
3:Eu
3+ and ZnO:Zn have been plotted at a primary beam energy of 3 keV. The sample current was adjusted to 1 μA at V
shield = −50 V and a focus voltage of 1.6 kV. The effect of focus voltage was substantial for Y
2O
3:Eu
3+, as seen in
Figure 3a, whereas the SE-yield in the case of ZnO:Zn was independent of the focus voltage. At the best focus voltage of 1.93 kV, the diameter of the spot size on the sample was 2.5 mm. The spot size increased to 9 mm at a focus voltage of 1.6 kV. From the curves in
Figure 3, we conclude that Y
2O
3:Eu
3+ is charging negatively. The absolute value of the charge increased when the spot sizes decreased, i.e., when the current density increased.
Figure 3a indicates that at high current density the shield voltage had to be decreased to collect all SEs. In some cases we had to decrease the shield voltage to −100 V to ensure complete collection. This proves that the charging voltages of Y
2O
3:Eu
3+ and Y
2O
2S:Eu
3+ phosphor layers are indeed limited, as described above.
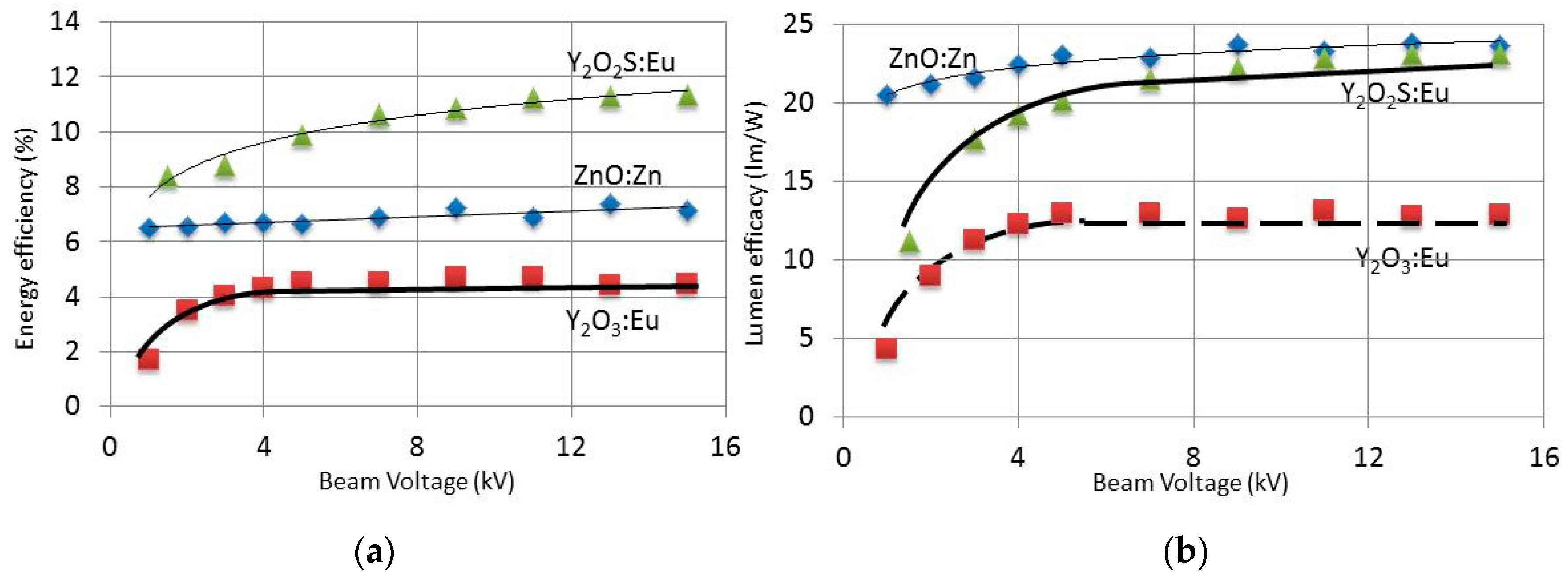
Figure 4a,b shows the energy efficiency and luminous efficacy, respectively, of ZnO:Zn, Y
2O
3:Eu
3+ and Y
2O
2S:Eu
3+ as a function of e-beam voltage. These results were presented, discussed and compared with literature data previously [
19]. We will suffice in making some comments on these results.
Figure 4a shows that the energy efficiency of Y
2O
2S:Eu
3+ is the highest, whereas ZnO:Zn has the highest luminous efficacy (
Figure 4b). The reason for this difference is due to cutting off the 707 nm emission peak of Y
2O
2S:Eu
3+ by convoluting the spectrum and the eye sensitivity curve in measuring luminance, which is the physical quantity for determining luminous efficacy. Another interesting result indicated in
Figure 4 is the levelling off of the efficiency curves for Y
2O
3:Eu
3+, whereas the curves of ZnO:Zn and Y
2O
2S:Eu
3+ show a monotonic increase.
