
A First Principles Study of H2 Adsorption on LaNiO3(001) Surfaces


Abstract
:1. Introduction
2. Models and Computational Methods
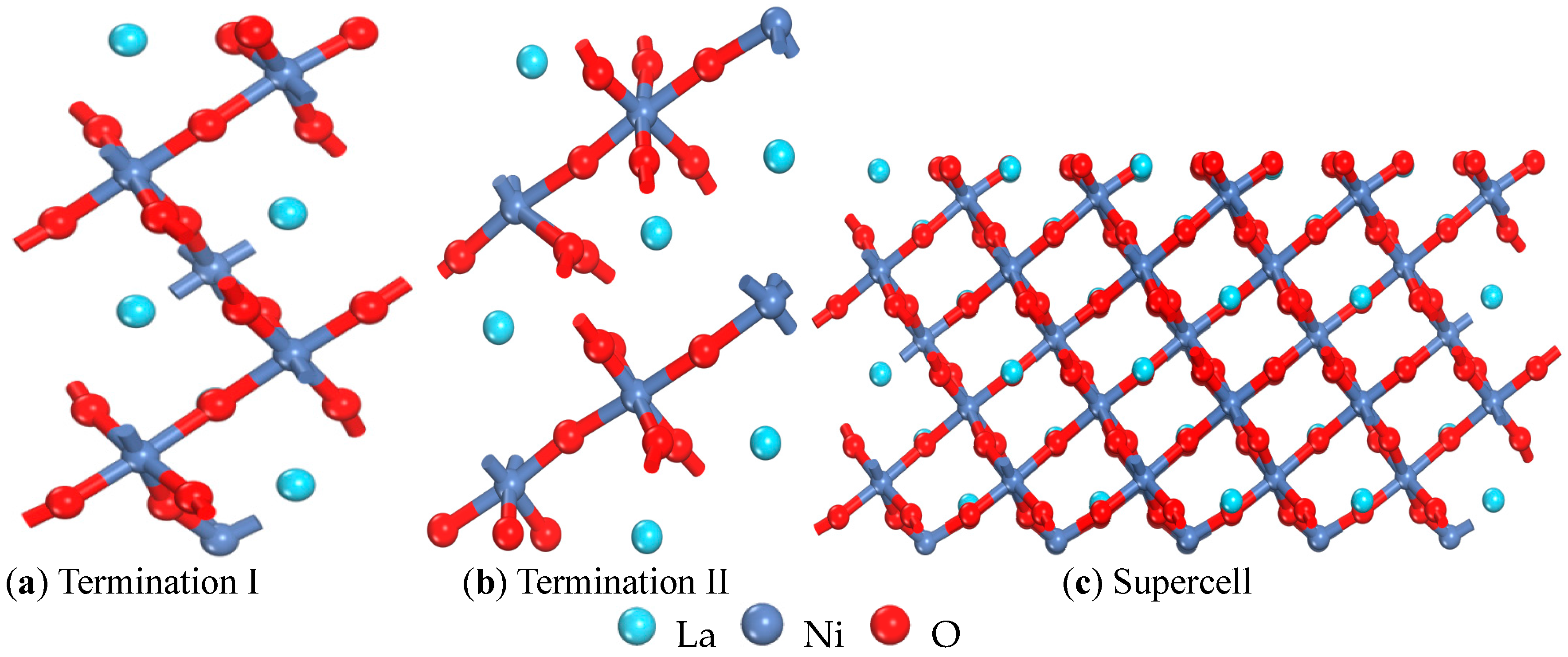
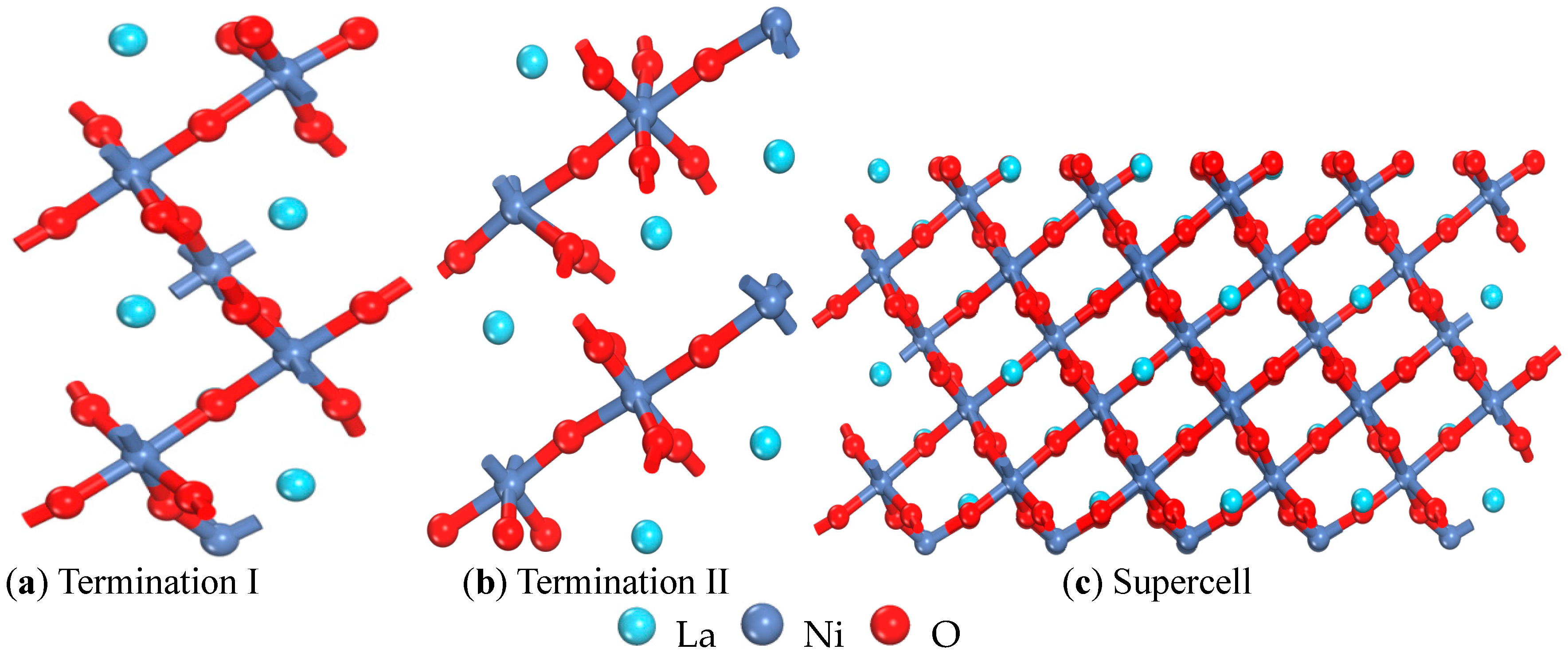
2.1. Calculation Parameters and Models
2.2. Calculations of Surface Energy
2.3. Calculations of Adsorption Energy and Dissociation Energy on the LaNiO3(001) Surface
3. Results and Discussion
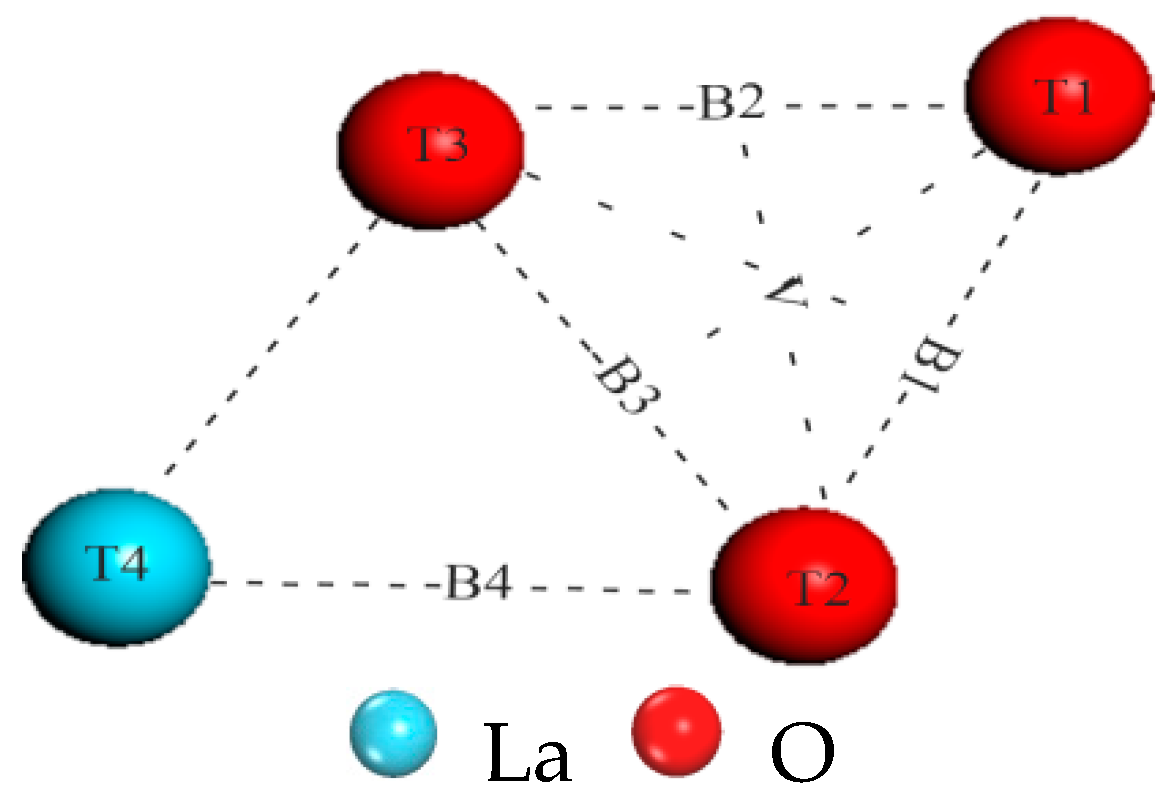
3.1. Analysis of Surface Adsorption Sites
3.2. Chemical Process of Dissociation and Adsorption for H2 Molecules
3.3. Analysis of Charge Population
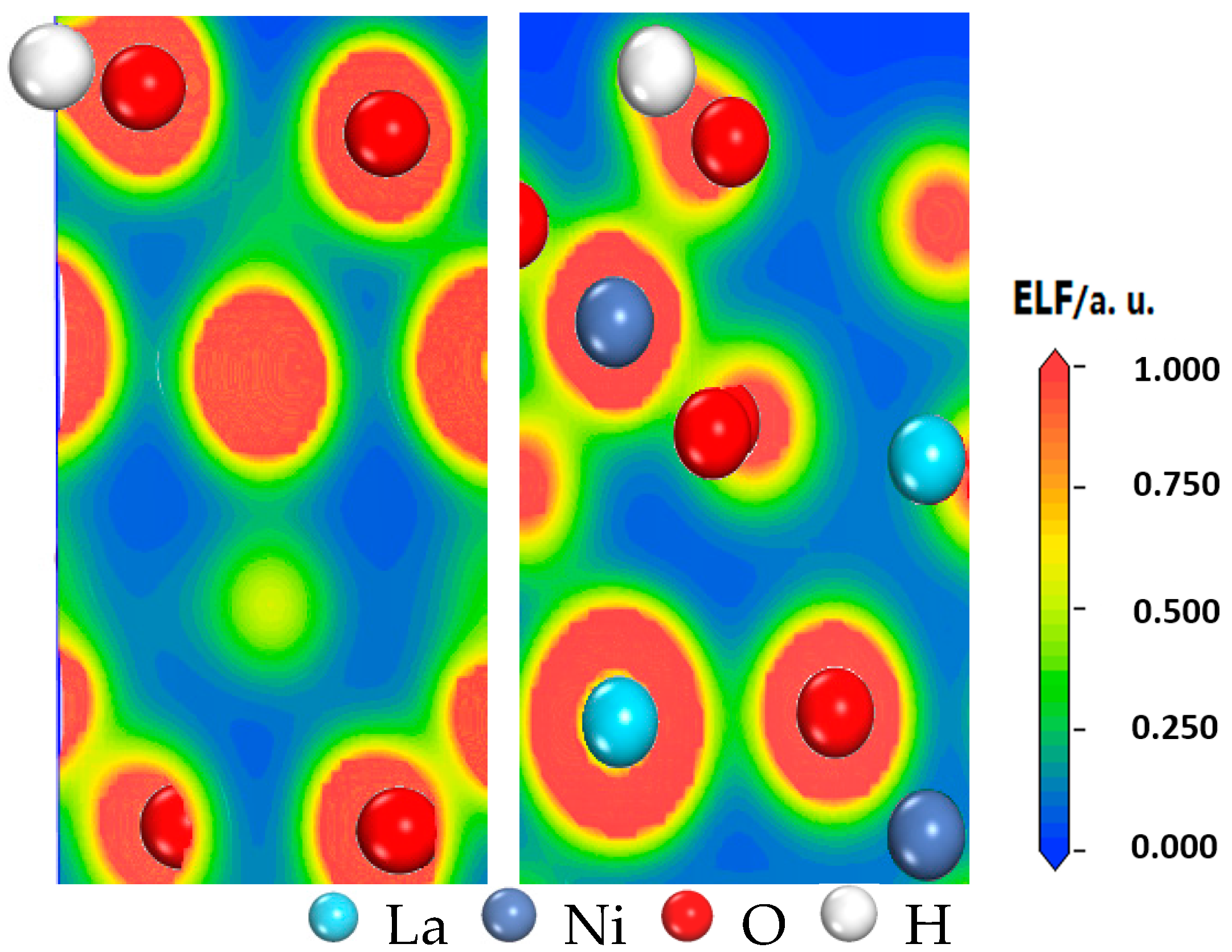
3.4. Analysis of Electron Localization Function
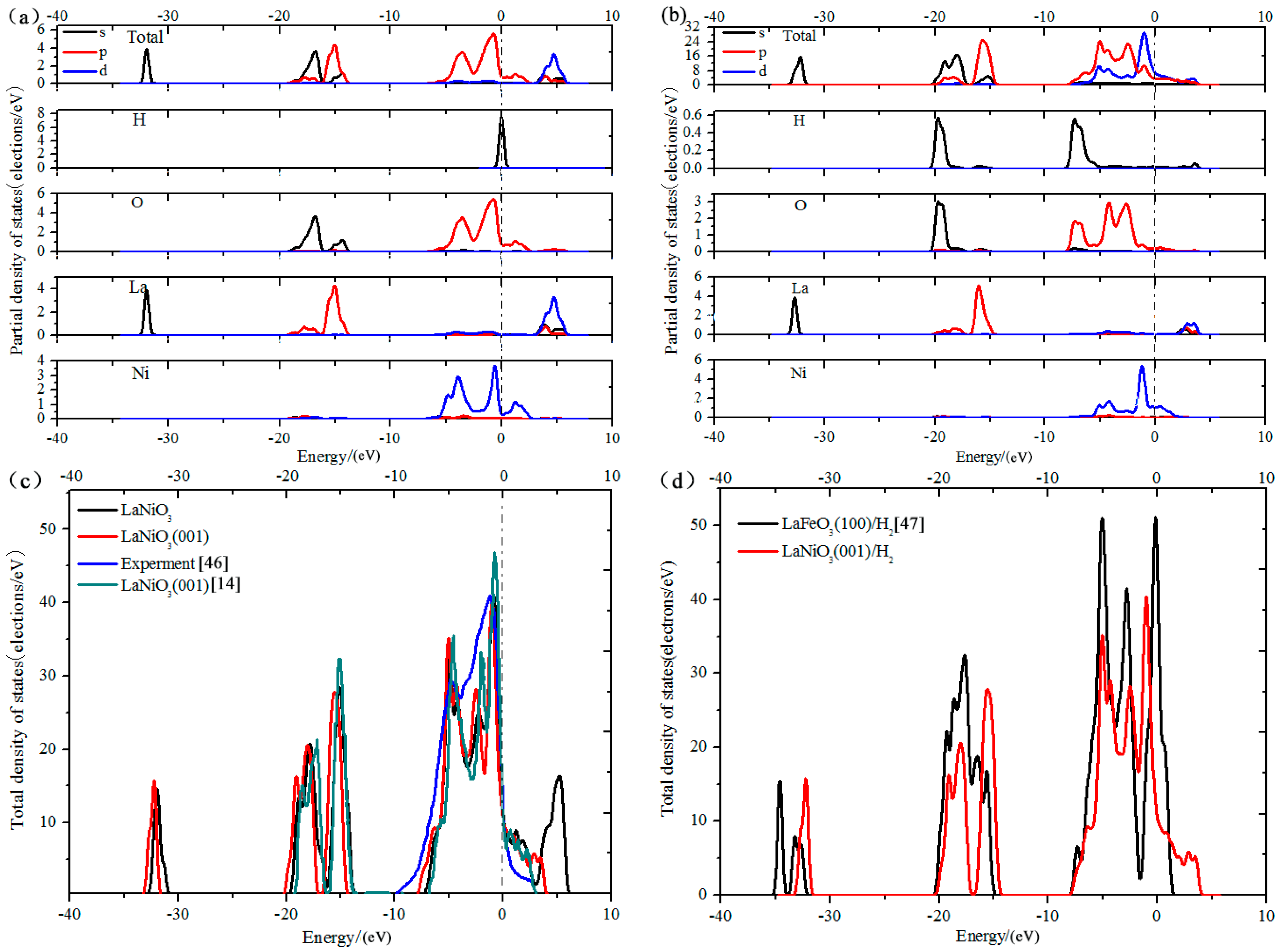
3.5. Analysis of Density of States
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Deng, G.; Chen, Y.; Tao, M.; Wu, C.; Shen, X.; Yang, H.; Liu, M. Electrochemical properties and hydrogen storage mechanism of perovskite-type oxide LaFeO3 as a negative electrode for Ni/MH batteries. Electrochim. Acta 2010, 55, 1120–1124. [Google Scholar] [CrossRef]
- Luo, B.C.; Wang, X.H.; Tian, E.; Wu, L.W.; Li, L.T. First-principles effective Hamiltonian simulation of ABO3-type perovskite ferroelectrics for energy storage application. J. Appl. Phys. 2016, 120, 074106. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Z.; Chen, Y.; Cheng, N.; Hui, Q. Hydrogen storage in perovskite-type oxides ABO3 for Ni/MH battery applications: A density functional investigation. Ind. Eng. Chem. Res. 2012, 51, 11821–11827. [Google Scholar] [CrossRef]
