A Review of Thermal Co-Conversion of Coal and Biomass/Waste


Abstract
: Biomass is relatively cleaner than coal and is the only renewable carbon resource that can be directly converted into fuel. Biomass can significantly contribute to the world's energy needs if harnessed sustainably. However, there are also problems associated with the thermal conversion of biomass. This paper investigates and discusses issues associated with the thermal conversion of coal and biomass as a blend. Most notable topics reviewed are slagging and fouling caused by the relatively reactive alkali and alkaline earth compounds (K2O, Na2O and CaO) found in biomass ash. The alkali and alkaline earth metals (AAEM) present and dispersed in biomass fuels induce catalytic activity during co-conversion with coal. The catalytic activity is most noticeable when blended with high rank coals. The synergy during co-conversion is still controversial although it has been theorized that biomass acts like a hydrogen donor in liquefaction. Published literature also shows that coal and biomass exhibit different mechanisms, depending on the operating conditions, for the formation of nitrogen (N) and sulfur species. Utilization aspects of fly ash from blending coal and biomass are discussed. Recommendations are made on pretreatment options to increase the energy density of biomass fuels through pelletization, torrefaction and flash pyrolysis to reduce transportation costs.1. Introduction
Energy is the cornerstone to economic stability and development. Since the industrial revolution, fossil fuels have stimulated economic growth especially in the developed world. Only recently have we realized that this accelerated economic growth has not occurred without a penalty. Carbon dioxide emissions from fossil fuels combustion, combined with land-use changes, have driven the concentration of this most significant greenhouse gas (GHG) to levels in our atmosphere not seen for at least 800,000 years, and probably many millions of years [1]. Although somewhat controversial, the scientific evidence of global warming and climate change and their link to anthropogenic activities has been established. The concentration of GHG in the atmosphere and its consequences is expected to be exacerbated given the impressive world population growth as well as economic development expected in developing countries with a direct effect on competition for energy resources. The quest for substitutes to fossil fuels, the need to mitigate the negative environmental effects of fossil fuels utilization and the necessity to safely and economically dispose wastes have encouraged the development of alternative sources of energy and promotion of low quality fuels. Co-conversion of coal and biomass/wastes for energy purposes and chemicals are among these alternatives.
Coal fired power plants are still the largest source of electricity generation in the Unites States (contributing to about 42% of net electricity generation), and will keep their lead until 2040 [2]. According to the International Energy Agency (IEA), coal will continue to be a global energy solution throughout the 21st century [3]. The world's coal stock is still enormous. Coal has a low cost and a high energy density. It is expected to contribute significantly in the future energy needs in many nations [4,5], especially in fast-developing countries such as China and India. China alone uses nearly twice as much coal as all countries from the Organization for Economic Co-operation and Development (OECD) combined, while India has since 2011 become the third largest coal consumer in the world after china and the Unites States [5]. Conversion of coal-to-liquids and chemicals could become significant in the petrochemical industry. However, coal has a major drawback: it is responsible for the emission of environmentally harmful compounds [sulfur, nitrogen (N) and heavy metals]. Combustion of fossil fuels, which accounts for about 75% of total GHG emissions, together with land-use change is the main source of anthropogenic GHG emissions [4–7]. Combustion of coal alone accounts for 30% to 40% of the share of GHGs from fossil fuels combustion [4–7]; therefore, coal utilization deserves special attention given the likelihood of continued use for electricity generation and the potential for coal to partially replace petroleum for chemicals and transportation fuels as has recently occurred in China.
Biomass is any organic matter recently derived from plants through photosynthesis. It is a versatile feedstock encompassing forestry and agricultural residues, organic waste by-products, energy crops, sewage sludge and biosolids, woody plants and municipal green waste [8] and can be used simultaneously to generate heat, power, gaseous liquid fuels, materials, and chemicals. The estimation of the amount of biomass that can be generated as energy with minimum disturbance to the supply for food crop is controversial, estimated to about 35 EJ/year to 1135 EJ/year by Hoogwijk et al. [9] and to about 100–600 EJ/year by Slade et al. [10]; the former corresponds to about 6% to 189% of the current global primary energy usage while the later corresponds to about 17%–100%. Therefore, biomass is a precious resource which, if harnessed sustainably could be a meaningful contributor to meeting the future world energy needs. On a security point of view, large proportions of oil and gas reserves are concentrated in politically unstable regions, therefore, increasing the diversity in energy sources is important for many nations to secure a reliable and constant supply of energy [11], while contributing to a reduction in GHG emissions.
Biomass in general has a high content of hydrogen (H), making it suitable as a blend to compensate the often-low H content of coal. Biomass as gasification feedstock, although giving a high hydrogen yield, has the disadvantage of low energy density because of its high oxygen and moisture content. This shortcoming is compensated for when blended with a higher energy content coal. Other challenges such as the seasonal limitation of biomass are somewhat mitigated through co-conversion with coal. The higher tar release (due to excessive volatile release and low gasification temperature from biomass gasification) is also reduced as blending with coal increases the temperature, enhancing tar cracking. Blending biomass and coal as feedstock can reduce the shortcomings of each fuel and boost the efficacy of the overall system.
The most attractive benefit of co-converting coal and biomass is certainly the reduction of GHG emissions and environmental pollution. Biomass, compared to coal, has lower sulfur, N and heavy metals. It is also carbon-neutral if produced sustainably, thus co-conversion of biomass and coal can make significant contributions in mitigating GHG emission and other emissions. Compared to carbon capture and storage (CCS), co-converting biomass with coal is relatively cost-effective, given the high energy penalty of CCS which can range from 15% to 40% of the energy output [12]. Even if CCS were to be implemented as a means for GHG mitigation, it would not, alone, be able to meet the 50% emission reduction target by 2050 suggested by the International Panel on Climate Change (IPCC) [13]. Life cycle analyses of co-conversion are currently mostly limited to co-firing coal and biomass for combustion, while co-fired gasification investigations are scarce. Co-firing coal and biomass is currently among the most efficient renewable energy and climate change technologies available due to the high power generation efficiencies that are generally not achieved in smaller-scale dedicated biomass facilities. Considering biomass as a carbon-neutral resource, CO2 emission should decline proportionally to the amount of coal offset by biomass [14]; however, thermal conversion of biomass also prevents some biomass waste from occupying landfills and thus also reduces the landfill methane emission. Co-firing also results in lower N2O emission compared to coal alone [15], as both CH4 and N2O have higher global warming potentials (GWP) than CO2 (21 and 310, respectively) [16]. The overall outcome of co-firing is therefore a percentage reduction of CO2 equivalent higher than the percentage of biomass in the co-firing blend. The analysis of Mann and Spath [17] for combustion and gasification are illustrative. Pollutants from co-firing and co-gasification are different (e.g., SO2 from co-firing versus H2S and COS from co-gasification; NOx from co-firing versus NH3 and CHN from co-gasification), making co-combustion not suitable to appraise pollutants release during co-gasification. However, the relatively low sulfur and nitrogen contents of biomass compared to coal imply that lower amounts of H2S, NH3 and HCN are expected when co-gasifying.
Co-conversion of coal and biomass offers a better prospect for cleaner coal utilization and a way to dispose wastes/biomass in an economical, safe, and environmentally-acceptable manner. Some research on these systems has been performed, however, the wide variability of parameters (type of feedstock, type of gasifier, gasification conditions) in these systems requires additional research to identify the most important parameters and clarify how they affect the co-conversion efficiency and product yield.
This present work critically examines literature on thermal co-conversion (liquefaction, pyrolysis, gasification, combustion) of coal and biomass from pre-processing of feedstock to product utilization. Given the complexity of gasification compared to other processes, it will be the focal process during this analysis.
2. Feedstock Preprocessing and Feeding
Carbonaceous solid fuels, which include coal and biomass, are complex collections of organic polymers consisting of aromatic chains combined by hydrocarbons and other atoms such as oxygen, N, sulfur, potassium (K) and sodium (Na). Coals are mainly formed as the results of slow metamorphosis of biomass over a long period of time. The degree of that metamorphosis is among the criteria used to determine coal rank. For instance, the lower degree of metamorphosis is found in lignites and the maximum are found in low-volatile bituminous and anthracite coals. Intermediary stages are the subbituminous and other bituminous ranks [18]. Among thermal conversion processes, gasification is the most demanding in terms of end-use products (synthesis gas), which can be affected by the pre-processing steps for biomass fuels.
Gasification produces synthesis gas (syngas) and by-products such as ash and tars. Whether the syngas is used for power generation or for liquid fuel production, a clean gas free from tar and other contaminants is required. Biomass gasification has the drawback of producing a high amount of tar, therefore, tar avoidance or removal becomes important. Updraft gasifiers (fixed bed) produce more tar than fluidized beds, and fluidized beds more than entrained flow gasifiers [16–20]. Therefore, fixed bed and fluidized bed gasifiers produce the “dirtiest” syngas compared to entrained flow gasifiers [16–20] thus entrained flow gasification could be more suitable for co-gasification. Nonetheless, most large biomass gasifiers operate with fluidized bed technology.
Obtaining a relatively clean gas suitable for liquid fuel production from carbonaceous fuels requires operation at high temperature, which can be easily met with entrained flow gasifiers. At such temperatures (≈1573–1973 K), a gas with very low concentration of tar, methane, and carbon dioxide is obtained, which significantly decreases the gas cleaning cost.
Modern entrained flow gasifiers using solid feed operate at a pressure of between 15 bar and 60 bar. These elevated pressures are advantageous for gas to liquid synthesis. Pressurizing synthesis gas requires more energy than pressurizing ox0ygen; the reason being that the volume of synthesis gas is about double the volume of oxygen required for its production [19]. Improved ash handling also occurs with entrained flow gasifiers under slagging conditions. However, to render the biomass mineral matter, the gasifier should operate in a non-slagging mode such that the dried ash produced could be released back to the forest. This is challenging as some toxic minerals from coal may be applied to the soil. Ash from coal contains considerable amount of toxic elements including traces of mercury and lead.
The high reaction rates of entrained flow gasifiers demand very small feedstock input size (∼100 μm), easily achievable for friable materials like coal, but more challenging and energy-consuming for biomass due to its fibrous structure and hygroscopic nature. Therefore, the viability of entrained flow gasification of biomass relies on its pretreatment. Moreover, entrained flow gasifiers operate at high pressures and need injection of the fuel, which is usually introduced into the pressurized reactor through an alternating pressurized and depressurized lock system. The periodic procedure is suited for high bulk density fuels and moderate pressures. At low bulk densities (e.g., biomass) and higher pressures (>50 bar; e.g., operating pressure for subsequent liquid processes), the amount of lock gas can exceed the fuel weight. Therefore, a well-designed feeding system to withstand back pressure from the gasifier is required and this may be as expensive as the gasifier itself [20]. The fibrous nature of the biomass also prevents it from fluidizing and fluffs are formed that may plug piping. Among the pretreatment options available to palliate these biomass shortcomings are pelletization, torrefaction and fast pyrolysis [21].