The Y
2O
3:Eu
3+ particles in this experiment were nanometre-sized with an average diameter of 300 nm (range of 100–500 nm), while the other two materials were micrometre-sized [
19]. In the case of large monocrystalline phosphor particles without any defects, one expects that the CL efficiency would be proportional with V
n, where V is the beam voltage and the exponent n is about 3.5. This follows from the fact that the penetration depth is proportional with V
1.66 and the electron penetration volume is more or less spherical [
20,
26]. At low beam voltages the efficiency rises steeply in some cases; however, as soon as the penetration depth approaches the particle size, electrons can escape from the first particle and may enter a second particle. This is an inefficient process since it also creates SEs and BSEs. Moreover, the photons suffer multi-scattering events before escaping from the layer, which decreases the light output because the absorption coefficient in the layers is not exactly zero. These losses are more pronounced in the layers of the nanometre-sized Y
2O
3:Eu
3+ than in the layers of Y
2O
2S:Eu
3+ and ZnO:Zn and may explain the stronger levelling off for Y
2O
3:Eu
3+. An alternative explanation is that there may be a thin surface dead layer on these materials, due for example to contaminant pick-up or due to slow back reaction with the atmosphere to more stable phases. For the coarse powders this dead layer contributes less to overall performance as the beam voltage (and therefore penetration) increases. This is not observed for the nanosized-Y
2O
3:Eu
3+, because its particle size is so small that even at low voltages the beam penetrates all the way through the particles.