- Wærnhus, I.; Grande, T.; Wiik, K. Electronic properties of polycrystalline LaFeO3. Part II: Defect modelling including Schottky defects. Solid State Ion. 2005, 176, 2609–2616. [Google Scholar] [CrossRef]
- Hansmann, P.; Yang, X.; Toschi, A.; Khaliullin, G.; Andersen, O.K.; Held, K. Turning a nickelate Fermi surface into a cupratelike one through heterostructuring. Phys. Rev. Lett. 2009, 103, 016401. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Moetakef, P.; LeBeau, J.M.; Ouellette, D.; Balents, L.; Allen, S.J.; Stemmer, S. Low-dimensional Mott material: Transport in ultrathin epitaxial LaNiO3 films. Appl. Phys. Lett. 2010, 96, 062114. [Google Scholar] [CrossRef]
- Boris, A.V.; Matiks, Y.; Benckiser, E.; Frano, A.; Popovich, P.; Hinkov, V.; Wochner, P.; Castro-Colin, M.; Detemple, E.; Malik, V.K.; et al. Dimensionality control of electronic phase transitions in nickel-oxide superlattices. Science 2011, 332, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.; Mizutani, N.; Yao, X.; Zhang, L. Structural, dielectric, and ferroelectric properties of compositionally graded (Pb, La) TiO3 thin films with conductive LaNiO3 bottom electrodes. Appl. Phys. Lett. 2000, 77, 1041–1043. [Google Scholar] [CrossRef]
- Kleperis, J.; Wójcik, G.; Czerwinski, A.; Skowronski, J.; Kopczyk, M.; Beltowska-Brzezinska, M. Electrochemical behavior of metal hydrides. J. Solid State Electrochem. 2001, 5, 229–249. [Google Scholar] [CrossRef]
- Hsiao, C.; Qi, X. Substrate-dependant chemical stability and conductivity of LaNiO3-Χ thin films. Thin Solid Films 2013, 529, 356–359. [Google Scholar] [CrossRef]