Torrefaction is a thermal treatment at a temperature of 473–573 K, near atmospheric pressure and in the absence of oxygen. Ciolkosz and Wallace [22] have explored the key features for biomass torrefaction for bioenergy production. This mild thermal treatment not only destroys the fibrous structure and tenacity of the biomass (wood), but is also known to increase the calorific value and to invert its hydrophilic nature [23,24]. Therefore, Torrefaction increases its handling characteristics for storage, transportation and gasification purposes. Torrefaction can significantly decrease the power need for the processing operation (up to 85%) and at the same time the capacity of the milling plant can increase considerably (by a factor of 2–6) while nearly 85%–90% of the original biomass energy is retained in the solid product [23]; issues such as bridging and blockage of feeding systems as well as poor pulverization characteristics are resolved [25,26]. The ground torrefied biomass is pneumatically fed if a dried-coal fired system is used or slurry fed if slurry-coal fired system is in place.
Fast pyrolyis is a rapid heating process at about 773 K in an inert atmosphere. Rapid condensation of the vapors in one to a few seconds occurs, and more than half of the biomass mass can be obtained as liquid pyrolysis oil at the expense of low char and gas yield. In these conditions, the pyrolysis gases contain less than 10% of the biomass energy, which is consumed on-site as energy input to the pyrolysis process. The brittle char obtained is ground to a fine powder and suspended in the liquid pyrolysis oil to form the bioslurry, with an overall energy content of up to 80%–90% of the original biomass energy [27,28], and considerable volume reduction.
The dispersed nature of the biomass feedstock requires the pretreated fuel to be transported to a central plant where it is co-converted with coal. Important parameters in deciding which of the two pretreatment options to consider are the energy density and post processing of the pretreated fuel.
Bioslurry is denser (and less bulky) than torrefied biomass. Thus besides feeding bioslurry under pressure being easier; pneumatically feeding powdered torrefied wood requires pressurizing with an inert gas that reduces the calorific value and complicates the gas cleaning process. Another advantage of the fast pyrolysis process is that to avoid soil nutrients depletion, pyrolysis can be designed to optimize and obtain only an energy-rich bio-oil containing 60% to 70% of the original biomass energy [29], while the remaining char may be immediately returned to the soil. This approach may well be more efficient since it avoids transporting the ash from the gasification plant to the forest. Moreover, it ensures that almost all the ash extracted by biomass is returned to the soil and prevents possible toxic elements that might be present in coal ash to be applied to the soil. Additional savings could result from the simplicity associated with handling bio-oil at the plant site. Handling bio-oil at the plant site will require fewer and comfortable metering and handling equipment compared to handling solid feedstock.
3. Conversion Parameters
3.1. Feedstock
Biomass fuels usually have a low as-received fixed carbon value compared to coals while the volatile matter values are higher, therefore, when co-gasifying coal and biomass, a low release of carbon-containing compounds such as CO2, CO, CH4 and other light hydrocarbons are expected in comparison, to gasifying coal. This is contrary to results obtained by [30–35]; their experimental results show that increasing biomass ratio in the blend either increases concentration of carbon-containing compounds [CO2, CO, light hydrocarbon gases (C1 and C2) and tars] while H2 either decreases or stays constant. A screening of the experimental data can clarify this apparent contradiction. Kumabe et al. [33] added biomass on a carbon basis, not on a total weight basis; however, the hydrogen content in the biomass used was high enough so increasing the biomass ratio in the fuel blend would have increased the ration of hydrogen in the gas, instead a reduced concentration of hydrogen in the gas was observed. Using the ultimate analysis of the fuels the relative atomic compositions were CH1.548O0.737N0.002 for biomass (Japanese Cedar) and CH0.597O0.159N0.013 (Mulia coal). The authors observed an increase in CO2, a decrease in H2 and a constant concentration of CO. They also acknowledge the relatively high air-fuel ratio as compared to common gasification conditions and a lower temperature. Therefore, the lower temperature was more favorable to exothermic reactions, which were enhanced by the high air-fuel ratio, likely oxidizing part of hydrogen produced to steam, and hydrocarbons and a portion of carbon monoxide to carbon dioxide. Similar analysis can be made for [30–32,35]. This analysis also seems to explain why Cascarosa et al. [36] observed an increase of gas yield but a decrease of the gas calorific value when mixing 1% meat and bone meal (MBM) with coal in a fluidized bed gasifier.
Many co-conversion studies related to co-gasification studies have focused on parametric studies of the gasifier [31–35,37–43], mostly omitting the mechanisms by which mixtures of coal and biomass interact and how coal and biomass thermally degrade. The interaction between coal and biomass during thermal co-conversion is an issue yet to be solved; some devolatilization and pyrolysis results of coal and biomass blends have revealed no or very little synergy between the two fuels [44–56] while others have revealed significant synergy [43,57–71]. It is important to segregate the synergistic effect from the catalytic effect engendered by biomass mineral matters; most of the studies that found synergistic effects between the two fuels failed to segregate these two phenomena. Devolatilization/pyrolysis conditions as well as the type of feedstock [72–75] dictate the yield and composition of products during this stage, and therefore, influence the resulting char and the environment in which the char decomposes. The results of Yang et al. [74] for a packed bed reactor revealed that, at low heating rate (10 K/min), temperature of up to 1173 K, inert environment and atmospheric pressure, cellulose contributed mostly to the release of CO, whereas hemicelluloses contributed mostly to CO2, and lignin to H2 and CH4. Their explanation of this observation is that for lignin decomposition, H2 is released during cracking and deformation of the aromatic C=C and C–H bonds while CH4 is released during cracking of methoxyl groups; for cellulose which is high in carbonyl groups, CO is released and for hemicelluloses rich in carboxyl, CO2 is released. Very few studies have focused attention on pyrolysis/devolatilization conditions that pertain to gasification (high heating rate, high temperature, and pressurized atmosphere). Moreover the mechanism by which biomass components (cellulose, hemi-cellulose and lignin) interact under heating, the effect of mineral matter, as well as how these individual components and biomass as a whole would interact when blended with coal has not been elucidated. The effect of feedstock can thus influence the conversion behavior during the early stage of fuel decomposition (devolatilization/pyrolysis) as well as during char gasification.
3.1.1. Devolatilization/Pyrolysis
The generalized biomass pyrolysis model based on superimposed cellulose, hemicelluloses and lignin kinetics [76,77] has been questioned by recent developments in biomass thermal decomposition under pyrolysis conditions. Although Raveendran et al. [78] found no detectable interaction between biomass components during pyrolysis, and therefore agreed with the superimposed model, they acknowledged the influence of biomass ash on both pyrolysis characteristics and the product distribution. A study by Couhert et al. [79] demonstrated that because of the interactions between the structural components and the role of mineral matters in biomass, an additive law does not allow the gas yields of biomass to be correlated with its fractions of cellulose, hemicelluloses and lignin, thus suggesting a deeper analysis of the effect of biomass components interactions and ash on its decomposition. However, it is well known that cellulose is mainly responsible for volatiles, lignin is the main contributor of char while hemicellulose contributes to almost equally to both [74,80]. All these studies have been conducted at temperatures less than 1200 K; studies on behavior of biomass components at temperatures of 1600 K and higher reflecting the conditions of an entrained flow gasification process are scarce. Understanding the role of biomass ash and biomass components on volatile yield and composition and on char formation during thermal decomposition would facilitate the knowledge of how blends of coal and biomass behave under thermal decomposition.
The mineral matter in biomass (especially the ion-exchangeable alkali metals) is expected to further promote catalytic activity when blended with coal. Whether the influence of biomass has a greater impact during gasification than pyrolysis is unclear, but the type of feedstock appears to be a major factor in this partitioning. This remark is highlighted by Storm et al. [81] who did not find any interaction during co-pyrolysis of coal and straw, and instead found a lower release of pyrolysis products from blend of coal and sewage sludge as compared to unmixed fuels. Biagini et al. [44] found that thermal decomposition of biomass is not significantly affected by the presence of coal; in the same manner, coal is not affected by the release of volatile matter from biomass. It should be noted that in their study, the flow of nitrogen in a TGA did sweep the volatile products away from the devolatilizing fuels, therefore, the devolatilizing gas could not easily interact with the char, and thus the additive behavior of the products prevailed. These observations are well contrasted with results of [43,47,82,83] who identified some synergy.
Since blending coal and biomass/waste (heat input base) leads to an increase of the volatile products (CO, CH4 and CnHm) the energy content of the gas is expected to be higher than when gasifying coal alone because of the high heating value of CH4 and CnHm. The energy conversion efficiency is therefore supposed to be higher than when coal is gasified alone [31,33,35]. Kumabe et al. [33] reported an increase in conversion efficiency with increase in biomass ratio of the feedstock as well. Franco et al. [30] obtained similar results and observed that higher gas yield was observed when 40% of pine waste was used by mass in the coal-biomass blend. However, they noted that the higher energy conversion efficiency and heating value was obtained when higher amount of polyethylene (PE) was present in the mixture. This was explained by PE having a high content of C and H as compared to coal and pine waste. For mixtures of coal with equal amounts of pine and PE wastes, the gasification results were found to lie between those obtained for compositions with the same amount of only one waste.