The comparison method enables a reliable determination of the CL efficiency of charging phosphor layers, because of the adjustment of the current density with a non-charging target. Another feature of the measuring technique is the evaluation of the radiance (and or luminance) in both reflection and transmissions modes, which means that the method is indifferent to variation of the layer thickness. The comparison method was devised to study the behaviour of double layers of phosphor particles for FEDs to enhance the luminance. The idea was to increase the light output of a phosphor screen by depositing a high-voltage phosphor on top of a low-voltage phosphor. Double and triple layers of phosphor particles were successfully used in so-called Penetrons to alter the CL-colour upon changing the beam voltage [
27]. Mixing of the phosphor layers was suppressed in Penetrons by fabricating a phosphor stack with separation layers. For luminance enhancement, this approach is impossible: we only found a modest increase of the CL by depositing Y
2O
3:Eu
3+ on top of ZnO:Zn due to mixing of the top and bottom layers [
19].
2.2. Cathodoluminescence in a SEM
Although the analysis of CL in a SEM and TEM is a standard technique, we shall describe in this section a simple measuring technique for the decay time of CL in a SEM that we have developed at Brunel University London [
20]. Before doing so, we shall briefly introduce CL-microscopy with a SEM.
Figure 5 compares SE- and CL-images of ZnS:Cu,Cl and ZnO:Zn particles.
Figure 5(a1,b1) shows SE-images, while
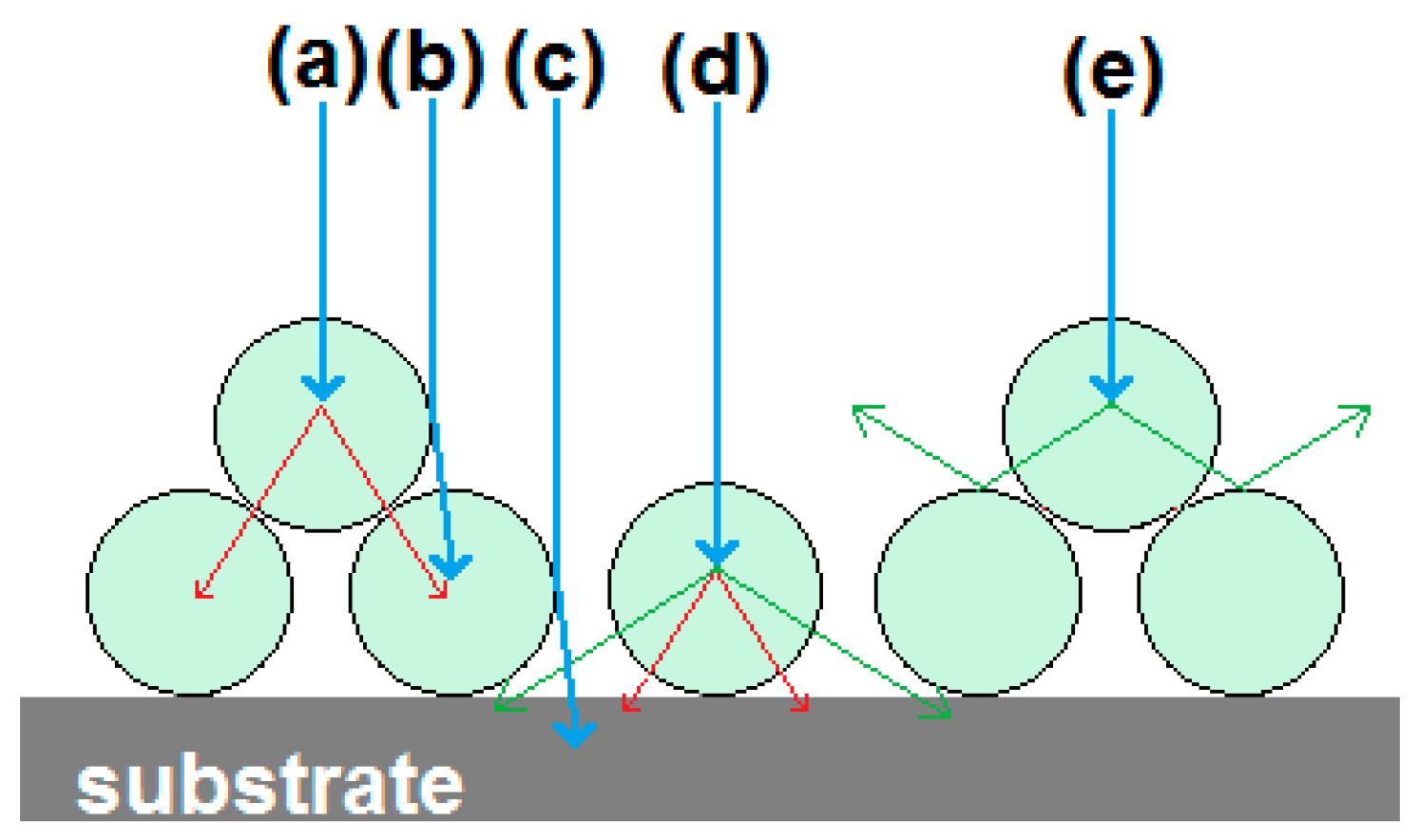
Figure 5(a2,b2) shows panchromatic CL-images. The SE- and CL-images show different features of the crystals, which helps in analysing these specimens. An eye-catching difference between the SE- and CL-images is the high brightness of some particles on top of others in the CL-images, for example the particles indicated by arrow 1 in
Figure 5(a2). This effect is explained in
