
CO2 Fixation by Membrane Separated NaCl Electrolysis

 ,
,
Abstract
:1. Introduction
2. Experimental Section
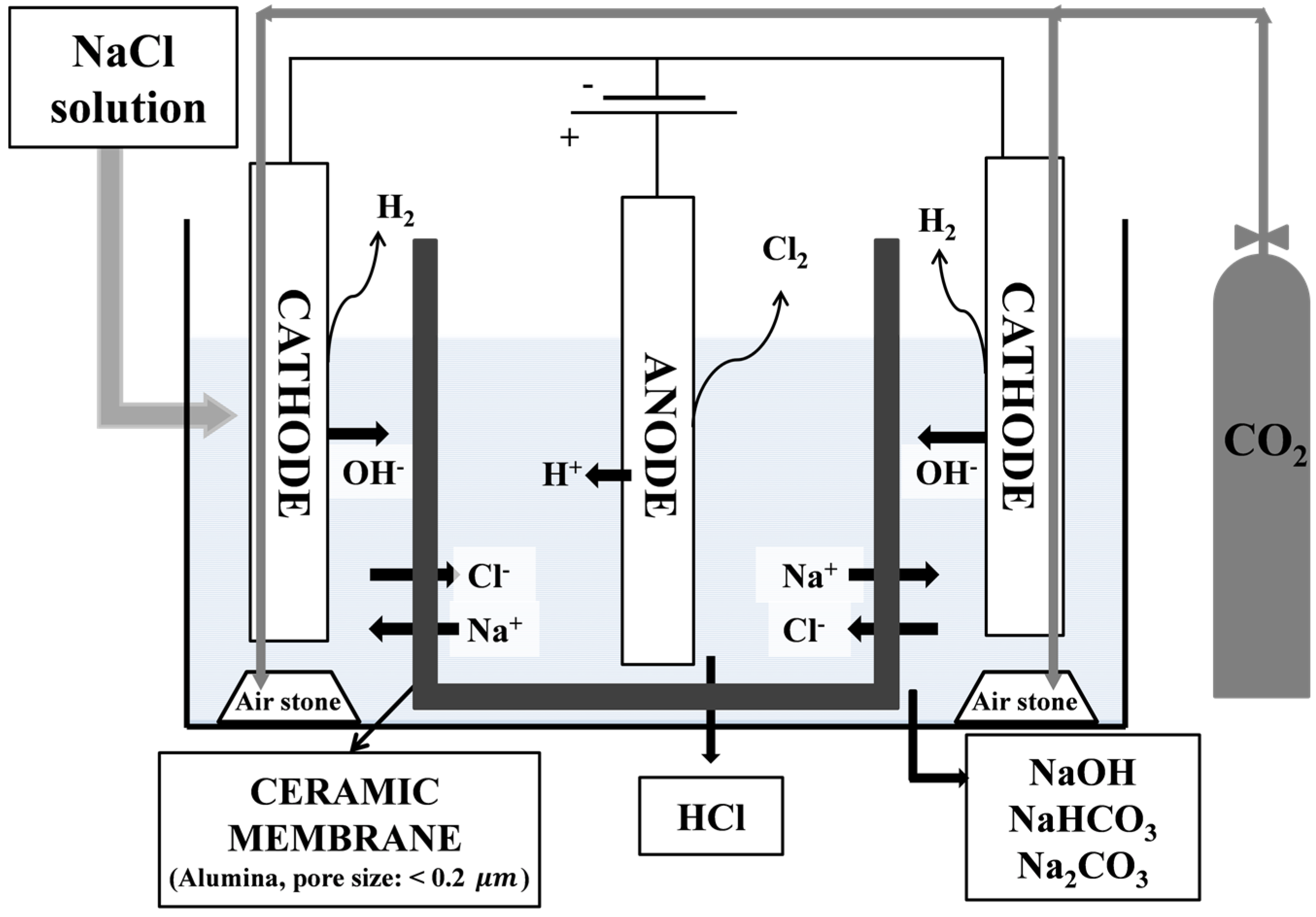
2.1. Materials and Electrolysis Device

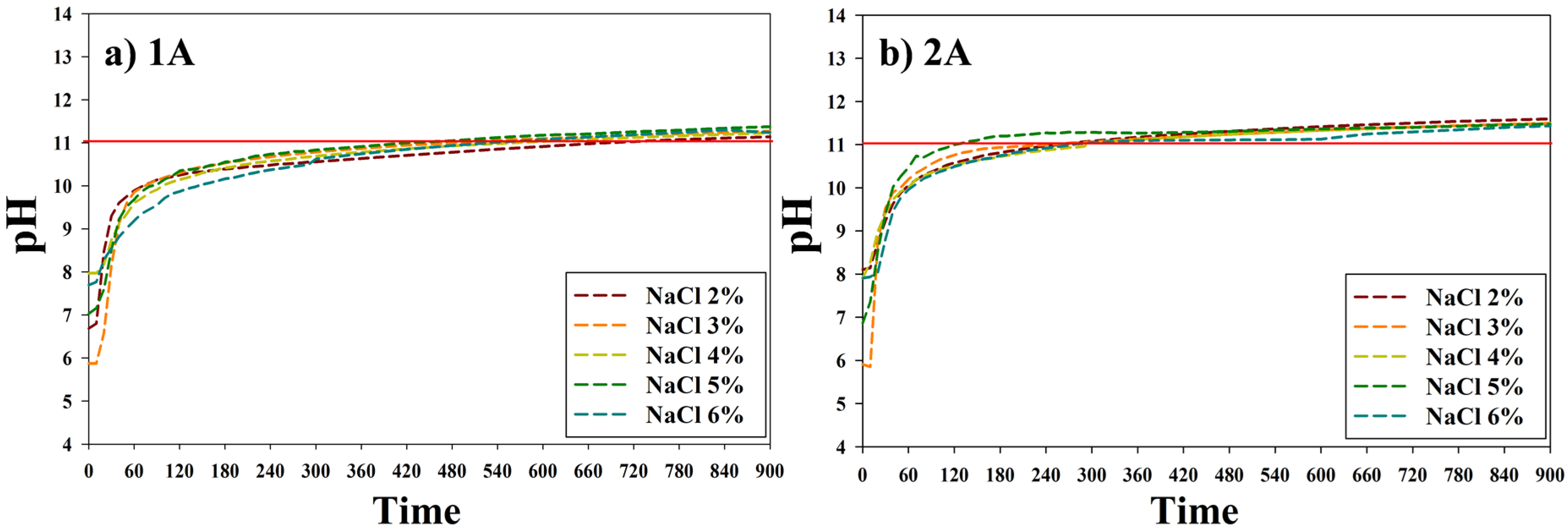
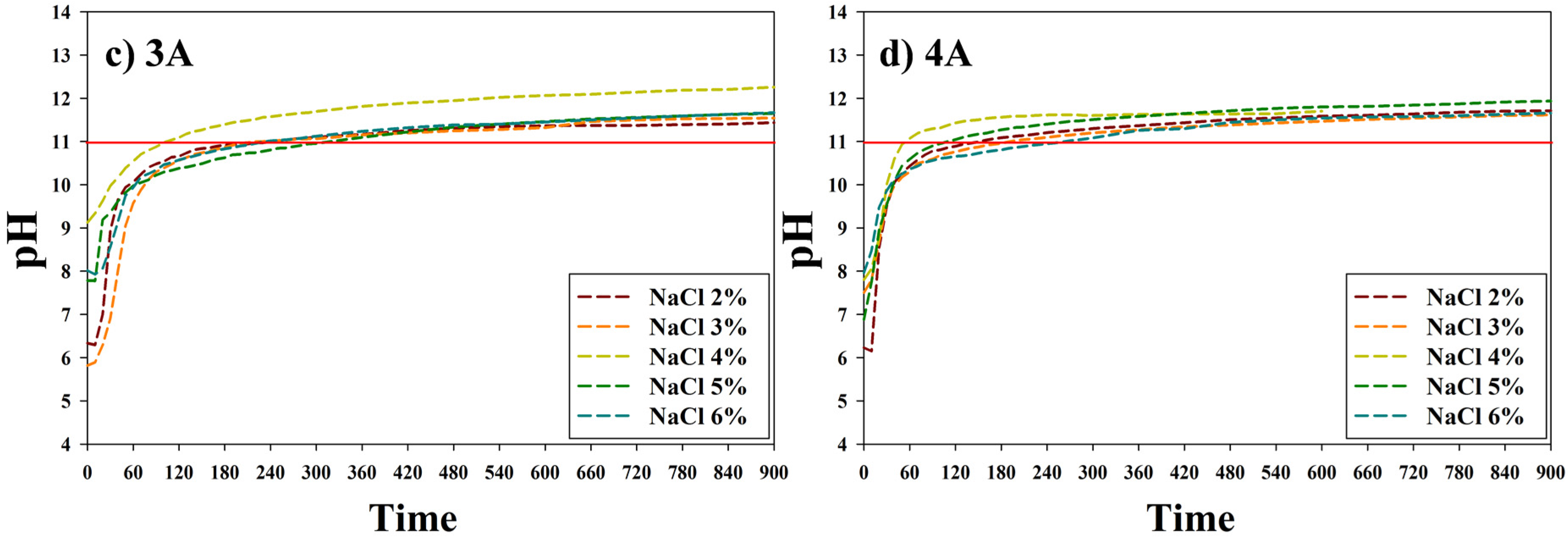
2.2. Electrolysis of NaCl Solution
2.3. CO2 Gas Capture Using Sodium Hydroxide
2.4. Precipitation of CaCO3 by the Titration
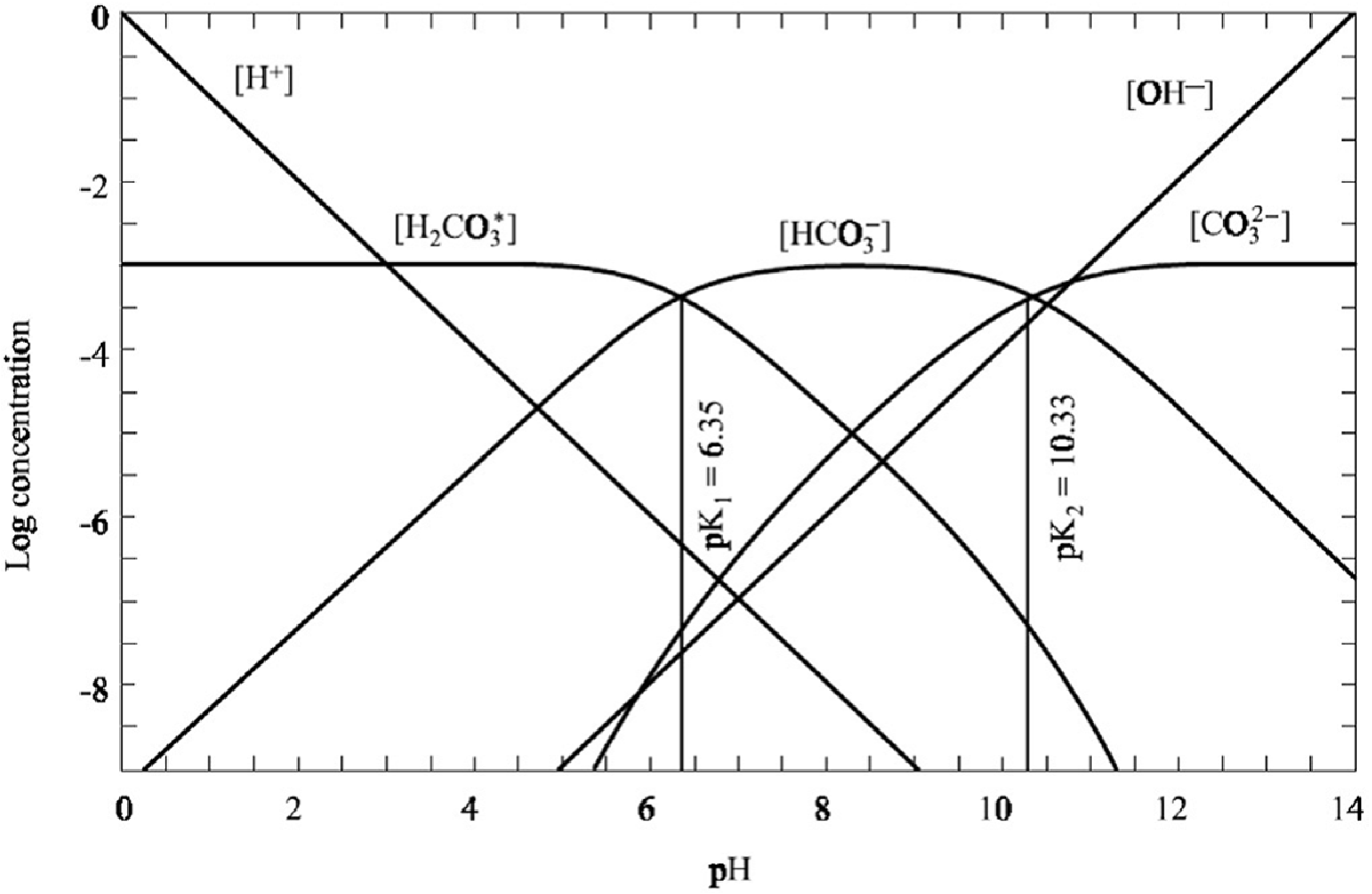
2.5. Characterization of CaCO3
3. Results and Discussion
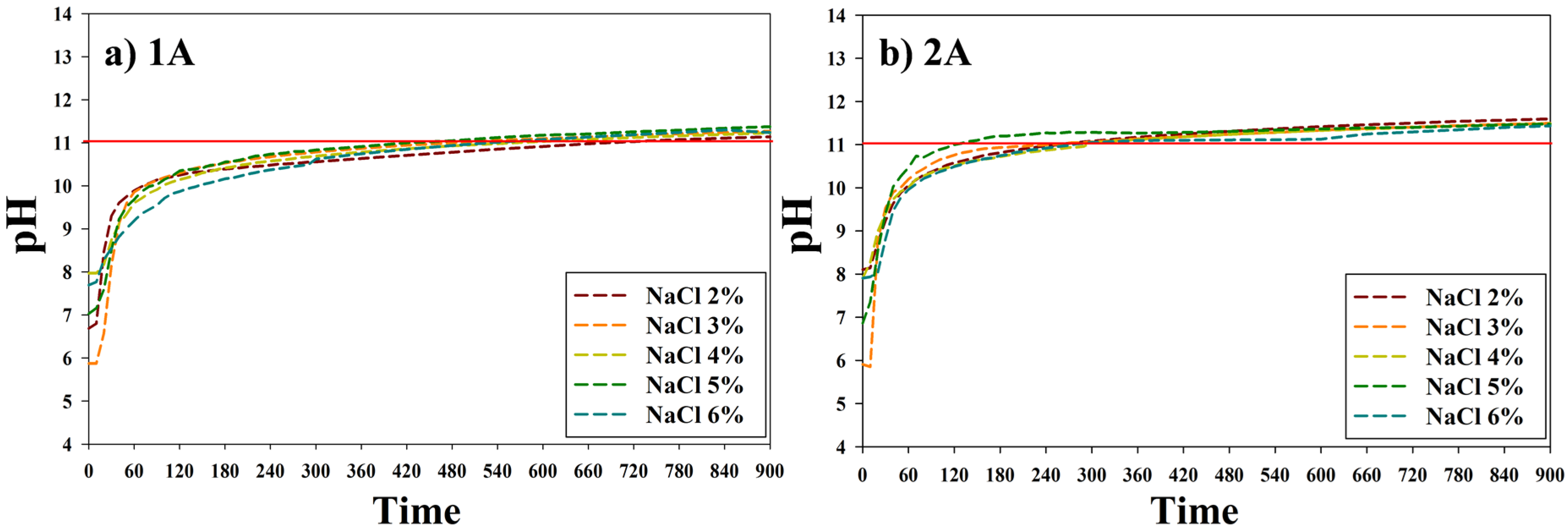
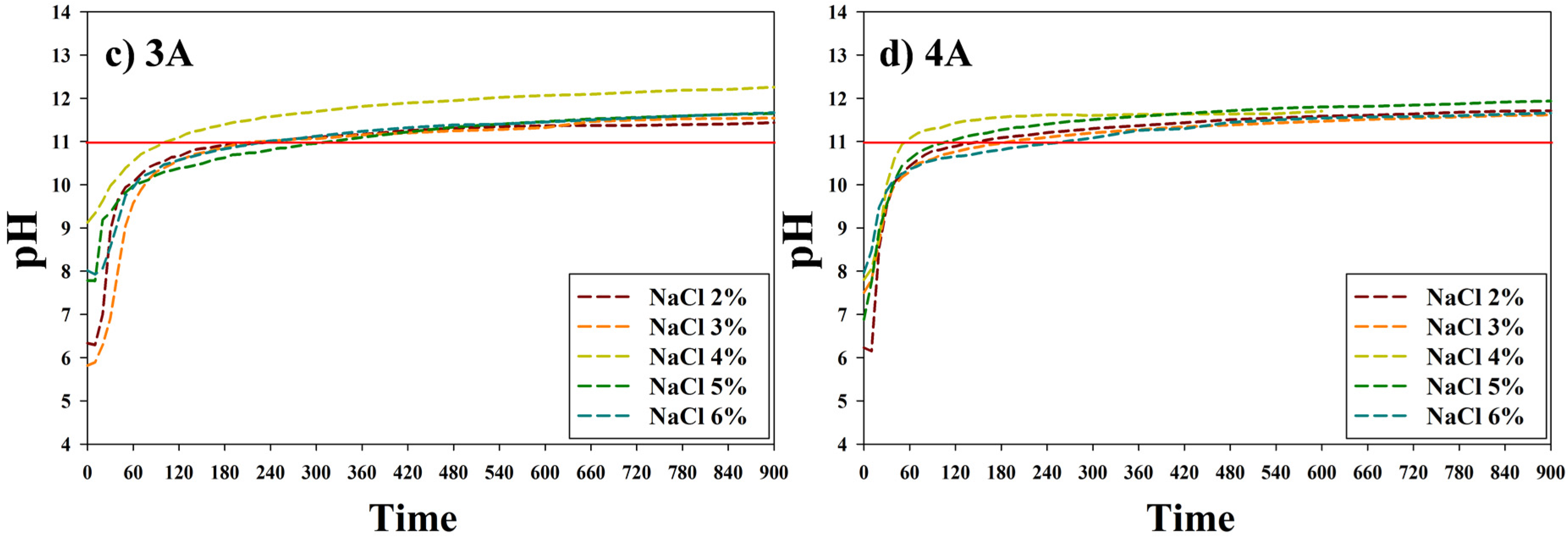
3.1. The Metal Carbonate Produced by the CO2 Fixation.
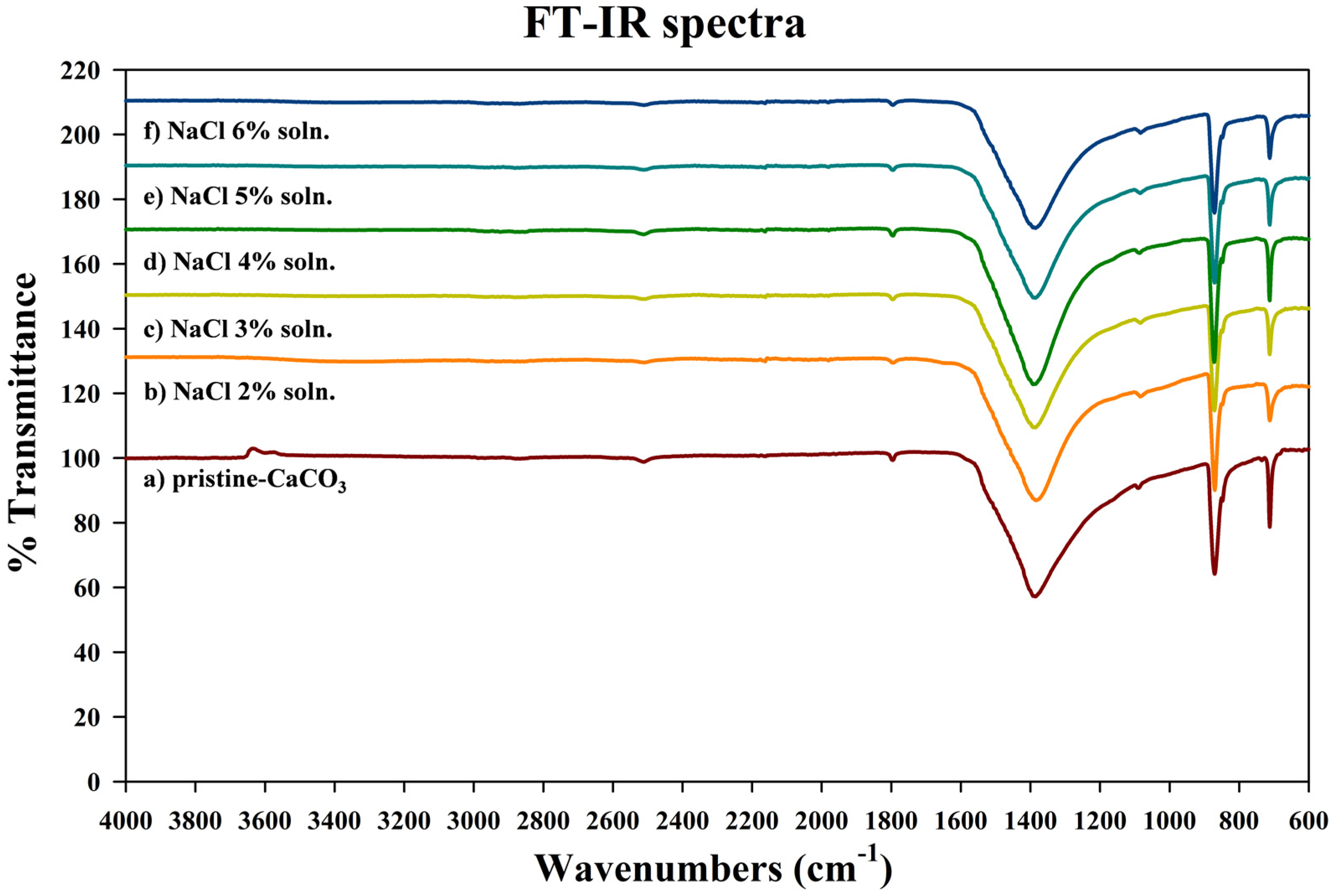
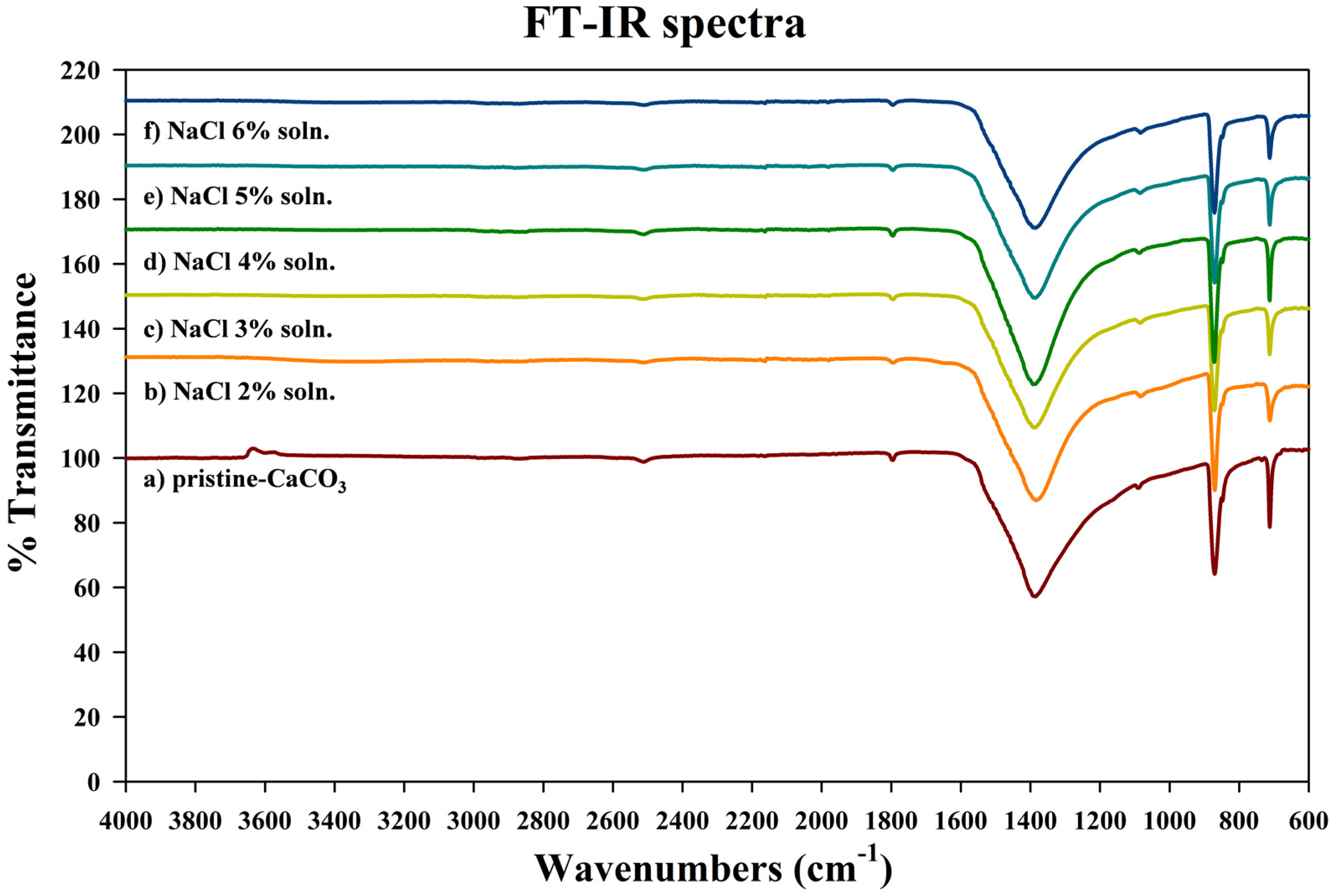
3.2. Characteristics of the Metal Carbonate: FT-IR Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber (cm−1) | Function Groups | Vibration |
|---|---|---|
| 2513 | Combination | 1080 cm−1 and 1440 cm−1 |
| 1796 | Combination | 1080 cm−1 and 713 cm−1 |
| 1418 | C–O | Stretching mode (1490 cm−1, 1420 cm−1 in vaterite; 1418 cm−1 in calcite; 1465 cm−1 in aragonite) |
| 1080 | O–C–O | Stretching mode |
| 876 | C–O | In-plane bending |