3.1.2. Synergistic Effect during Coal and Biomass/Waste Co-Conversion
3.1.2.1. Synergy through Catalytic Effect
The merits of alkali (K+ and Na+) and alkaline earth (Ca2+) metals as catalysts for coal gasification have been extensively investigated [84–87], given the high content of these elements in some biomass, co-gasification of coal and biomass is expected to exhibit some catalytic behavior, whose significance will depend on the type of biomass and coal being used. In fact, Na is the principal alkali metal in lignite (low rank coal) and is said to be bonded to the oxygen anions in the carboxyl groups, while the amount of K is low [88]. In bituminous coals (high rank), K is said to be contained exclusively in illite or closely related clay structure [89], while Na is generally present as NaCl, usually as solution in moisture adsorbed in coal pores and capillaries [90]. As for calcium (Ca), it is present as exchanged ion with the hydrogen of the carboxyl group of low-rank coals; from low-rank coals to higher rank coals, Ca is systematically changed from carboxyl-bound to calcite [91], with decreasing catalytic activity [92]. The K present in the illite of bituminous coal is known to be transformed into a K-aluminosilicate glass [89] whereas the NaCl is highly volatilized. Therefore, mineral matters of high-rank coals have little catalytic activity during coal gasification. Moreover, the associated ion-exchangeable cations in low-rank coals are well distributed, at the atomic level. Therefore, any catalytically active ions such as Ca and Na are highly dispersed [93]. This is applicable to biomass fuels as well and sheds some light on why biomass mineral matter exerts some catalytic activity when blended with coal during thermal conversion. Ren et al. [94] compared co-gasification of MBM blended with a high-rank coal (anthracite), and MBM blended with a low-rank coal (lignite). Acid washed MBM samples blended with anthracite or lignite had a lower carbon conversion compared to the raw MBM blend. Thus, biomass mineral matter (Na, K and Ca) demonstrated catalytic influence during co-gasification, as reported by other authors [95,96]. They also found that the catalytic effect of MBM minerals was not perceivable on lignite but was significant for anthracite. This is in accordance with McKee et al. [97] and Srivastava et al. [98] who demonstrated the increased catalytic activity of alkali metals on gasification with increasing coal rank. The reaction rates of a blend of waste birch wood and Daw Mill coal was significantly increased and the char yields were lower than a simple blending prediction in a pressurized fluidized bed oxygen atmosphere gasifier with temperatures varying from 973 K to 1173 K at a pressure of 0.4 MPa [67]. Brown et al. [96] came to the conclusion that switchgrass char and switchgrass ash displayed catalytic activity in mixtures with coal char produced from Illinois No. 6 coal. The coal char gasification rate increased by eight fold at 1168 K in a mixture of 10:90 of coal char and switchgrass ash. In general, biomass is more reactive than any coal [99]. Its char is therefore continuously consumed during gasification, leaving very little remains at the end of the process, whereas coal char is less reactive and continuously accumulates in the bed during the course of the gasification. Blending biomass and coal takes advantage of both the high reactivity of biomass and its catalytic effect. However, some negative catalytic effects have been evidenced by Habibi et al. [100]. They observed an antagonism between switchgrass and a subbituminous coal while the mixture of switchgrass and fluid coke showed a synergy. They explain the antagonism by a deactivation of mineral catalysts due to sequestration of the mobile alkali elements by reaction with aluminosilicate minerals in coal to form inactive alkali aluminosilicates.
The catalytic effect of biomass in co-gasification with coal is also expected to play an important role for the abatement of environmentally harmful species containing sulfur and nitrogen. Biomass species with a high content of K, Na and Ca can form sulfate and capture sulfur from the gas phase when co-processed with coal [101]. During co-firing of straw and coal, Pedersen et al. [101] observed a net decrease of NO and SOx emission, due to the decrease of fuel- nitrogen conversion to NO and due to retention of sulfur in the ash. Sjöström et al. [67] also identified a lower ammonia yield during co-gasification of birch wood and Daw Mill coal. Cordero et al. [68] also showed enhancement in desulfurization when blending coal with different types of biomass during co-pyrolysis compared to coal pyrolysis. Haykiri-Acma and Yaman [57] showed that the addition of hazelnut shell to lignite contributed to the sulfur fixing potential of the resulting char in the form of CaS and CaSO4 during co-pyrolysis of these feedstocks.
3.1.2.2. Synergy through Other Mechanisms
The synergistic effect of co-conversion of coal and biomass can be viewed in terms of:
- -
increase in total volatiles (tars and light gases) release and decrease in char yield;
- -
overall decrease in pollutants (oxides of nitrogen and their precursors and well as SOx and their precursors).
The increase in total volatiles released and corresponding decrease in char production has received the most attention for synergistic evaluations. In the literature on thermal co-conversion of coal and biomass/waste, the non-catalytic mechanism responsible for this observation has been attributed to free radical/hydrogen transfer. However, the existence of non-catalytic synergistic effect remains controversial. Some researchers [43,57–71,101–108] have shown evidence of synergy between biomass and coal while others [44–56] found no evidence of synergy. Trevor and Kandiyoti [109] pointed out that the way biomass components are intermeshed affects the distribution of pyrolysis products. This important remark suggests that different biomass fuels may exhibit different behavior during co-conversion with coal, since the interlacing of biomass components (cellulose, hemicellulose and lignin) is unique for different biomass species.
It is obvious that interactions occurring between coal and biomass/waste during thermal conversion have not been sufficiently investigated and require further evaluation to elucidate the mechanism and conditions by which the two fuels interact when blended and converted.
3.1.3. An Analysis of Conditions Favorable to Synergy by Free Radical/Hydrogen Transfer during Thermal Conversion of Coal and Biomass/Waste Blends
3.1.3.1. Synergistic and Non-Synergistic Activities of Coal-Biomass Conversion Processes
3.1.3.1.1. Direct Liquefaction
As depicted in Figure 1, liquefaction is a process by which a solid fuel (coal or biomass) is thermally ruptured into small radical fragments that are then stabilized by hydrogen atoms provided by a solvent or a hydrogen rich gas; the solvent also promotes bonds breaking, thereby allowing the process to occur at lower temperatures.
This process has been thoroughly investigated. There is clear evidence of the interaction during co-liquefaction of some fuels. The earliest depiction of synergy mechanisms during co-liquefaction is probably by Coughlin and Davoudzadeh [102]. In their experiment, they co-liquefied lignin and Illinois No. 6 bituminous coal using a series of catalysts with tetralin and phenol as solvents. They concluded that when coal and lignin react together in a solvent, they depolymerize under mild conditions to produce a filterable liquid product with yields greater than would be predicted based on a simple side-by-side, concurrent liquefaction of both components independently. One interesting finding was that the overall conversion of coal plus lignin increased as the fraction of lignin in the reaction mixture increased, while the opposite held true for increased proportion of coal in the reaction mixture. However, they noted that the overall conversion was not improved further by increasing lignin ratio beyond 0.7. Feng et al. [103] came to the same observation when catalytically co-liquefying coal and waste plastics. They used a mixture of subbituminous coal (Black Thunder), PE and polypropylene in a tubing reactor with catalysts: HZSM-5 zeolite catalyst, ferrihydrite treated with citric acid, co-precipitated A12O3-SiO, and a ternary ferrihydrite—A12O3-SiO containing 10% A12O3-SiO. They also measured synergy in terms of enhanced oil yield. Mastral et al. [104] also conducted co-liquefaction of a low rank coal (SAMCA coal, from Utrillas, Spain) and waste tires in tubing bomb reactors without solvent. They showed that the addition of tire to coal during co-liquefaction gives a positive interaction not by increasing the total conversion, but by influencing the mechanism of stabilization of the generated coal radicals. This assertion was made due to the high yield of asphaltenes, confirming the lower yield of light hydrocarbons (C1–C3) in the experiment as compared to the theoretical estimation. Another investigation by Mastral et al. [105] revealed that the slight synergy found can be due to the small free radicals from vulcanized rubber decomposition, which are able to stabilize coal radicals to light products. Taghiei et al. [106] also conducted co-liquefaction of PE and mixed plastic waste with a bituminous coal, a subbituminous coal and lignite in a tubing bomb reactor. Co-liquefaction of mixed waste plastics with a bituminous coal exhibited higher oil yields than obtained by liquefaction of either the waste plastics or the coal alone implying synergistic effects. Co-liquefaction of a 50:50 mixture of mixed waste plastic and an iron ion-exchanged subbituminous coal with the addition of 1.0 wt% of HZSM-5 zeolite catalyst gave a total conversion of over 90% and oil yield of approximately 70%. The oil yield of the mixture appeared to have been increased approximately 10% by synergistic effects. Co-liquefaction of iron ion-exchanged Beulah lignite with medium-density PE with no added solvent gave very good oil yields and total conversions at 723 K, indicating that the plastic plays the role of a hydrogen donor solvent for the coal under those conditions. It is interesting that the total conversion of lignite and plastic is possible at 723 K without a solvent. Kanno et al. [107] also observed synergy in the conversion of Yallourn coal (lignite) and PE under pressurized hydrogen using 1-methylnaphthalene and tetralin as solvent. The conversion and the oil yield were larger than the additive values of respective fuels processed separately and the observed values of residues for both Yallourn coal and PE during the co-liquefaction were evidently smaller than the estimated values. Anderson co-liquefied tires and a low-rank coal in a magnetically stirred autoclave under hydrogen pressures of 10, 5 and 1 MPa and nitrogen pressure of 0.1 MPa. They noted that the synergy was observed only at low hydrogen pressures. The oils obtained in the co-liquefaction showed a more aromatic nature than those obtained when each material was processed separately, a higher boiling point was also observed, suggesting that radicals from rubber and coal reacted between each other instead of reacting with hydrogen radicals. Although the existence synergy has been agreed upon and the mechanism has been somehow well understood during co-liquefaction, it has not been the case for other thermal conversion processes.
3.1.3.1.2. Pyrolysis
Pyrolysis is the decomposition of an organic material by heating in the absence of oxygen, but with inert gases or less reactive gases (argon and nitrogen) producing a viscous dark liquid (oil), gases and leaving a charring material composed mainly of carbon. Devolatilization on the other hand is the first step of combustion or gasification. It differs from pyrolysis with the presence of a reactive gas (O2, H2O, CO2, etc.) during heating, and both processes will be referred to as pyrolysis.
Unlike liquefaction, no solvent is added to the organic feedstock during pyrolysis. Therefore, the thermal breakdown of the organic feedstock is not chemically enhanced, nor is there any hydrogen donor (solvent as in the case of liquefaction) to stabilize the radicals involved during thermal bond breaking.