Figure 6. When particles are directly on top of the absorbing substrate, then any primary electrons that penetrate them are lost to the substrate, whereas when they are sitting on other phosphor particles this energy will generate additional CL (cf. rays (a) and (d)). Primary electrons may also be backscattered either onto other phosphor particles as in ray (b) or into the substrate as in ray (c). Additionally, CL that is emitted towards the substrate has a better chance of being scattered to the detector if the particle is located on top of other particles, rather than if it is in direct contact with the carbon-loaded substrate pad (cf. rays (d) and (e)).
In SE-images, one can often observe surface-tilt contrast, which is caused by the reduced penetration depth (and hence enhanced SE-emission) of the electron beam when hitting a tilted surface such as the sides of a phosphor grain, indicated by arrow 2 in
Figure 5(a1) [
20,
28]. CL-images may show contrast enhancement similar to that in SE-images, indicated by arrow 3 in
Figure 5(b2). We have explained this contrast enhancement in terms of the ratio between the particle diameter and the electron penetration depth [
20].
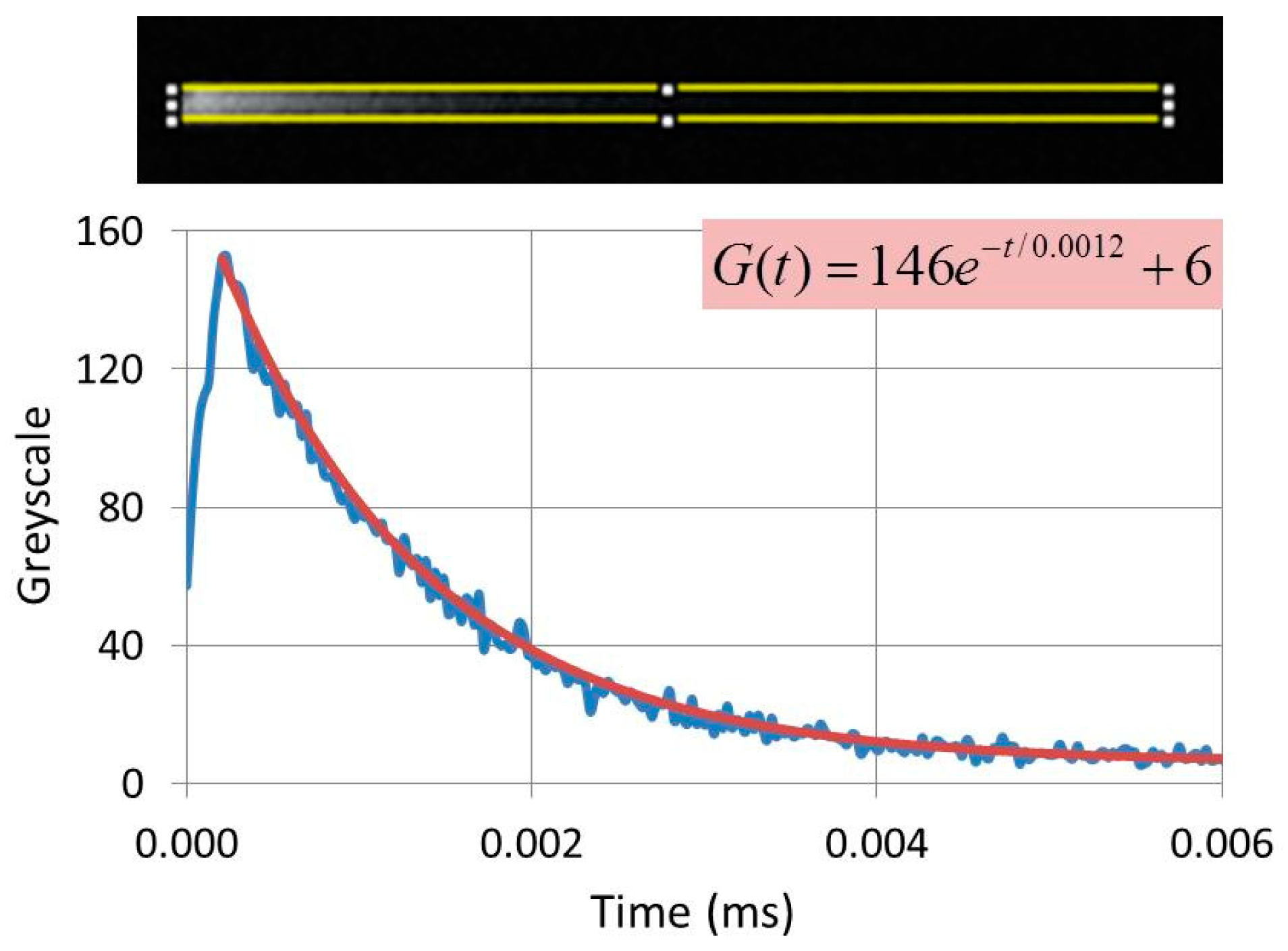
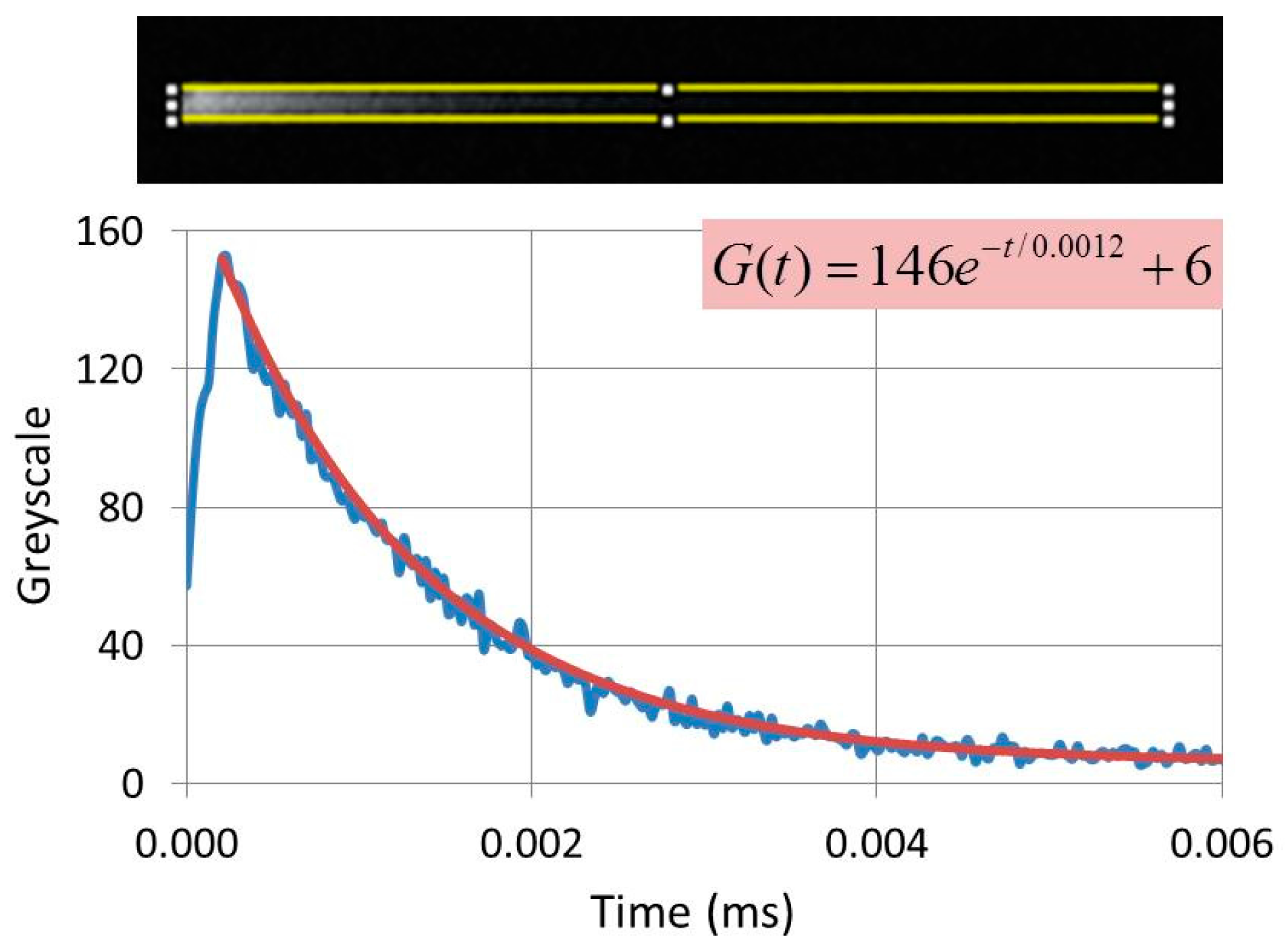
The CL-image of phosphor materials can be smeared out at high scan rates of the SEM, when a particle continues to emit light after the beam has moved onto to a subsequent pixel. Smearing out is detrimental to the picture quality and usually the scan rate is decreased to suppress this effect as much as possible. Smearing out causes comet-like structures, which offer the opportunity to measure the loss of brightness along the tail of these features and, hence to determine the decay time of the materials, as indicated in
Figure 7. The images shown in
Figure 7 are from reference 20 and have been slightly modified.