- Kamble, R.B.; Tanty, N.; Patra, A.; Prasad, V. Field emission properties and strong localization effect in conduction mechanism of nanostructured perovskite LaNiO3. Appl. Phys. Lett. 2016, 109, 083102. [Google Scholar] [CrossRef]
- Tietz, F.; Raj, I.A.; Ma, Q.; Baumann, S.; Mahmoud, A.; Hermann, R.P. Material properties of perovskites in the Quasi-Ternary system LaFeO3–LaCoO3–LaNiO3. J. Sol. St. Chem. 2016, 237, 183–191. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Guan, L.; Liu, B.; Jin, L.; Guo, J.; Zhao, Q.; Wang, Y.; Fu, G. Electronic structure and optical properties of LaNiO3: First-principles calculations. Solid State Commun. 2010, 150, 2011–2014. [Google Scholar] [CrossRef]
- Guan, L.; Zuo, J.; Jia, G.; Liu, Q.; Wei, W.; Guo, J.; Dai, X.; Liu, B.; Wang, Y.; Fu, G. Structural stability and electronic properties of LaO- and NiO2-terminated LaNiO3(001) surface. Appl. Surf. Sci. 2013, 264, 570–573. [Google Scholar] [CrossRef]
- Zhu, M.W.; Wang, H.L.; Lei, H.; Zhang, Y.J.; Jia, N.; Wang, Z.J. Strain-induced modification in microstructure and electrical properties of polycrystalline LaNiO3-δ films. Appl. Phys. A 2016, 122, 364. [Google Scholar] [CrossRef]
- Amoresi, R.A.C.; Felix, A.A.; Lustosa, G.M.M.M.; Gasparotto, G.; Simões, A.Z.; Zaghete, M.A. Role of morphological characteristics on the conductive behavior of LaNiO3 thin films. Ceram. Int. 2016, 42, 16242–16247. [Google Scholar] [CrossRef]
- Forslund, R.P.; Mefford, J.T.; Hardin, W.G.; Alexander, C.T.; Johnston, K.P.; Stevenson, K.J. Nanostructured LaNiO3 perovskite electrocatalyst for enhanced urea oxidation. ACS Catal. 2016, 6, 5044–5051. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Park, J.; Kim, D.; Byeon, S.; Kim, D. XANES study on Ruddlesden-Popper phase, Lan+1NinO3n+1(n = 1, 2 and ∞). J. Synchrotron Rad. 2001, 8, 704–706. [Google Scholar] [CrossRef]