As stated earlier, the literature on synergy during co-pyrolysis of solid fuels is controversial. Among co-pyrolysis studies that resulted in no synergy are that of Vuthaluru [111] who conducted co-pyrolysis of a subbituminous (Collie coal) coal with waste wood and with wheat straw using a thermo gravimetric analyzer (TGA). The samples were heated at 20 K/min from ambient to 1523 K under atmospheric pressure using argon as the carrier gas. No synergy was found. Collot et al. [37] also did not find synergy in the co-pyrolysis of Daw Mill coal (bituminous) and Silver Birch Wood in a fluidized bed reactor. In a fixed bed reactor the degree of synergy was insignificant at 1123 K and 1273 K with a heating rate of 10 K/min, using helium as the carrier gas under pressure from 1 bar to 25 bar. It is however worth noting that they observed a slight increase (though not significant) of tar in the fixed bed reactor. Storm et al. [81] pyrolyzed blends of biomass (straw), sewage sludge of a high-volatile bituminous coal (Gottelborn hard coal) in a bench scale entrained flow reactor at 1473 K and atmospheric pressure, using nitrogen as the carrier gas. Neither coal/biomass nor coal/sewage sludge tests showed synergy. Meesri and Moghtaderi [45] co-pyrolyzed a high-volatile bituminous coal (Drayton coal) and Radiata pine sawdust in a horizontal tubular reactor at slow heating rate (10 K/min) and temperatures of 473–1173 K, and in a drop tube furnace at high heating rate and temperatures of 1173–1673 K. The pyrolysis products obtained from co-pyrolysis during their experiments correlated well with the weighted sum of the individual fuels, indicating a lack of synergy. Idris et al. [112] investigated the pyrolysis behavior of blends of a Malaysian subbituminous coal and empty fruit bunches, kernel shell, mesocarp fibers of palm tree in a TGA using nitrogen as the carrier gas. The heating was varied at 10, 20, 40 and 60 K/min and the temperature ranged from 298 K to 1173 K. No synergy was found. Another co-pyrolysis study that failed to depict synergy is that of Moghtaderi et al. [46]. The same high-volatile bituminous coal used in [45] was blended with Radiata pine and pyrolyzed. Two sets of experiments were conducted: slow heating conditions (10–50 K/min) and a temperature range of 473 K to 673 K and a high heating condition (10,000 K/min) and temperature range of 1173–1673 K with nitrogen as the carrier gas in both cases. No synergy was depicted in their investigation. Vamvuka et al. [83] modeled the devolatilization kinetics of lignite blends with olive kernels, forest residues and cotton residues using TGA in nitrogen gas. The heating rate in their experiment varied from 10 K/min to 100 K/min and the temperature ranged from 298 K to 1123 K. They concluded that the co-pyrolysis kinetic data followed a weighted average of the pyrolysis data of the individual fuels. In a similar study, Vamvuka et al. [47] modeled the co-pyrolysis kinetics of a high-volatile bituminous or lignite coals, blended with olive kernel and straw. Experimental conditions were similar to [83] and they came to the same conclusion: no synergy. Despite the lack of synergy in the co-pyrolysis studies cited above, many other investigators have found synergy during coal/biomass, coal/waste pyrolysis.
Jones et al. [113] conducted devolatilization studies of blends of some coals (Wujek, bituminous; Kaltim Prima, bituminous; Turoszow, lignite) with a variety of biomass (pinewood, cellulose, lignin, xylan and polywax model compounds) in pyrolysis-gas chromatography-mass spectrometry (GC-MS), TGA, and static batch pyrolysis reactors at slow and moderate heating rates and a temperature range of 673–1173 K. Synergy was observed only in the batch reactor while the py-GC-MS and TGA displayed additive behavior. Hayashi et al. [114] conducted co-pyrolysis of Yallourn coal (lignite), Taiheiyo coal (subbituminous), with fine powder of homo-polypropylene, low-density PE or high-density PE in a Curie-point pyrolyser. They found an increase of total volatiles (tar and light gases) and a decrease of char for all combinations of coal-polyolefin as compared to the sum of corresponding individual yields. Oney et al. [115] conducted co-pyrolysis of Seyitömer lignite and safflower seeds blends both in a TGA and in a fixed bed reactor. In the TGA, the sample was heated at 5 K/min from ambient temperature to 1073 K. In the fixed bed reactor, the samples were heated at a rate of 7 K/min to 723, 773, 823 and 973 K. In both reactors, nitrogen was used as the carrier gas. Their investigation revealed considerable synergy in the fixed bed reactor especially when the proportion of coal in the blend was <33%. Park et al. [64] conducted co-pyrolysis of Sawdust and a subbituminous coal in a TGA under nitrogen at 1.2 bar and in a fixed bed reactor. In the TGA, the samples were heated at 15 K/min from ambient to 1173 K, in the fixed bed reactor (the heating rate and temperature was not provided). They observed that char yield was lower than would be obtained from additive behavior. They also noted that the difference was more pronounced around 873 K and disappeared as the temperature approached 1073 K and higher. These observations are in accordance with Zhang et al. [59]; they pyrolyzed blends of Dayan coal (lignite) and legume straw in a free fall reactor under atmospheric pressure at temperatures ranging from 773 K to 973 K with nitrogen as the carrier gas. Their investigation revealed that synergy was pronounced at lower temperatures (around 873 K) but reduced as temperature increased and was less pronounced around 993 K. Gao et al. [116] made a similar observation. Park et al. [64] also noted as Oney et al. [115] as well as Gao et al. [116], that synergy was more pronounced at higher biomass blending ratio (around 70%). Ulloa et al. [117] conducted co-pyrolysis of blends of subbituminous (Bitsch Coal), bituminous (Lemington coal) coals, with Radiata Pine sawdust in a TGA under nitrogen at different heating rates (10, 30 and 50 K/min) to a maximum temperature of 1473 K. Their experiment depicted interaction between biomass and the two coals at temperatures higher than 673 K and up to 1473 K. Their explanation of the interaction was that the synergy between devolatilization of biomass and lignin (which spans from 673 K to 1173 K) coincides with that of coal. However, they identified the role of biomass mineral matter as catalytic agent favoring decomposition of coal/biomass blends by decreasing the formation of char and favoring the generation of more volatile material. Another comprehensive study of coal and biomass co-pyrolysis and co-combustion is that of Kubacki [118]. This investigation involved coals of different ranks with biomass (straw) in different reactors: TGA, Pyroprobe-GC/MS, batch pyrolysis reactor, and a heated wire mesh pyrolysis reactor coupled to a GC/MS. He found that the type of biomass can lead to a small change of the rate of coal pyrolysis; slight synergistic effects were seen for the TGA study, where co-pyrolyzed coals in blends often had lower peak temperatures compared to coal alone, and higher volatile matter yields were produced. Analysis of the gases evolved was consistent with higher gas yields. This effect was present for certain biomass (e.g., oat straw) even after minerals were removed, and so this is not purely the result of catalytic ash components. Regarding pollutants emission, it was [118] noted that synergy effects contributing to reduction in pollutants during co-gasification of coal and biomass is not solely attributed to interactions of volatiles in the vapor phase, rather, there is a strong linkage between the pyrolysis and combustion steps and as such, the two steps needs to be studied together. We can extend this remark to devolatilization/gasification as well. From this observation, we can infer that synergy seems to be dictated by the pyrolysis/devolatilization step. Therefore, any synergy observed during the gasification/combustion step is only the consequence of what has already taken place during pyrolysis/devolatilization:
cracking of tar to produce more gases in case of gasification;
more reactive char for gasification and combustion.
3.1.3.1.3. Gasification and Combustion
As mentioned in the previous section, synergy during gasification or combustion cannot be isolated from the pyrolysis/devolatilization step. Synergy during gasification/combustion is usually expressed in terms of the high amount of gases (gasification), high reactivity of the char (gasification and combustion), reduced pollutants (gasification and combustion). These observations occur as a consequence of the interactions occurred during pyrolysis/devolatilization. Kubacki [118] mentioned it in his thesis and suggested that pyrolysis and combustion need to be studied together.
3.1.3.2. Synergistic Effects by Free Radical/Hydrogen Donation
3.1.3.2.1. General Assessment
The following explanation for synergy between coal and biomass/waste has been postulated [119]:
The weak covalent bonds in biomass and the higher content of oxygen compared to coal causes its early reaction, therefore, releasing volatiles which break down and release free radicals or undergo combustion reactions with oxygen present in the system. The free radicals from biomass react with coal and enhance its decomposition. The gases resulting from cracking of the heavy volatiles from biomass, as well as light volatiles molecules from biomass are rich in hydrogen which react with coal's free radicals as hydrogen donors, therefore, preventing their recombination reactions and reduce the amount of less reactive secondary chars. This explanation is in accordance with similar conclusions made by Squire et al. [120], who found that polymers which undergo extensive tar-forming thermolysis have limited supplies of hydrogen available to donate thus tend to produce more char than polymers with abundant hydrogen. Coal volatiles are also commonly of high calorific value, thus their combustion further increases the temperature in the region around the particle. The high temperature is then favorable for cracking high molecular weight gaseous compounds (tar) produced by biomass and coal; the higher temperature is also favorable for endothermic conversion of biomass and coal char into gases. The overall effect is a net increase in volatiles yield, less char, and a higher fuel conversion rate. Similar suggestion have been reported in the literature; Ahmaruzzaman and Sharma [121] observed synergy in the thermal decomposition of polypropylene, petroleum vacuum residue and biomass blend. They explained the synergistic interaction by the generation of reactive free radicals from polypropylene cracking, which react with the thermal decomposition products from petroleum vacuum residue and biomass.
Thus, hydrogen transfer between biomass and coal seems to be a plausible explanation of the synergistic effect between the two fuels. The hydrogen transfer mechanism between coal and solvents during coal liquefaction can provide some clue in the investigation of synergistic effects between coal and biomass during pyrolysis/devolatilization. In fact, co-liquefaction of coal and lignin [102], as well as coal and tire [105] have displayed significant synergies owing to the same mechanism. Similar mechanisms have been found during pyrolysis of solvent swollen coals [122–125] and coal impregnated with polyolefin [114]. Miura et al. [122] concluded that “to supply the radicals to coal effectively and to increase the total volatile matter, the formation rate of the radicals must match that of the radical fragments from coal at first. Furthermore, both radicals must be close enough spatially to react with each other, because the reaction rates among the radicals are much faster than the transport rate of the radicals.” One other important observation they made was the temperature dependence of the interaction between coal and solvent. The total volatiles yield of the solvent swollen coal increased with the increase of temperature and reached a maximum value. Further increase in temperature caused a decrease in the total volatiles yield of solvent swollen coal until it became equal to that of the raw coal. Thus, it can be inferred that synergy between coal and biomass is highly dependent on the process condition (temperature), the closeness (interaction) between coal and biomass particles, and the matching of the release of coal and biomass radical intermediates. The first two conditions imply that synergy may depend on the type of reactor being used. Therefore, it is expected to find more synergy in reactors operating at lower temperatures and favoring tight, intimate contact between fuel particles (fixed bed and fluidized bed) and little synergy in reactors that operate at higher temperatures and that segregate the fuel particles (entrained flow). However, these assumptions are somewhat contradicted by the results of Zhang et al. [59] and Lapuerta et al. [43]. The former [59] found synergy during co-pyrolysis of lignite and straw in a free fall reactor. The char collected was subjected to a CO2 reactivity test; results from the co-pyrolysis chars were about twice as high as those of coal char alone, and even higher than those of biomass alone while the latter [43] found synergies in a drop-tube reactor. Collot et al. [37] concluded in their pyrolysis experiments that neither intimate contact between fuel particles nor their relative segregation led to synergistic effects, making the case of fuels' interaction condition for synergy even more puzzling.