The CL-images in
Figure 7(b1,b2) show comet-like structures: the length of the tail is a measure of the decay time. The greyscale of the comet was analysed with ImageJ software and the experimental grayscale curve was fitted to an exponential curve
G(t), as shown in
Figure 8. This curve can generally be written as
where the index
i labels the transitions that contribute to the light generation; τ
i is the time constant of transition
i, being the 1/e-value of the decay time (τ
1/e); t indicates the time;
gi is the maximum value of the exponential
i at
t = 0; and
BG is the background correction. This latter correction depended on the gain setting of the PM tube and was near zero in most cases.
The CL-images shown in
Figure 7 are panchromatic, which implies that the decay times determined with this technique are “overall decay times”: these cannot be compared to spectral selective decay times, as indicated in Equation (6). In the case of
Figure 8 the emitted light is mainly from the
5D
0→
7F
2 (C
2) transition of Eu
3+ at 611 nm, because the contribution of other transitions is in the order of a few per cent only and may be neglected. However, if the concentration of Eu
3+ in Y
2O
3 is lowered to 1% and the dwell time of the e-beam on the phosphor particle is shortened, the Eu
3+ 5D
0→
7F
2 (C
2) transition gets much more saturated than the
5D
1→
7F
1 (C
2) transition at 533 nm [
26]. In that case the grey curve
G(t) can be represented satisfactorily by two exponentials and by curve fitting we were able to determine the decay times of both transitions [
20]. However, in many cases, the grey curves
G(t) can be represented by a single exponential by optimizing the scanning conditions, as in the case of
Figure 8 and the examples that have been summarized in
Table 1.
Deviations of G(t) from exponential decay may also be an indication of the presence of trapping defects that slow the release of energy into the matrix.
It can be concluded that the technique described above is well-suited for measuring overall decay times of phosphors in the range between 1 μs and 0.1 s; this range is mainly limited by the scan rate of the SEM. This measuring technique is attractive, since it allows the determination of decay times of individual phosphor particles, which may have micrometre or nanometre dimensions [
20]. Finally, no additional investments are needed to make these measurements with a (FE)SEM that is equipped with a panchromatic CL-detector such as a PM tube.
2.3. Cathodoluminescence Analysis in a TEM
In this section we shall present some work that we have recently carried out in the TEM facility of Brunel University London. The main characteristics of this TEM are described in the next section. The work studied the synthesis and luminescence of nanosized (Lu
1-xGd
x)
2O
2S:Tb
3+ phosphors between
x = 0 and
x = 1 with 0.1 and 2 mol% Tb
3+. This work will be published in total elsewhere [
29]; however, an analysis of one sample in the TEM will be presented here. The concentration of Gd
3+ in (Lu
1-xGd
x)
2O
2S:Tb
3+ was varied in steps of 0.1 (mol ratio Gd
3+). We found that during annealing of the hydroxy carbonate precursor with sulphur at 900 °C that the sulphurisation reaction was incomplete for the samples with
x < 0.7: those samples contained a mixture of Lu
1-xGd
x)
2O
3:Tb
3+ and (Lu
1-xGd
x)
2O
2S:Tb
3+, while the sample at
x = 0 was pure Lu
2O
3:Tb
3+. The hydroxy carbonate precursor was made by homogeneous precipitation with urea in aqueous solutions [
30]. From XRD-analyses the concentration of the oxide and oxysulphide phases in the various samples could be determined, because the oxides were cubic crystals and the oxysulphide phase was hexagonal. SEM and STEM pictures of (Lu
0.5Gd
0.5)
2O
2S:2%Tb
3+ are shown in
Figure 9.
It can be seen in
Figure 9 that the range of particle sizes was considerable, from about 100 nm to about 1.5 µm. The drift corrector square, which is indicated in
Figure 9, delivers the correction signals to prevent the sample from drifting during recording of the CL-spectra; this feature guarantees that the spectra are recorded exactly at the desired positions. The concentration of oxide phase in this 50/50 sample was found to be 52%.
Figure 9a,b shows two types of crystals: cuboid crystals and particles with angles of 120°. It is tempting to assign the cuboid particles to the oxide phase and the other type to the hexagonal oxysulphide.
To verify this assignment, we analysed the (Lu
0.5Gd
0.5)
2O
2S:2%Tb
3+ sample with energy dispersive X-ray spectroscopy (EDS) in a FESEM and recorded CL spectra of the crystals that are denoted by SI.1, SI.2 etc. in
Figure 9b.