- Chiou, W.C., Jr.; Carter, E.A. Structure and stability of Fe3C-cementite surfaces from first principles. Surf. Sci. 2003, 530, 87–100. [Google Scholar] [CrossRef]
- Choi, Y.; Mebane, D.S.; Lin, M.C.; Liu, M. Oxygen reduction on LaMnO3-based cathode materials in solid oxide fuel cells. Chem. Mater. 2007, 19, 1690–1699. [Google Scholar] [CrossRef]
- Evarestov, R.A.; Kotomin, E.A.; Mastrikov, Y.A.; Gryaznov, D.; Heifets, E.; Maier, J. Comparative density-functional LCAO and plane-wave calculations of LaMO3 surface. Phys. Rev. B 2005, 72, 214411. [Google Scholar] [CrossRef]
- Chen, Y.H.; Zhang, B.W.; Yang, L.; Zhang, M.L.; Zhang, C.R.; Kang, L.; Luo, Y.C. First-principle study of H2 adsorption on LaFeO3(110) surface. J. Nanomaterials 2014, 2014, 758985. [Google Scholar]
- Kresse, G.; Hafner, J. First-principles study of the adsorption of atomic H on Ni(111), (100) and (110). Surf. Sci. 2000, 459, 287–302. [Google Scholar] [CrossRef]
- Huda, M.N.; Ray, A.K. An Ab initio study of H2 interaction with the Pu(100) surface. Phys. B Condens. Matter 2005, 366, 95–109. [Google Scholar] [CrossRef]
- Lie, G.C.; Clementi, E. Molecular-dynamics simulation of liquid water with an Ab Initio flexible water-water interaction potential. Phys. Rev. A 1986, 33, 2679–2693. [Google Scholar] [CrossRef]
- Chen, Y.H.; Zhang, B.W.; Zhang, C.R.; Zhang, M.L.; Long, K.; Luo, Y.C. First-principle study of H2 adsorption on Mg3N2(110) surface. Phys. Lett. 2014, 31, 063101. (In Chinese) [Google Scholar]