| 848 | C–O | In-plane bending (only appeared in vaterite and aragonite) |
| 713 | C–O | Out-plane bending (750 cm−1 in vaterite; 713 cm−1 in calcite; 707 cm−1, 692 cm−1 in aragonite) |
3.3. Characteristics of the Metal Carbonate: FE-SEM Image Analysis

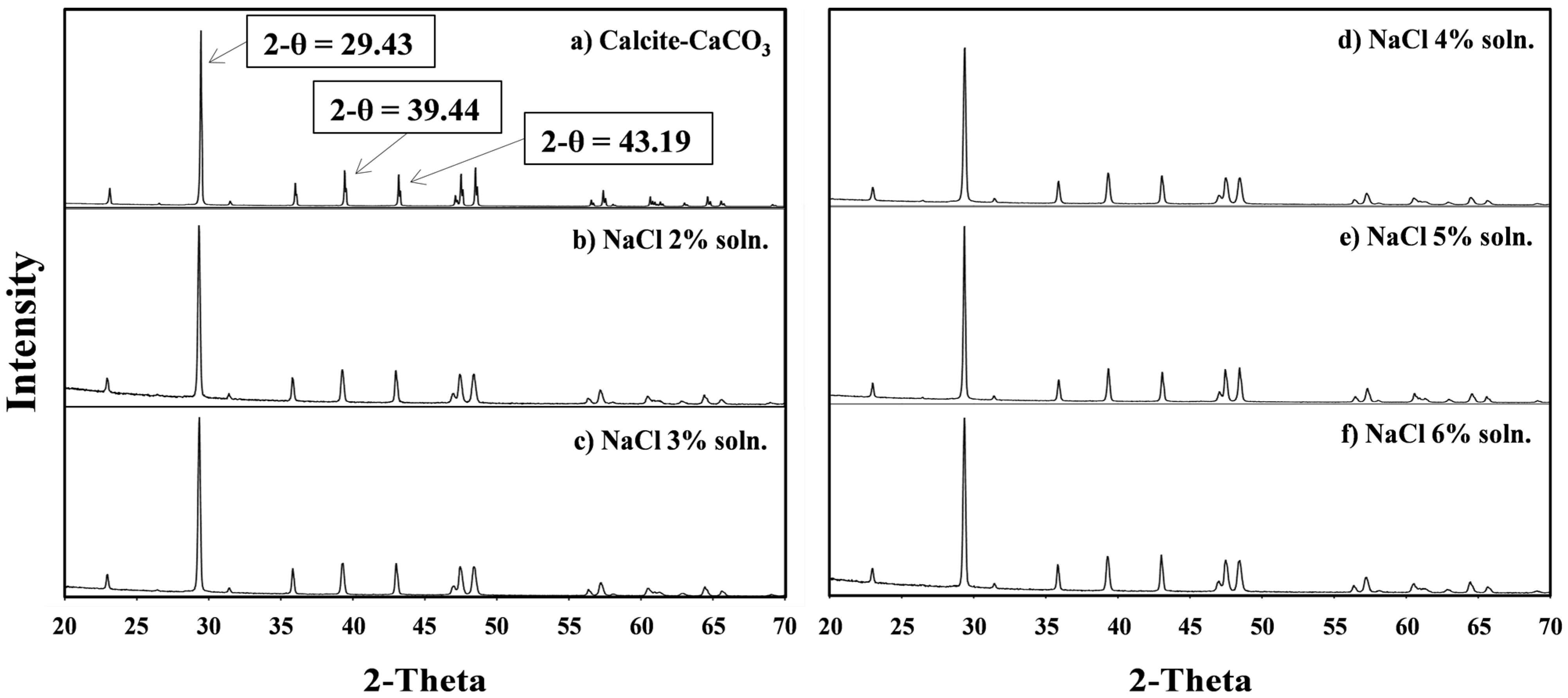
3.4. Characteristics of the Metal Carbonate: The XRD Pattern Analysis
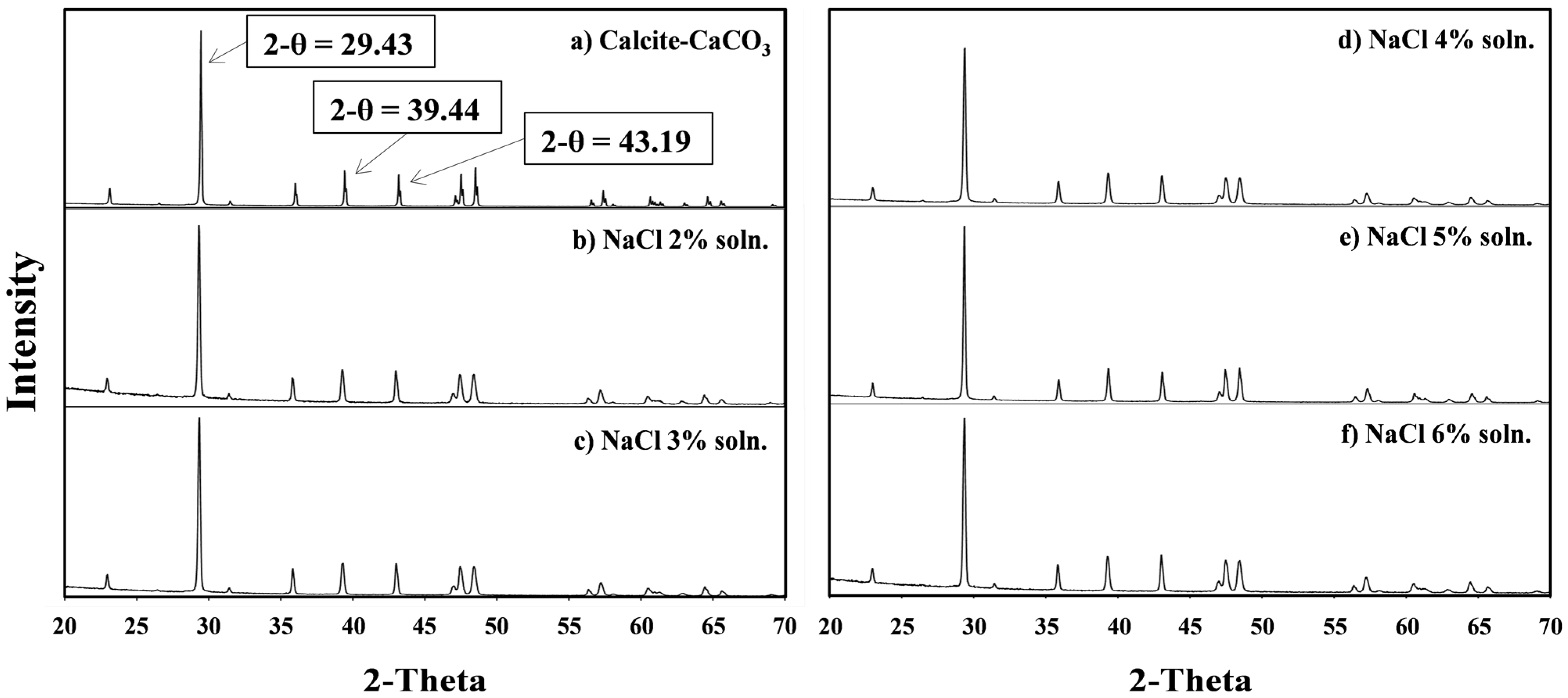
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yu, K.M.K.; Curcic, I.; Gabriel, J.; Tsang, S.C.E. Recent advances in CO2 capture and utilization. ChemSusChem 2008, 1, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Khoo, H.H.; Tan, R.B. Environmental impact evaluation of conventional fossil fuel production (oil and natural gas) and enhanced resource recovery with potential CO2 sequestration. Energy Fuels 2006, 20, 1914–1924. [Google Scholar] [CrossRef]
- West, T.O.; Peña, N. Determining thresholds for mandatory reporting of greenhouse gas emissions. Environ. Sci. Technol. 2003, 37, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Zeebe, R.E.; Zachos, J.C.; Caldeira, K.; Tyrrell, T. Carbon emissions and acidification. Sci. N. Y. Wash. 2008, 321, 51. [Google Scholar] [CrossRef] [PubMed]