The temperature dependence of the interaction between the coal and biomass seems to be consistent with the results of some researchers [75,112–114], confirming that synergy is effective up to some optimum temperature and then disappears at higher temperatures.
A thorough understanding of free radical formation and evolution during pyrolysis seems to be the key to the possible interaction between coal and biomass. While extensive studies have been conducted on free radicals release during coal pyrolysis [115–124], such studies involving direct in situ observation of radicals released during biomass or biomass components (cellulose, hemicelluloses and lignin) pyrolysis are lacking. Understanding the mechanism of free radicals release during biomass/biomass components pyrolysis can pave the way to improvements in the modeling of coal and biomass blends during co-pyrolysis/co-gasification, and the synergistic effect between coal and biomass; therefore, providing an insight to the prediction of co-conversion of coal and biomass.
3.1.3.2.2. Hydrogen and Free Radicals Interaction
There is no doubt that hydrogen donation is a key factor in improving the yield of oil and increasing the conversion of the feedstock during liquefaction. Investigation of hydrogen transfer from a liquid carrier (tetralin) to coal was first described as a free radical reaction with thermal breakage of the coal molecules by Curran et al. [126]. They found that the degree of coal conversion was a function of the amount of hydrogen transferred and relatively independent of the solvent composition employed. Neavel [127] noted that the presence or lack thereof of a hydrogen donor vehicle had little effect on the thermal rupture of the coal fragment. This thermal cleavage results in the formation of highly reactive free radicals. In non-donor solvents, these free radicals react with surrounding molecules to form higher molecular weight compounds that become insoluble in subsequently employed extracting solvents. However, when a hydrogen donating solvent is employed, hydrogen is abstracted from the solvent and stabilizes pyrolysis-formed free radicals. Products are then relatively low molecular weight substances capable of being dissolved in most solvents. Progressively more donor hydrogen is abstracted for each increment of conversion as the reaction proceeds [127]. Vernon [128] conducted liquefaction of coal model compound dibenzyl and observed that when heated for 30 min at 723 K with an excess of a good donor solvent, tetralin, the conversion was 47% by mass and the only major product was toluene; very little benzene and ethyl benzene were produced. When the same experiment was carried out in the presence of high pressure hydrogen (× 11 MPa) the conversion increased to 58%. He then concluded that even molecular hydrogen could serve as a donor vehicle during liquefaction reactions. Li et al. [129] concluded that the primary factor inhibiting coal liquefaction is the consumption of hydrogen free radical (H●) from solvent or H2 and condensation of free radicals from coal pyrolysis after a period of reaction. So the essential approach for increasing oil yield and conversion of coal is to provide enough H● to stabilize the free radicals from coal pyrolysis. Unlike Neavel [127], McMillen [130] offered that the direct transfer of hydrogen atoms from solvent derived radicals to substituted positions in aromatic rings is substantial as preliminary steps in depolymerization of coal structures. Wei et al. [131] believe molecular hydrogen enhances the thermolysis, hydrogenation and hydrocracking of coal model compounds, as oppose to hydrogen donating solvents which inhibit these reactions. Although investigators opinions regarding liquefaction process may vary, there is a consensus that free radicals released during cleavage of the coal molecule needs to be stabilized by some hydrogen donor shuttle to suppress cross-linking reactions.
3.1.3.3. Enhanced Conversion of Solvent Swollen Coal
As the previous section described, the transfer of hydrogen to coal radicals is essential in achieving a higher yield of oil during liquefaction. It will be shown that improved pyrolysis is also achieved by effective transfer of hydrogen to free radicals. It has been suggested that the contact at the molecular level between the solvent and the fuel functional groups is essential in addition to the matching of the dehydrogenation rate of the solvent and the primary decomposition rate of coal. Miura et al. [132] ensured the contact at the molecular level was achieved by swelling the coal with solvent at 373–523 K under a pressurized atmosphere. The observed increase of both the total volatile matter and the tar yield was brought about by not only the effective hydrogen transfer from tetralin to the coal but by the effective utilization of small radicals such as OH● and H● from the coal for stabilizing the coal fragments. Mae et al. [133] used a similar technique by pyrolysing a pyridine vapor swollen coal. The suppression of cross-linking reactions significantly increased the volatile and tar yield. Awan et al. [134] conducted studies on tetralin treated coals. Similarly, he obtained a significant increase in tar and volatile yield and explained this observation in terms of the suppression of the cross-linking reactions of coal fragments due to the penetration of tetralin in to the micropores of coal, as well as the effective H● radicals transfer from tetralin to coal fragments during pyrolysis. Several experiments of this nature have been conducted to demonstrate the effectiveness of hydrogen transfer in the efficient release of gases during pyrolysis [123,132,135].
Non-catalytic synergistic effects observed in the release of volatiles and tar during coal biomass/waste co-conversion is evidenced by what precedes. The mechanism is a complex one, since in the case of biomass and coal, the release of hydrogen from biomass should match the free radical release from coal and the two fuels should be spatially close for the transfer to happen. The discrepancy observed in the scientific community about this topic is probably due to these constraints.
3.1.4. Effects of Co-Conversion of Coal and Biomass/Waste on Tar Release
Tar can be defined as the organics, produced under thermal or partial-oxidation regimes of any organic material, and generally assumed to be largely aromatic [136], condensing at ambient to lower temperatures. Tar is one of the most challenging constituents of the syngas. The tar content of the product gases from gasification of biomass is one of the major factors affecting the subsequent process stages. The type of feedstock used and gasification temperature are the main factors affecting tar yield during gasification [99]. Synergistic effects between coal and biomass particles are expected to lower the yield of tar compared to independent gasifying of these feedstocks. Kumabe et al. [33] obtained a slight decrease in tar yield by varying Japanese cedar (biomass) ratio of the feedstock from 0 to 100 when co-gasifying with Mulia (Indonesia) coal with air and steam in a downdraft fixed bed gasifier at 1173 K. Pinto et al. [137] observed a decrease in tar yield for a mixture of 80% coal (high-ash coal from Puertollano, Spain) and 20% pine wood waste compared to coal alone in a fluidized bed gasifier operating at atmospheric pressure and a temperatures of 1123–1173 K using a mixture of oxygen and steam as the gasifying agent. However, the addition of PE waste in the feed instead led to an increase in tar release; a probable reason for this behavior being the polymeric nature of PE which breaks into smaller fractions by thermal cracking, contributing to a greater amount of tars. A ternary blend of coal, PE and pine resulted in less tar release than when mixing coal and PE. They also found that dolomite was an efficient catalyst for reducing the tar yield during co-gasification of coal and biomass/waste mixtures containing PE. The presence of dolomite in the fluidized bed has the advantage of decreasing tar content and augmenting gas yield [35]. No evidence of synergetic effect however was found by Collot et al. [37] with Polish coal and forest residue mixture. Aznar et al. [32] and Andre et al. [35] also identified an increase in tar yield with an increase in biomass content of the feedstock, suggesting that synergetic effect during coal and biomass co-gasification might be highly dependent on biomass type as well as gasification conditions. One of the challenges emphasized by Mettler et al. [138] in their reviews of fundamental challenges of biomass pyrolysis for biofuels is the difficulty still encountered by researchers to identify and characterize nascent liquid intermediaries evolved during biomass pyrolysis. Therefore, the pertinence of these intermediates during co-conversion with coal is unknown. Further investigation is still needed to clarify the issue of synergy during coal and biomass/waste co-conversion.
3.2. Pressure
Pressure is among the conversion factors that depend on the reactor condition. Bahng et al. [139] provides a good update on available technologies for thermal conversion of biomass. The effects of pressure on biomass/waste and coal co-gasification have not been thoroughly investigated, which is the same as the effect of pressure on biomass gasification. However, many researchers have studied the effect of pressure on coal gasification alone. The pressure effect on gasification is depicted at different stages of the gasification process.
3.2.1. Effect of Pressure on Pyrolsys/Devolatilization
During the devolatilization phase of coal gasification, increasing the pressure decrease the total volatile yield. Lee et al. [140] conducted pyrolysis of a bituminous coal at pressures up to 0.39 MPa with a heating rate of 1000–10,000 K/s. They concluded that increasing pressure reduced the volatile release rate. The same observation is confirmed by Sun et al. [141] when pyrolysing two Chinese coals at 1173 K peak temperature and a heating rate of 20 K/min. They observed a significant decrease in total volatiles when increasing the operating pressure from 0.1 MPa to 1.3 MPa. However, their work emphasized that temperature affects the effect of pressure during coal devolatilization. They delineated two temperature ranges: one between 833 K and 953 K where the effect of pressure on devolatilization is insignificant, and one between 833 K and 953 K where increased pressure significantly reduces the devolatilization rate for Chinese bituminous and anthracite coals. Seebauer et al. [142] observed a similar trend on bituminous coal at pressure between 0.1 MPa and 4 MPa. Collot et al. [37] obtained the same trend on pyrolysis of Daw Mill coal at pressures ranging from 1 bar to 25 bar and temperatures of 1123–1273 K in a helium environment using a hot-rod reactor. It is important to note that the volatile decrease is sharp between 1 bar and 10 bar and remains almost constant for subsequent increase of pressure. However, when gasifying the same type of coal in a CO2 environment, keeping other parameters identical as in the pyrolysis, they instead observed an increase in total volatile at 1273 K and a decrease at 1123 K with increasing pressure. Gibbins et al. [143] obtained a decreased yield in a helium atmosphere while in a hydrogen atmosphere, an increase of total volatiles was observed during pyrolysis in a wire mesh reactor. Shan [144] cited by Wall et al. [145] computed the data for total volatile yield as a function of pressure from different authors and derived Figure 2.
The pressure effect during devolatization of biomass is similar to that of coal. Pindoria et al. [146] observed a decrease in total volatile yield as pressure increased from 0.25 MPa to 7 MPa during pyrolysis of Eucalyptus sawdust in a hot-rod reactor heated at 10 K/s to a peak temperature of 723 K. Collot et al. [37] also obtained a decrease in total volatiles yield on silver birch with pressure varying from 2 bar to 25 bar and temperatures of 1123–1273 K in a helium environment with a hot-rod reactor; it's important to note that the volatile decrease is sharp between 2 bar and 10 bar and remains almost constant for subsequent increases of pressure. When gasifying silver birch wood in a CO2 atmosphere, keeping all other parameters identical as in pyrolysis, the total volatile at 1273 K did not show any change with increased pressure while at 1123 K, a slight decrease was observed. All studies demonstrated that the effect of pressure on total volatile yield is less pronounced at higher pressures.