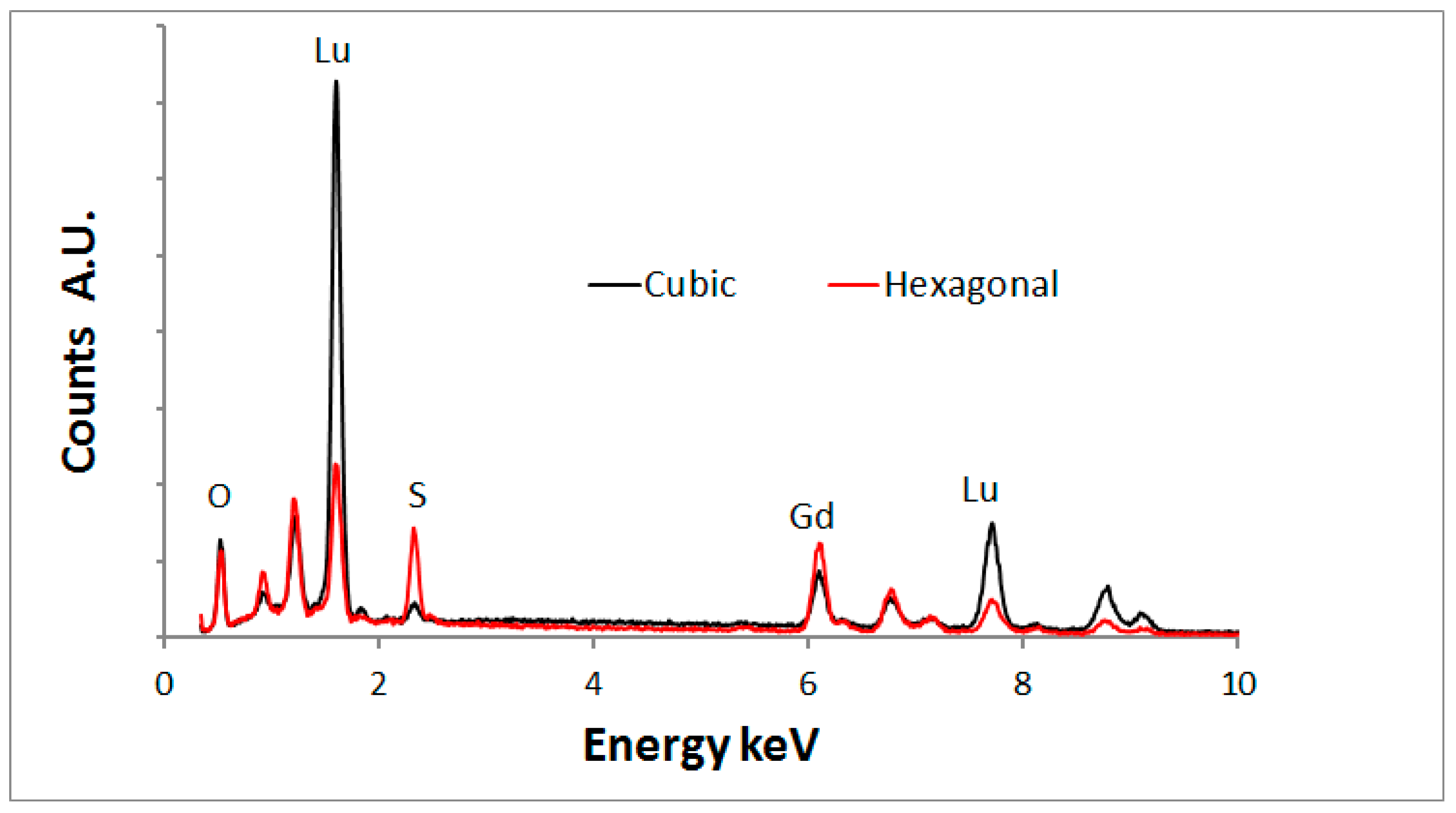
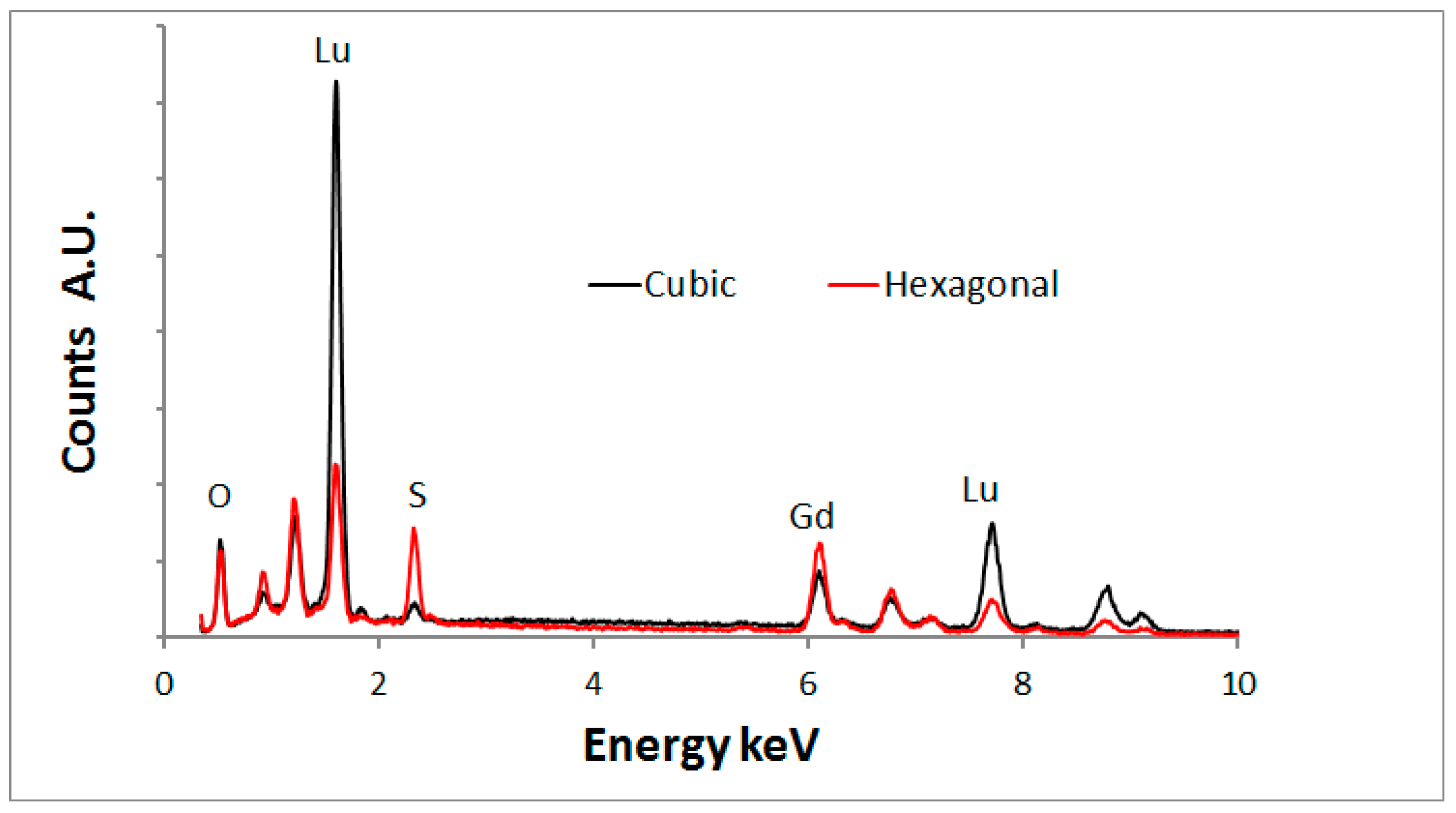
Figure 10 shows an overlay of the EDS- spectra of the cuboid and hexagonal types.