- Kubas, A.; Berger, D.; Oberhofer, H.; Maganas, D.; Reuter, K.; Neese, F. Surface adsorption energetics studied with “Gold Standard” wave-function-based Ab initio methods: Small-molecule binding to TiO2(110). J. Phys. Chem. Lett. 2016, 7, 4207–4212. [Google Scholar] [CrossRef] [PubMed]
- Tamijani, A.A.; Salam, A.; de Lara-Castells, M.P. Adsorption of noble-gas atoms on the TiO2(110) surface: An Ab initio-assisted study with van der Waals-corrected DFT. J. Phys. Chem. C 2016, 120, 18126–18139. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Morgan, D.; Kleis, J.; Rossmeisl, J. Ab initio defect energetics in LaBO3 perovskite solid oxide fuel cell materials. ECS Trans. 2009, 25, 2761–2767. [Google Scholar]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Hanson, J.C.; Frenkel, A.I.; Kim, J.Y.; Pérez, M. Experimental and theoretical studies on the reaction of H2 with NiO: Role of O vacancies and mechanism for oxide reduction. J. Am. Chem. Soc. 2002, 124, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Bao, S.N.; Zhuang, Y.Y. The geometric structure of ethylene adsorption on Ni(110) surface. J. Phys. 2003, 52, 202–207. (In Chinese) [Google Scholar]
- Guo, X.J.; Li, L.; Liu, Z.Y.; Yu, D.L. Hardness of covalent compounds: Roles of metallic component and d valence electrons. J. Appl. Phys. 2008, 104, 023503. [Google Scholar] [CrossRef]
- Kong, X.P.; Wang, J. Copper(II) adsorption on the kaolinite(001) surface: Insights from first-principles calculations and molecular dynamics simulations. Appl. Surf. Sci. 2016, 389, 316–323. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, P.X.; Zhu, J.C.; Liu, Y. First-principles study of NiAl alloyed with Co. Comput. Mater. Sci. 2016, 111, 34–40. [Google Scholar] [CrossRef]