- Abbott, T.M.; Buchanan, G.W.; Kruus, P.; Lee, K.C. 13c nuclear magnetic resonance and Raman investigations of aqueous carbon dioxide systems. Can. J. Chem. 1982, 60, 1000–1006. [Google Scholar] [CrossRef]
- Inoue, R.; Ueda, S.; Wakuta, K.; Sasaki, K.; Ariyama, T. Thermodynamic consideration on the absorption properties of carbon dioxide to basic oxide. ISIJ Int. 2010, 50, 1532–1538. [Google Scholar] [CrossRef]
- Siefert, N.S.; Litster, S. Exergy and economic analyses of advanced IGCC-CCS and IGFC-CCS power plants. Appl. Energy 2013, 107, 315–328. [Google Scholar] [CrossRef]
- Baciocchi, R.; Corti, A.; Costa, G.; Lombardi, L.; Zingaretti, D. Storage of carbon dioxide captured in a pilot-scale biogas upgrading plant by accelerated carbonation of industrial residues. Energy Procedia 2011, 4, 4985–4992. [Google Scholar] [CrossRef]
- Gough, C. State of the art in carbon dioxide capture and storage in the UK: An experts’ review. Int.J. Greenh. Gas Control 2008, 2, 155–168. [Google Scholar] [CrossRef]
- Holloway, S. Underground sequestration of carbon dioxide a viable greenhouse gas mitigation option. Energy 2005, 30, 2318–2333. [Google Scholar] [CrossRef]