Figure 10 indicates that the hexagonal type crystals contain much more sulphur than the cubic crystals, which proves that the hexagonal crystals are oxysulphide. A small sulphur signal was detected from the cubic crystals. It is unlikely that this is a part of the crystal structure and it is assumed that the surface of these cubic crystals is contaminated with sulphur.
Figure 10 shows also that the lutetium signal is substantially higher than the gadolinium signal for the oxide crystals, whereas the oxysulphide crystals show the opposite. This phenomenon can be explained in terms of segregation of lutetium and gadolinium during the annealing with sulphur. A discussion of this effect is beyond the scope of this article; it will be described in a forthcoming publication [
29].
These CL-spectra that were recorded of the crystals shown in
Figure 9b were compared with photoluminescence (PL) spectra that were recorded separately from the powder samples. This comparison is shown in
Figure 11.
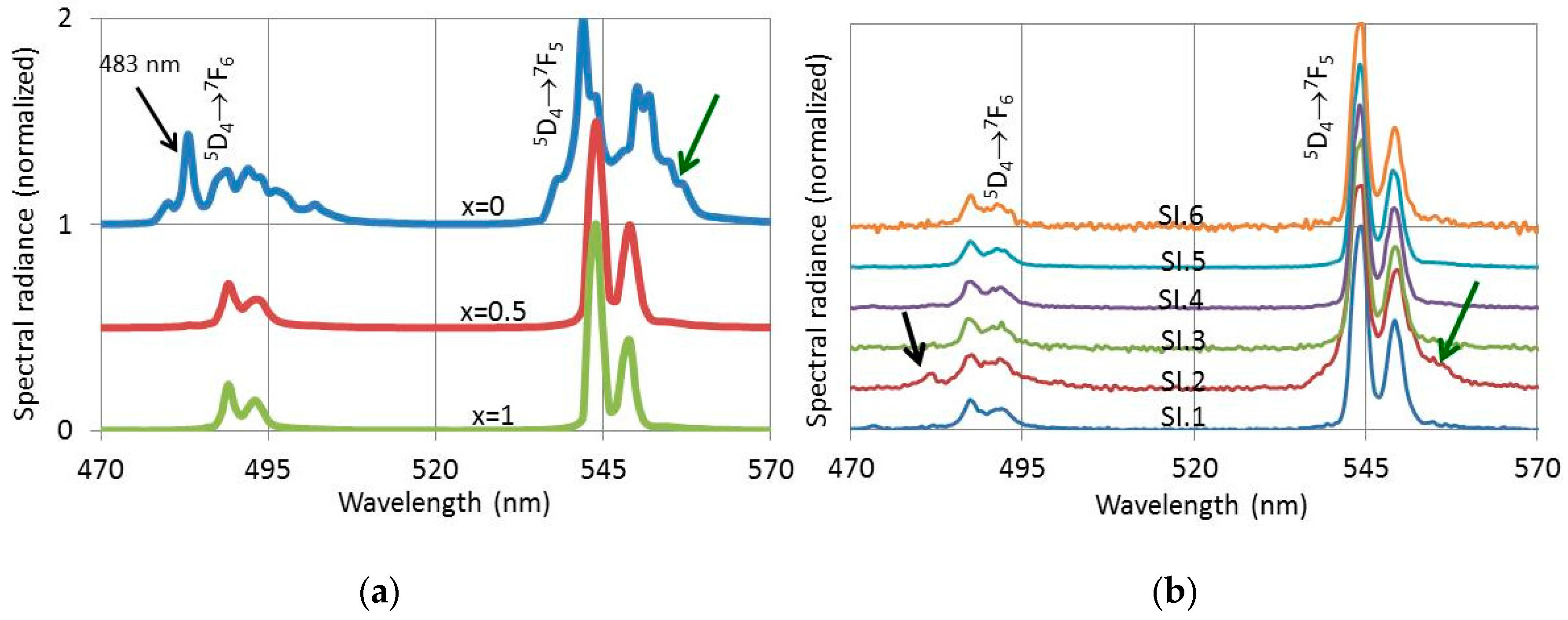
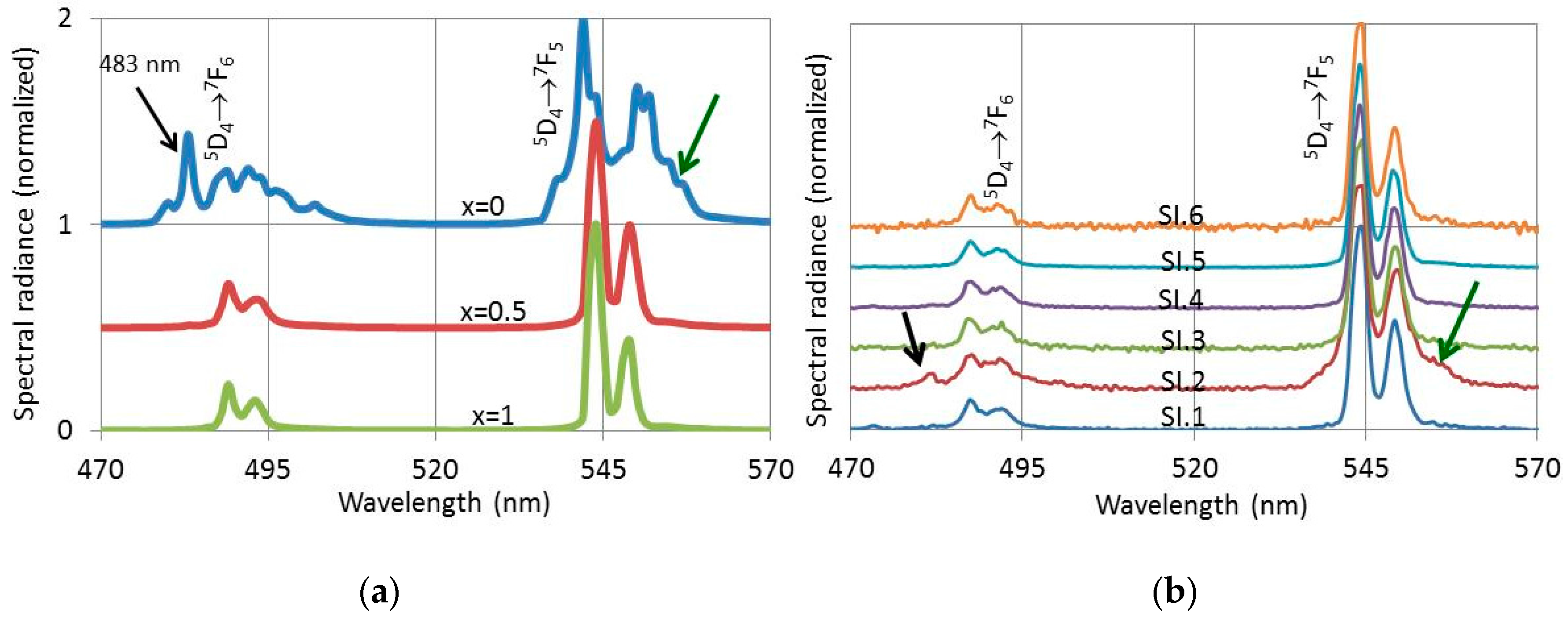
Figure 11 presents PL- and CL-spectra of (Lu
1-xGd
x)
2O
2S:2%Tb
3+ between 470 nm and 570 nm. The PL spectra for
x = 0, 0.5 and 1 in
Figure 11a were excited with 291 nm UV light, while the CL-spectra in
Figure 11b were recorded with the Gatan spectrometer in the TEM at 200 keV at the various spots indicated in