- Ding, S.S.; Huang, W.Q.; Yang, Y.C.; Zhou, B.X.; Hu, W.Y.; Long, M.Q. Dual role of monolayer MoS2 in enhanced photocatalytic performance of hybrid MoS2/SnO2 nanocomposite. J. Appl. Phys. 2016, 119, 205704. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Burdett, J.K.; McCormick, T.A. Electron localization in molecules and solids: The meaning of ELF. J. Phys. Chem. A 1998, 102, 6366–6372. [Google Scholar] [CrossRef]
- Lee, S.; Chen, R.; Balents, L. Metal-insulator transition in a two-band model for the perovskite nickelates. Phys. Rev. B 2011, 84, 165119. [Google Scholar] [CrossRef]
- Sarma, D.D.; Shanthi, N.; Barman, S.R.; Hamada, N.; Sawada, H.; Terakura, K. Band theory for ground-state properties and excitation spectra of perovskite LaMO3 (M = Mn, Fe, Co, Ni). Phys. Rev. Lett. 1995, 75, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.G.; Li, W.W.; Li, Y.W.; Zhu, M.; Zhu, Z.Q.; Chu, J.H. Electronic properties of nanocrystalline LaNiO3 and La0.5Sr0.5CoO3 conductive films grown on silicon substrates determined by infrared to ultraviolet reflectance spectra. Appl. Phys. Lett. 2009, 94, 221104. [Google Scholar] [CrossRef]
- Lv, X.X. Hydrogen Storage Property Study of H2 Molecule Adsorption on LaFeO3 (100) Surfaces. Masters’ Dissertation, Lanzhou University of Technology, Lanzhou, China, 2012. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Termination I (001) | Termination II (001) | (001) Ref. [15] | (110) | (101) | (011) | (111) | (100) | (001) |
|---|---|---|---|---|---|---|---|---|