The decrease in total volatiles is mostly due to the decrease in tar as pressure is increased. Pressure suppresses the formation and release of tar and shifts the molecular weight of tar to a lighter fraction while promoting secondary reactions that decompose tar. Therefore, decreases in the tar output occur as well as increases in the total yield of light gases [147]. This observation is also well illustrated by Suuberg [148].
A high pressure inhibits the release of larger tar molecules that may easily evaporate at lower pressures, which is a plausible explanation why tar yield decreases and most of the tars shift toward lower molecular weights. Secondary reactions may explain why the effect of pressure on total volatiles is more pronounced at higher temperatures. The secondary repolymerization of the tar and the auto-hydrogenation phenomenon occurring at elevated pressure may also explain the increase of hydrocarbon gas yields, particularly methane.
Arendt and van Heek [145] obtained a significant decrease in tar yield during pyrolysis of five German hard coals in an inert gas (He) atmosphere as well as hydrogen atmosphere, with pressure varying from 0.1 MPa to 9 MPa and a temperature range of 1223–1273 K. They found that with increasing pressure, tar repolymerised and cracked more significantly, resulting in increased yields of char and hydrocarbon gases.
Similar results were obtained by Güell et al. [146] on pyrolysis of Pittsburgh No. 8 and Linby (UK) coals. Their experiment was conducted in a wired-mesh reactor with pressure ranging from atmospheric to 150 bar and peak temperatures of 973–1123 K under a helium and hydrogen atmosphere. Like total volatile yield, the effect of pressure on tar yield is less pronounced at higher pressures. Biomass reveals similar behavior; which is depicted by [142] on pyrolysis of eucalyptus wood waste.
3.2.2. Effect of Pressure on Char Characteristics
The effect of pressure on char formation and reactivity seems to be more complex. Reactivity of char is in fact highly dependent on the pyrolysis condition and pressure is one of the most important factors; it significantly affects the ash formation during char gasification as well. Under gasification conditions, one of the main parameters used for analyzing char reaction rate is its internal surface area. Higher pyrolysis pressures lead to lower surface area of char, and therefore, lower reactivity during gasification. Lee et al. [149] tested the reactivity of Illinois No. 6 bituminous coal chars obtained from different pyrolysis pressures and concluded that the char reactivity decreased slightly as the pyrolysis pressure increased. Sha et al. [150] also obtained a decrease in char's reactivity with increasing pyrolysis pressure in the range of atmospheric to 2 MPa on lignite, subbituminous and bituminous coals devolatilized at 1173 K in nitrogen. A previous study by Mühlen et al. [151] revealed similar observations; they investigated the reactivity of German bituminous coal chars pyrolysed under pressures of 0.2–4 MPa at 1173 K in helium. The reactivity of the chars was then evaluated at 4 MPa and 1173 K in hydrogen using a TGA.
Experiments with biomass show comparable results. Cetin et al. [152,153] tested the reactivity of chars from pyrolysis of Radiata pine, spotted gum (Eucalytus maculata) and sugar cane bagasse. They found that char particles produced at 20 bar reacted three times slower than the char generated at atmospheric pressure, thus the apparent gasification reactivity of biomass char decreases with increase pyrolysis pressure. Pindoria et al. [154] derived analogous results on the combustion reactivity of eucalyptus sawdust chars obtained at a peak temperature of 1123 K in helium, hydrogen and carbon dioxide atmosphere.
Several assumptions have been postulated to explain this observation. It's believed that high pressure may inhibit the transport effect during pyrolysis such as to reduce the release of volatile matters, causing large deposition on the pore surfaces, which creates secondary reactions, and therefore, deactivates active sites on the resulting char. Another assumption is that higher pyrolysis pressure enhances fluidity of coal, leading to a better mobility, alignment and ordering of carbon layers with reduction of micropores and subsequent loss of gasification reactivity. Concurrent influences of these two phenomena are also possible.
4. Co-Conversion and Emissions
4.1. Effect of Blending Coal and Biomass/Waste on NOx and N2O Precursors (HCN, NH3 and HNCO)
During combustion, nitrogen contained in solid fuels is converted to nitric oxides [NOx (NO and NO2)], gaseous nitrogen (N2) and nitrous oxide (N2O). The amount of fuel nitrogen retained in the char is generally insignificant. Life cycle assessment of biomass combustion in a heating furnace has revealed that 38.6% of the environmental impact is attributed to NOx, 36.5% to particulate matters, 2% to CO2 and 22.9% to all other pollutants [155]. It is known that NOx pollutants are formed in a two-step process: formation of the precursors (usually HCN and NH3) then combustion of the precursor. Therefore, a better understanding of the mechanism of formation of NOx precursors is essential to its in situ mitigation. Moreover, in oxygen-deprived environment (pyrolysis and gasification) NOx precursors are the main N2 compounds formed.
The origins, forms and modes of occurrence of nitrogen compounds (NH3 and HCN) during coal pyrolysis and gasification have been thoroughly investigated. Nitrogen in coal is mostly in pyrrolic and pyridinic forms [156–159]. Quaternary forms of nitrogen are also present, especially in low-rank coals. Amine functionalities are observed in low-rank coals [160], while they are present in biomass in diverse forms: proteins [161,162] (polymer of amino acids), alkaloids (heterocyclic nitrogen compounds), non-protein amino acids, nucleic acids, inorganic nitrogen and chlorophyll [163]. Chlorophyll, the green coloring matter in leaves and green stems, contains N in a magnesium-containing pyrrole derivative. Some biomass fuels are rich in nitrogen (sewage sludge, meat and bone mill, chicken liter, soya bean cake, slaughterhouse wastes, veterinary sanitation wastes, some agricultural wastes especially from legumes family [164]) impelling the need to investigate the release of NOx and N2O precursors during co-pyrolysis and co-gasification with coal. It is uncertain whether results from coal nitrogen investigations could be applied to biomass nitrogen, since this element is bound differently in the two types of fuel.
Reaction pathways involving nitrogen during pyrolysis/gasification is complex due to the overlapping of reaction stages, the influence of reaction conditions (residence time, temperature, pressure and gasification agent) and the differences between fuels. During pyrolysis, part of the fuel bound nitrogen (FBN) is released with the devolatilizing gases while the remainder is retained in the solid char to be released during subsequent gasification/combustion of the char [165,166]. The allocation of fuel-N to volatiles and char is dictated by temperature [160,167–170], particle size [168], fuel type [160], residence time [168,170], heating rate [171], and pressure [171]. Temperature, fuel type, and particle size, are the dominant factors in this allocation while heating rate and pressure only have a minor effect.
4.1.1. Fuel Nitrogen Behavior during Biomass Conversion
Given the predominance of proteins as the main nitrogen-containing compound in biomass, some researchers [172–177] have used amino acids as model compounds to study the behavior of nitrogen during thermal decomposition of biomass. Ren and Zhao [175] studied the pyrolysis and O2 and CO2 gasification behavior of phenylalanine, aspartic acid and glutamic acid in a TGA at temperatures ranging from 0 K to1073 K. The distinct structures of these amino acids cause them to behave slightly differently under argon pyrolysis as well as O2 and CO2 gasification. Ren et al. [176] found that the structure of the amino acids, the mineral matter content of biomass [178,179], the pyrolysis/gasification condition and the particle size [161,180] affects significantly the fate of nitrogen during pyrolysis of biomass. The thermal decomposition of amino acids and proteins proceeds mainly through dehydration with formation of cyclic amides, with diketopiperazine (DKP) being the common one [172–174,177].
Compared to coal, biomass has a higher content of oxygen, which enhances the direct release of NO and isocyanic acid (HNCO) as part of the devolatilizing gases during pyrolysis. The presence of CO2 suppresses the release of HNCO while enhancing formation of NH3 and NO [180] during gasification of amino acids, whereas HCN, HNCO and NO are promoted with the presence of O2.
Becidan et al. [181] found that HNCO can react with steam to give ammonia and carbon dioxide:
HNCO may also lose its hydrogen to give NCO, which then reacts with NO to give N2O and CO, it is therefore considered a precursor of N2O:
However, HCN is the main precursor of N2O [186,187] according to the following reactions:
The effect of particle size on FBN release is insignificant at lower temperatures (<673 K) but becomes considerable as the temperature increases. As the particle size increases, the yield of nitrogen species generally reduces [180]. This observation correlates with the fact that volatile release from a particle increases as the particle size decreases. With smaller particle sizes, volatile escape from the particle is fast enough to limit secondary reactions, resulting in high yield of volatile nitrogen.
Biomass also usually has a higher moisture content compared to most coals. Moisture can react with isocyanic acid to give ammonia and carbon monoxide, this reaction is known to be enhanced by the presence of CaO [188,189], which is present in most biomass:
Therefore, larger particles with longer diffusion path may favor reaction of HCN with H2O and enhance formation of NH3 as has been observed by some researchers [161,180]. It has also been observed that HNCO and NO release is higher for smaller particles. A probable explanation is that the temperature gradient within smaller particles is negligible during pyrolysis, therefore, the product of cracking of cyclic amides are quickly released as HNCO and NO [180]. Large particles retain more nitrogen in the solid phase than small particles because volatiles rebound to the solid phase during the transport out of the particles [190].
The effect of temperature on biomass fuel nitrogen has also been thoroughly investigated. Becidan et al. [181] observed an abrupt increase in N-conversion to HCN with temperature. They inferred that this observation is the result of tar and volatile cracking as well as secondary reactions. A similar inference has been made by Tian et al. [191]. NH3 also increase with temperature but reaches a maximum around 1023–1173 K as has been observed by many researchers [181,191]. It has also been observed that further increases in temperature cause a decrease in NH3 concentration in the gas [165,181,192,193] at temperatures higher than 1073–1173 K. This behavior could be explained by the conversion of ammonia into N2 and H2 as suggested by [165]:
It is further confirmed based on a thermodynamic point of view that NH3 destruction is endothermic, hence could be favored at higher temperatures [194] confirming the decrease of NH3 beyond a particular temperature.
Fuel oxygen usually forms OH radicals and other oxygen-containing radicals during the pyrolysis stage of solid fuel conversion. These radicals may promote secondary reactions including conversion of HCN to NH3. High heating rates suppress these secondary reactions, and hence favor formation of HCN [193]. This argument can be verified with slow heating rate pyrolysis where more NH3 is observed than HCN [194,195].