Figure 9b. The spectra in
Figure 11 have been normalized towards the maximum peak of the Tb
3+ 5D
4→
7F
5 manifold at 544 nm and 542 nm for the
x = 0 spectrum in
Figure 11a. This approach facilitated the visualisation of the difference between the oxide spectrum for 100% Lu (
x = 0) and the oxysulphide spectra (
x = 0.5 and
x = 1). It should be realized that the PL-efficiency of the oxysulphide is about 18 times larger than that of the oxide. From the CL-spectra represented in
Figure 11b it can be concluded that the spectrum SI.2 has a clear oxide character, because it has an emission peak at 483 nm and the Tb
3+ 5D
4→
7F
5 manifold at 545 nm is clearly broadened. These are the distinguishing features of the pure oxide spectrum at
x = 0 in
Figure 11a. Because of the large difference in efficiency between oxide and oxysulphide, we conclude that crystal SI.2 is a pure oxide, although the CL spectrum recorded at SI.2 also shows oxysulphide characteristics. The oxysulphide characteristics of the SI.2-spectrum in
Figure 11b are due to X-ray excitation of the neighbouring oxysulphide crystals. These X-rays are generated by the beam at SI.2 and cannot easily be eliminated.
Both EDS and CL spectroscopy indicate clearly that the cuboid crystals in
Figure 9 are an oxide phase, whereas the hexagonal type crystals are oxysulphide.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}