- Damen, K.; Faaij, A.; Turkenburg, W. Health, safety and environmental risks of underground CO2 storage—Overview of mechanisms and current knowledge. Clim. Chang. 2006, 74, 289–318. [Google Scholar] [CrossRef]
- Mazzoldi, A.; Hill, T.; Colls, J.J. CFD and gaussian atmospheric dispersion models: A comparison for leak from carbon dioxide transportation and storage facilities. Atmos. Environ. 2008, 42, 8046–8054. [Google Scholar] [CrossRef]
- Burgherr, P.; Hirschberg, S. Comparative Assessment of Natural Gas Accident Risks; Paul Scherrer Institute: Villigen PSI, Switzerland, 2005. [Google Scholar]
- Lackner, K.S.; Butt, D.P.; Wendt, C.H. Progress on binding CO2 in mineral substrates. Energy Convers. Manag. 1997, 38, S259–S264. [Google Scholar] [CrossRef]
- Stasiulaitiene, I.; Fagerlund, J.; Nduagu, E.; Denafas, G.; Zevenhoven, R. Carbonation of serpentinite rock from Lithuania and Finland. Energy Procedia 2011, 4, 2963–2970. [Google Scholar] [CrossRef]
- Fagerlund, J.; Nduagu, E.; Romão, I.; Zevenhoven, R. CO2 fixation using magnesium silicate minerals part 1: Process description and performance. Energy 2012, 41, 184–191. [Google Scholar] [CrossRef]