Higher heating rates appear to increase the HCN/NH3 ratio for both coal and biomass fuels [195–197]. However, the main product of most biomass thermal decomposition experiments under high heating rates is NH3, since biomass fuels contain significant amounts of quaternary and amine functionalities favoring NH3 formation [160]. At low heating rates NH3 is generally the dominant product, for both coals and biomass [198–200]. Minor amounts of isocyanic acid (HNCO) have been measured in fluidized bed pyrolysis [201] where the heating rate is usually low, while it has not been detected in entrained flow reactor experiments where the heating rate is usually high [199]. Contradicting results are however observed in literature where the dominant nitrogen specie during pyrolysis of biomass is HCN [202]. These authors (Yuan et al. [202]) investigated the fast pyrolysis of rice straw, chinar leaves, pine sawdust and soybean cake in a high frequency furnace. Volatile nitrogen of rice straw, chinar leaves and sawdust was dominated by HCN while that of soybean cake was dominated by NH3. They hypothesized that the presence or absence of lignin in those fuels could explain the difference observed. They suggested that the “microshells” formed by lignin around hemicellulose and cellulose in the cell wall of rice straw, chinar leaves and pine sawdust amplifies polymerization reactions between protein, cellulose and hemicellulose, resulting in formation of heterocyclic nitrogen which further decomposes into HCN. Whereas in soybean cake, the content of lignin and holocellulose is low, therefore, little heterocyclic nitrogen is formed, resulting in most of the fuel nitrogen being released as NH3.
The presence of Ca significantly reduces the emission of N2O probably by interfering with HCN chemistry [203]. In fact, it has been found that some alkali and alkaline earth metals (AAEM) can catalyze the conversion of fuel-N in the gas or solid phase. Therefore, Ca and K can promote formation of NH3 and N2 and suppress HCN [199,204]. These effects of Ca and K are in accordance with the reactions in Equations (3) and (2) listed earlier; since HCN is a precursor of N2O, and HCN is suppressed by Ca and K, then the presence of these elements reduces emission of N2O.
Pressure only has a minor effect on the chemistry of biomass nitrogen. Although many investigations have been conducted on the effect of pressure on coal nitrogen release, very few have been done for biomass. The equivalence ratio as well as the gasifying agent are also negligible factors in biomass nitrogen chemistry. However, it has been observed that CO2 as a gasifying agent reduces formation of HCN and suppresses formation of HNCO [180]. It was mentioned earlier that these two gases are precursors of N2O; it can be inferred that CO2 gasification of biomass could reduce formation of N2O. Jiachun et al. [165,194] found that within the range of 0.2–0.4, the equivalence ratio has a negligible effect on FBN conversion.
4.1.2. Fuel Nitrogen Behavior during Coal Conversion
Hydrogen cyanide is the primary product from devolatilization of coal nitrogen. Unlike ammonia, HCN has been observed in many experiments conducted at lower temperatures. The formation of HCN starts at 673 K and its profile can be divided into two parts: the release at an early stage as a result of the decomposition of the tar nitrogen and the elimination of CN-groups, and in a second high-temperature stage as a result of ring cleavage of hetero-aromatic compounds [205]. Pyrolysis of nitrogen model compounds (pyrrole and pyrimidine) of coal has yielded HCN as the major product [205–209]. NH3 is believed to be formed from secondary reactions of volatile nitrogen, HCN and HNCO. However, some literature [210] on fuel nitrogen conversion argues that NH3 formation also results from primary pyrolysis. According to Li and Tan [211–213], the main route for NH3 formation is the direct hydrogenation of the nitrogen in the pyrolysing coal/char particle. Their theory suggests that the cracking reactions taking place inside the coal/char particles generates free radicals, particularly hydrogen required for the hydrogenation of the nitrogen heteroaromatic ring systems. These free radicals would attack nitrogen-containing ring systems, thus initiating the ring activation. Migration of the free radical to the nitrogen -containing site would result in full hydrogenation of the nitrogen site, with the release of NH3 [211–213]. This theory has yet to be verified and confirmed experimentally. Studies of HCN and NH3 release during pyrolysis of petroleum coke seem to strengthen the idea that most of the NH3 is formed directly from pyrolysis of the char. Furimsky and Ohtsuka [204] observed that the yield of HCN was significantly lower than that of NH3 during pyrolysis of petroleum coke in helium and gasification with O2 and air. Yuan et al. [210] did not observe any HCN during rapid pyrolysis in argon. This revelation reinforces the hypothesis about the significant amount of NH3 being formed directly from pyrolysis of the char. Moreover, the yield of NH3 did not decrease at higher temperatures as observed in many coal pyrolysis experiments. The explanation given by the authors is that, given the high degree of graphitization of petroleum coke, most of the N atoms exist in the grid of the macromolecules, in the form “centre-N” (N inside an aromatic ring) and “valley-N” (N at the edge of an aromatic ring). The three bonds connected to the N atom have similar bond strength, therefore, may be cracked simultaneously during rapid pyrolysis when a high-energy impact can be provided transiently. This phenomenon could lead to formation of N● and NHi● radicals that can easily combine with H atoms to form NH3. Careful analysis of the pyrolysis conditions should be taken when observing a yield of NH3 higher than HCN. For instance Yuan et al. [210] referenced the work of Xu and Kumagai [214] where a higher yield of NH3 was observed, and made a similarity to their observation. But the latter conducted pyrolysis in a H2 atmosphere at high pressure, which is obviously favorable for a hydrogenation reaction and formation of NH3.
Nitrogen thermochemistry during coal conversion also depends strongly on the factors already listed for biomass: fuel type, temperature, particle size, heating rate, pressure and residence time [190]. The effects of these parameters on coal are similar to what is observed during biomass conversion. It is worth mentioning that the type of fuel brings a significant difference between low-rank and high-rank coals. Low-rank coals exhibit a nitrogen conversion behavior closer to that of biomass, due to the presence of quaternary nitrogen and amine functionalities that form NH3. It has been noted that devolatilization of bituminous coals produces mainly HCN, while more NH3 evolves from low-rank coals and biomass [199,200,215]. However, the source of NH3 even in low-rank coals is still debated, since model compound studies of the thermal decomposition of compounds representative of the nitrogen containing species in coals, such as pyridine and pyrrole, do not result in NH3 formation. Amine type functional groups would provide a possible source for NH3 formation, but N(1s) X-ray photoelectron spectra of coals do not reveal the presence of significant quantities of such groups, even for low-rank coals [216]. Intra-particle conversion of HCN to NH3 has already been mentioned as an important route for large particles and low-heating rates. However, it is suspected that fast reactions of HCN with free radicals on the char surface could be a source of NH3 [212,213] even in smaller particles and high heating rates. Another possible route for the formation of NH3 is the reaction of HNCO with steam. In fact, the release of isocyanic acid during coal pyrolysis is controversial. HNCO has been observed in the volatile from many bituminous and subbituminous coals pyrolysis over all ranges of temperatures. There is, however, little evidence for HNCO in the pyrolysis gas from low-rank coals [216]. A high amount of steam is released during low rank coals pyrolysis compared to high-rank coals; this steam may react with isocyanic acid to give NH3:
This mechanism may explain why HNCO is observed in high-rank coals pyrolysis, but not in low-rank coals.
4.1.3. Fuel Nitrogen Behavior in Co-Conversion of Coal Blended with Biomass/Waste
Analysis made in the last two sections show that nitrogen thermochemistry is complex and depends on many parameters. Whether synergy is observed in the decomposition of coal and biomass/waste blend should surely influence the release of fuel nitrogen. Yuan et al. [217,218] found that synergy between coal and biomass enhanced the release of fuel-N as volatile, reducing the amount of residual nitrogen left in the char during co-pyrolysis. It is well known that nitrogen species released in the gas phase increases as the volatile release increases. Since synergy boosts volatile yield, it is expected that it will also augment the yield of nitrogen species in the gas phase. At higher temperatures, N2 was the dominant nitrogen species observed by Yuan et al. [217,218]. Nitrogen partitioning between the two main precursors HCN and NH3 is puzzling and still not well understood. However, there is clear evidence that the discrepancies observed by various investigators are due to experimental conditions. An illustration of the experimental conditions on contradicting release of HCN and NH3 is presented by Bassilakis et al. [196]. A series of US coals pyrolyzed in TGA (slow heating rate) showed NH3 as the major nitrogen compound in the volatile, while the same coals pyrolyzed in an entrained flow reactor (fast heating rate) showed mainly HCN as the main N compound and displayed a negligible amount of NH3. A similar illustration can be made with the works of Di Nola et al. [219,220] where pyrolysis of the same fuels is conducted in slow and fast heating rates conditions. Volatile nitrogen in the slow heating rate experiment is dominated by NH3 while in the fast heating rate experiment, it is dominated by HCN.
It has been observed that more nitrogen is retained in the char during low temperature co-pyrolysis than higher temperatures. The reason evoked is that blending coal and biomass/waste reduces the packing density of the mixture. Since heat transfer is dominated by conduction at lower temperatures (as opposed to radiation at higher temperatures), the heating rate of the fuel is reduced and contributes to more nitrogen being retained in the char [217,218].
In thermal conversion of solid fuels, the release of fuel-bound nitrogen in the volatile is desirable over its' remaining in the char. With staged combustion, nitrogen compounds in the volatile are more susceptible to nitric oxides' control than char nitrogen. Their combustion occurs in two zones. In the first zone, the fuel is fired with less than stoichiometric amount of air, creating a fuel-rich condition near the primary flame. In the second zone, the rest of the combustion air is introduced to complete the fuel consumption. The deficiency of O2 in the first zone and the low temperature in the second zone both contribute to a reduction in NOx formation. Staged combustion techniques can reduce NOx emissions by 20%–50% [221]. Many experiments conducted on pyrolysis of blends of coal and biomass/waste have revealed a higher yield of nitrogen species in the volatile [217,218,220]. Therefore, co-conversion of coal mixed with biomass/waste is advantageous in terms of NOx emission reduction. It is however important to mention that investigation of the mechanisms of NOx and N2O precursors release during co-conversion (co-pyrolysis and co-gasification) are scarce. Konttinen et al. [222] have provided some groundwork based on experiments for estimating the total amount of reactive volatile-nitrogen species during fuel pyrolysis as well as reactions of char nitrogen in oxidizing and reducing environments; the authors claim their data are also adequate for fuel blending.