| 1.97 | 1.84 | 2.03 | 4.23 | 2.25 | 5.04 | 6.61 | 6.10 | 6.67 |
| Type | Initial Position (H2) | rH–H (Å) | rH–O (Å) | rH–Ni (Å) | rH–La (Å) | Eads (eV) | Edis (eV) |
|---|---|---|---|---|---|---|---|
| Model I | T1 | 1.678 | 1.021 | 2.368 | 4.367 | −1.463 | 0.955 |
| T2 | 1.675 | 0.995 | 2.219 | 3.036 | −1.517 | 0.965 | |
| T3 | 1.636 | 0.994 | 2.359 | 2.919 | −2.074 | 1.105 | |
| T4 | 0.757 | 4.153 | 5.310 | 2.951 | 0.045 | 4.525 | |
| B1 | 3.104 | 0.982 | 2.257 | 3.210 | −2.106 | −1.678 | |
| B2 | 2.907 | 0.983 | 2.605 | 2.975 | −1.397 | −1.520 | |
| B3 | 3.047 | 1.003 | 3.035 | 2.901 | −2.822 | −1.638 | |
| B4 | 2.847 | 0.992 | 2.958 | 3.038 | −2.593 | 0.017 | |
| V | 3.061 | 0.982 | 2.250 | 3.225 | −2.816 | −1.648 | |
| Model II | T5 | 0.878 | 3.213 | 1.586 | 6.482 | −0.301 | 4.313 |
| Experiment | H2O [29] | 1.545 | 0.978 | - | - | - | - |
| H2 [27] | 0.752 | - | - | - | - | 4.48 |
| Atom | Electron Population (e) | Bond | Bond Population (e) | ||
|---|---|---|---|---|---|
| Before Adsorption | After Adsorption | Before Adsorption | After Adsorption | ||
| O1 | −0.64 | −0.72 | O3–Ni | 0.05 | 0.00 |
| O2 | −0.64 | −0.72 | O3–La | 0.25 | 0.05 |
| O3 | −0.64 | −0.81 | O1–O3 | −0.03 | −0.07 |
| La | 1.47 | 1.34 | H1–O1 | - | 0.10 |
| Ni | 0.56 | 0.40 | H2–O2 | - | 0.03 |
| H1 | - | 0.37 | H2–O3 | - | 0.63 |
| H2 | - | 0.34 | H1–O3 | - | 0.68 |
| Adsorption Site | Barrier from Reactant (eV) | Barrier from Product (eV) | Energy of Reaction (eV) |
|---|---|---|---|
| T3 | −0.869 | −2.833 | −1.964 |
| B3 | −1.282 | −3.789 | −2.507 |
| Atom | Before Adsorption (e) | After Adsorption (e) | ||||||
|---|---|---|---|---|---|---|---|---|