- Fagerlund, J.; Nduagu, E.; Zevenhoven, R. Recent developments in the carbonation of serpentinite derived Mg(OH)2 using a pressurized fluidized bed. Energy Procedia 2011, 4, 4993–5000. [Google Scholar] [CrossRef]
- Fagerlund, J.; Teir, S.; Nduagu, E.; Zevenhoven, R. Carbonation of magnesium silicate mineral using a pressurised gas/solid process. Energy Procedia 2009, 1, 4907–4914. [Google Scholar] [CrossRef]
- Gerdemann, S.J.; O’Connor, W.K.; Dahlin, D.C.; Penner, L.R.; Rush, H. Ex situ aqueous mineral carbonation. Environ. Sci. Technol. 2007, 41, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Khoo, H.; Bu, J.; Wong, R.; Kuan, S.; Sharratt, P. Carbon capture and utilization: Preliminary life cycle CO2, energy, and cost results of potential mineral carbonation. Energy Procedia 2011, 4, 2494–2501. [Google Scholar] [CrossRef]
- Dou, B.; Song, Y.; Liu, Y.; Feng, C. High temperature CO2 capture using calcium oxide sorbent in a fixed-bed reactor. J. Hazard. Mater. 2010, 183, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Song, H.-J.; Lee, S.; Park, K.; Lee, J.; Chand Spah, D.; Park, J.