| s | p | d | Charge | s | p | d | Charge | |
| O | 1.90 | 4.75 | - | −0.64 | 1.87 | 4.91 | - | −0.78 |
| O | 1.90 | 4.75 | - | −0.64 | 1.84 | 4.99 | - | −0.83 |
| La | 2.23 | 6.13 | 1.17 | 1.47 | 2.34 | 6.12 | 1.19 | 1.35 |
| Ni | 0.40 | 0.68 | 8.57 | 0.30 | 0.40 | 0.66 | 8.66 | 0.28 |
| H1 | 1.00 | - | - | - | 0.70 | - | - | 0.30 |
| H2 | 1.00 | - | - | - | 0.67 | - | - | 0.33 |
| Bond | Population (e) | Length (Å) | ||
|---|---|---|---|---|
| Before Adsorption | After Adsorption | Before Adsorption | After Adsorption | |
| O1–Ni | 0.40 | 0.22 | 1.832 | 2.168 |
| O2–Ni | 0.41 | 0.31 | 1.830 | 1.984 |
| H2–O1 | - | 0.67 | - | 0.981 |
| H1–O2 | - | 0.66 | - | 1.003 |
| O1–La | 0.24 | 0.25 | 2.247 | 2.504 |
| O2–La | 0.25 | 0.25 | 2.797 | 2.504 |
| H2–Ni | - | –0.19 | - | 2.250 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, C.; Chen, Y.; Wu, N.; Zhang, M.; Yuan, L.; Zhang, C. A First Principles Study of H2 Adsorption on LaNiO3(001) Surfaces. Materials 2017, 10, 36. https://doi.org/10.3390/ma10010036
Pan C, Chen Y, Wu N, Zhang M, Yuan L, Zhang C. A First Principles Study of H2 Adsorption on LaNiO3(001) Surfaces. Materials. 2017; 10(1):36. https://doi.org/10.3390/ma10010036
Chicago/Turabian StylePan, Changchang, Yuhong Chen, Na Wu, Meiling Zhang, Lihua Yuan, and Cairong Zhang. 2017. "A First Principles Study of H2 Adsorption on LaNiO3(001) Surfaces" Materials 10, no. 1: 36. https://doi.org/10.3390/ma10010036
APA StylePan, C., Chen, Y., Wu, N., Zhang, M., Yuan, L., & Zhang, C. (2017). A First Principles Study of H2 Adsorption on LaNiO3(001) Surfaces. Materials, 10(1), 36. https://doi.org/10.3390/ma10010036