-W.; Filburn, T.P. Simplified estimation of regeneration energy of 30 wt.% sodium glycinate solution for carbon dioxide absorption. Ind. Eng. Chem. Res. 2008, 47, 9925–9930. [Google Scholar] [CrossRef]
- Nduagu, E. Mineral Carbonation: Preparation of Magnesium Hydroxide [Mg(OH)2] from Serpentinite Rock. Master Thesis, The Åbo Akademi University, Åbo, Finland, 2008. [Google Scholar]
- Gilliam, R.J.; Decker, V.; Seeker, W.R.; Boggs, B.; Jalani, N.; Albrecht, T.A.; Smith, M. Low-Voltage Alkaline Production from Brines. U.S. Patent 12/703,605, 10 February 2010. [Google Scholar]
- Ferro, P.; Mishra, B.; Olson, D.; Averill, W. Application of ceramic membrane in molten salt electrolysis of CaO-CaCl2. Waste Manag. 1998, 17, 451–461. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters; John Wiley & Sons: Hoboken, NY, USA, 2012; Volume 126. [Google Scholar]
- Tlili, M.; Amor, M.B.; Gabrielli, C.; Joiret, S.; Maurin, G.; Rousseau, P. Characterization of CaCO3 hydrates by micro- raman spectroscopy. J. Raman Spectrosc. 2002, 33, 10–16. [Google Scholar] [CrossRef]
- Wu, G.; Wang, Y.; Zhu, S.; Wang, J. Preparation of ultrafine calcium carbonate particles with micropore dispersion method. Powder Technol. 2007, 172, 82–88. [Google Scholar] [CrossRef]
- Lyu, S.-G.; Sur, G.-S.; Kang, S.-H. A study of crystal shape of the precipitated calcium carbonate formed in the emulsion state. J. Korean Ins. Chem. Eng. 1997, 35, 186–191. [Google Scholar]
- Saylor, C.H. Calcite and aragonite. J. Phys. Chem. 1928, 32, 1441–1460. [Google Scholar] [CrossRef]
- Kontoyannis, C.G.; Vagenas, N.V. Calcium carbonate phase analysis using XRD and FT-Raman spectroscopy. Analyst 2000, 125, 251–255. [Google Scholar] [CrossRef]
- McMurdie, H.F.; Morris, M.C.; Evans, E.H.; Paretzkin, B.; Wong-Ng, W.; Ettlinger, L.; Hubbard, C.R. Standard X-ray diffraction powder patterns from the JCPDS research associateship. Powder Diffr. 1986, 1, 64–77. [Google Scholar] [CrossRef]
- Yoshioka, S.; Kitano, Y. Transformation of aragonite to calcite through heating. Geochem. J. 1985, 19, 245–249. [Google Scholar] [CrossRef]
- Rao, M.S. Kinetics and mechanism of the transformation of vaterite to calcite. Bull. Chem. Soc. Jpn. 1973, 46, 1414–1417. [Google Scholar] [CrossRef]
- Koga, N.; Kasahara, D.; Kimura, T. Aragonite crystal growth and solid-state aragonite-calcite transformation: A physico-geometrical relationship via thermal dehydration of included water. Cryst. Growth Des. 2013, 13, 2238–2246. [Google Scholar] [CrossRef]
- Svensson, R.; Odenberger, M.; Johnsson, F.; Strömberg, L. Transportation systems for CO2 application to carbon capture and storage. Energy Convers. Manag. 2004, 45, 2343–2353. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.S.; Lee, J.S.; Han, J.; Park, S.; Park, J.; Min, B.R. CO2 Fixation by Membrane Separated NaCl Electrolysis. Energies 2015, 8, 8704-8715. https://doi.org/10.3390/en8088704
Park HS, Lee JS, Han J, Park S, Park J, Min BR. CO2 Fixation by Membrane Separated NaCl Electrolysis. Energies. 2015; 8(8):8704-8715. https://doi.org/10.3390/en8088704
Chicago/Turabian StylePark, Hyun Sic, Ju Sung Lee, JunYoung Han, Sangwon Park, Jinwon Park, and Byoung Ryul Min. 2015. "CO2 Fixation by Membrane Separated NaCl Electrolysis" Energies 8, no. 8: 8704-8715. https://doi.org/10.3390/en8088704
APA StylePark, H. S., Lee, J. S., Han, J., Park, S., Park, J., & Min, B. R. (2015). CO2 Fixation by Membrane Separated NaCl Electrolysis. Energies, 8(8), 8704-8715. https://doi.org/10.3390/en8088704