4.2. Effect of Blending Coal and Biomass/Waste on the Fate of Sulfur Species
Sulfur compounds in the synthesis gas stream are converted to SO2 and SO3 during combustion. These two gases are major components of acid rain. For many chemical applications (ammonia synthesis, methanol synthesis, Fischer Tropsch, gas shift, etc.) sulfur compounds will poison the catalyst and corrode equipment. Understanding the mechanism of sulfur migration and release during pyrolysis of single fuels as well as blends is highly relevant in designing efficient desulfurization systems.
Sulfur in plants is mainly assimilated by the roots in the form of inorganic sulfate. It is then transported to the leaves where it reduced into sulfite, then into sulfide. Sulfide will further combine with organic molecules to form cysteine or methionine which are amino acids from which proteins are formed [223], as well as glutathione [224]. Sulfur is therefore a key element in plant's protein synthesis. Annual crops such as herbaceous biomass are characterized by high growth rates, thus the high production rate of proteins which require a rich sulfur supply [225]. Sulfur in coal originates primarily from two sources: the original plant material from which the coal is formed and the deposition environment where the coal is formed. Much of the sulfur in low-sulfur coals derives from the sulfur content of the plant material making up the original peat. Sulfur contents greater than a few tenths of a percent have long been known to derive mostly from the depositional environment [226,227]. There is also post-deposition deposit in the fracture network of the coal.
The mechanisms of release of sulfur species during thermal conversion of solid fuels are influenced by many factors: the type of fuel (coal rank, type of biomass) and the forms of sulfur present in the fuel and the experimental conditions. Sulfur in biomass is found in both organic and inorganic forms. It is generally accepted that the stability of organically-bound sulfur in biomass is low, resulting in its decomposition at lower temperatures (673 K) during the devolatilization. Due to their high stabilities, the inorganic sulfates will not be released during the devolatilization stage. The plant growth conditions, species, and the overall sulfur availability during growth dictates the sulfur content as well as the ratio between organic and inorganic sulfur. Unlike chlorine (Cl) and K, sulfur removal by pretreatment processes like aqueous leaching will only take out the sulfur not organically bound to the structure of the plant [228–232]. Dayton et al. [232] observed a bimodal distribution of SO2 release during unleached wheat straw combustion, with a peak during the devolatilization phase and a second more intense peak during the char combustion stage. The less stable organic form of sulfur was released during devolatilization while the more stable inorganic metal sulfates contributed to the second peak as a result combustion at high temperatures and high oxygen concentration.
Some trace elements found in biomass fuels dictate the reaction mechanisms involving thermal decomposition of sulfur. Among these elements, K, Ca, Cl and silicon (Si) have the most significant effect. A better understanding of these elements' inclusion in the plant is necessary to gain a strong insight on how sulfur will behave. Annual biomass usually has a high amount of K and Cl [233,234]. K and chlorine are usually not metabolized by plants and remain mainly in ionic form K+ and Cl−. During drying of the plant material, these elements will precipitate and become prone to removal by aqueous leaching. It has been shown that up to 90% of biomass K and Cl can be removed by leaching [232,235]. K and Cl are mainly recovered in the aqueous fraction during chemical fractionation of annual biomass. Thus, these elements may occur as easily soluble salts or loosely bound to organic exchange sites [236]. Plants assimilate Si mainly in the form of monosilicic acid [Si(OH)4] [237,238]. It forms a silicate network structure in the cell walls and contributes to reinforcing the structural integrity and also provides a measure of protection against microorganisms. Si may also be present in the form of SiO2 particles and clay minerals due to soil contamination [228]. Once incorporated in the plant structure, Si is immobile and cannot be removed by leaching. Some amount of Ca is also present in plant. It is less mobile than K and is bound to organic counter-ion complexes [239]. Annual biofuels exhibit a low mobility of Ca as confirmed by their chemical fractionation, since the fraction of Ca recovered in the aqueous leachate is low [240].
Thermodynamic equilibrium of biomass pyrolysis (Figure 3) has shown that sulfur is released mainly in the form of hydrogen sulfide (H2S) and carbonyl sulfide (COS) [230]. It has also depicted a high affinity between potassium and sulfur (Figure 4), however, competition with Cl and Si may prevent sulfur from reacting with K (Figure 3).
Cl is released in two steps: during pyrolysis at temperatures as low as 773 K in the form of HCl and during char burnout at temperatures higher than 973 K in the form of KCl and NaCl [231,241,242].
Knudsen et al. [229,230] did not find any K silicates in the char fraction during pyrolysis of biomass at 973 K. For temperatures higher than 973 K, the amount of K retained by Si in the char was significant. At first look, this observation conflicts with the thermodynamics. A logical explanation to this observation is that at lower temperatures, the cell wall structure is still intact and no interaction between K and Si is taking place. Where at higher temperatures, the cell wall has collapsed and allows K and Si to mix and react. The affinity between Cl and K is also high (Figure 3); this will indirectly affect the release of sulfur. K will preferentially be released in the gas phase as KCl for Cl rich biomass fuels, therefore interfering the capture of sulfur in the solid char/ash.
Si in coal however, might behave differently as it can have a beneficial effect on the sulfur retention at high temperature. In this case, Si may physically enwrap sulfates of lower thermal stability and prevent the dissociation [228,230].
In coal, both organic and inorganic forms of sulfur can be characterized. Inorganic sulfur in coal comprises of pyritic or sulfide sulfur, and sulfate sulfur. Elemental sulfur has also been identified in coal, but its amount is generally negligible. The organic sulfur is bound to the hydrocarbon matrix. This form of sulfur occurs in covalently bonded C–S compounds (thiophenes, thioethers and bis-thioethers). Zhao et al. [243] conducted a series of pyrolysis on inorganically desulfurized coals at temperatures ranging from 673 K to 1173 K. They observed that most of the organic sulfur escaped as H2S. Calkins [244] conducted pyrolysis of raw and pyrite removed Pittsburgh #8 coal in a pyroprobe GC/MS at temperatures of 523–1273 K. He observed H2S, COS, CS2, CH3SH, SO2, and mostly thiophene and thiophenic compounds in the gaseous product. Investigations on the pyrolysis of thiophene and thiophenic compounds have concluded that H2S is the major compound released [245–248]. Therefore, it can be concluded that H2S is the main compound resulting from pyrolysis of organic sulfur.
Many investigations have concluded that coal mineral sulfur could interact with the organic matrix during coal pyrolysis [249–253]. Decomposition of pyrite can release hydrogen sulfide, which can interact with the organic matrix, causing its retention in the char. It has also been proven that H2S evolved from decomposition of pyrite can be partially retained in the solid phase (char/ash) as alkali sulfides [251–253]. Organically associated Ca can recapture the H2S to form sulfidic sulfur at temperatures above 873 K. Pyrolysis at temperatures of 873–1073 K, of ion-exchanged acid-washed coals with calcium acetate solution has revealed a significant increase of the sulfidic sulfur in the char [253]. In fluidized bed gasification/combustion, the role of Ca-based sorbent such as limestone or dolomite in capturing H2S/SO2 has been well documented [254–258]. Given the high content of AAEM in some biomass fuels, sulfur capture by these metals could be an added advantage of co-conversion of coal and biomass based fuels.
Lu et al. [259] found that blending municipal solid waste (MSW) with coal during co-combustion significantly reduced SO2 emission in the flue gas. They attributed this observation to the high concentration of the particles in the dilution zone of the reactor and the high alkali metals content in the fly and recirculating ashes, enhancing the ability for SO2 capture in the ash. Similar observations were observed by Kazagic et al. [260] in a pulverized fuel entrained flow reactor; they also observed a reduction in sulfur retention with increasing temperature, which was explained due to the reaction of SO2 with CaO being more efficient at lower to moderate temperatures. For biomasses with a higher content of AAEM and relatively low content of sulfur compared to coal, increasing the ratio of biomass in the blend means increasing the AAEM/sulfur ratio in the blend. The increase of the AAEM/sulfur ratio in the fuel blend obviously improves the efficiency of sulfur retention in the ash; but they are other factors interfering with sulfur capture in the solid phase such as the Cl and silicone content of the fuels.
5. Ash Characteristics in Co-Fired Systems
One major issue encountered in the thermal conversion of solid fuels is the formation and deposition of ash causing slagging and agglomeration in furnaces and fouling of boiler heat transfer surfaces. Inorganic matters and impurities in coal and biomass contribute to ash formation during thermal conversion; the characteristics of ash formed depend on many factors:
the feedstock category (coal, coke, biomass and waste, etc.);
variation in a feedstock category:
- ○
the type of coal dictates its ash formation and behavior [261];
- ○
the type of biomass and the part of the plant being used as fuel dictate the amount and characteristics of the ash, e.g., barks, straw, grasses and grain generally have a higher mineral matter content compared to wood [262];
- ○
MSW and sewage sludge usually have a higher content of heavy metals compared to coal and biomass;
Reaction conditions (temperature, pressure), atmosphere (oxidizing, reducing), flow dynamics [261].
Biomass mineral matter is usually rich in AAEM and low in alumino-silicates, whereas the content of alumino-silicates is higher in coal. Because of the low melting temperature of AAEM and high melting temperature of alumino-silicates metals, biomass ash tend to melt at lower temperatures than coal ash. It's therefore important to know the behavior of the ash mixture from coal and biomass in the design of co-gasification systems. The wide variation of mineral matter content and quality observed in biomasses as well as in coals causes the design of co-gasification systems to be regarded on a case-by-case basis. There is a need for models of predicting the formation and characteristics of coal-biomass ash mixtures under different gasification conditions.
There is a large amount of literature on the behavior of coal ash [88,263,264], biomass ash [263,265–268], and coal-biomass ash mixture under combustion [101,263,269–273] but very few under gasification conditions. Literatures of combustion conditions cannot be applied directly to gasification since the thermo chemistry of each of these conversion methods is different. Higman and van der Burgt [274] suggests that for gasification application for instance, the ash melting characteristics should be determined under reducing conditions. However, the data obtained in oxidizing conditions can serve as a starting point and provide some hints when investigating the behavior of coal-biomass ash during co-gasification.
Investigating the behavior of coal and biomass blend's ash during co-gasification requires starting with an understanding of the different forms of mineral matters present in coal and biomass fuels, the possible chemical reactions they can undergo under a given temperature, pressure and gaseous atmosphere.
5.1. Forms of Mineral Matter in Solid Fuels and Influence on Ash Behavior
Mineral matter in solid fuels is generally grouped into two factions:
The inherent inorganic material, which exists as part of the organic structure of the fuel, and is most commonly associated with the oxygen, sulfur and nitrogen-containing functional groups. These organic functional groups can provide suitable sites for the inorganic species to be associated chemically in the form of cations or chelates. Biomass materials tend to be relatively rich in oxygen-containing functional groups, and a significant fraction of the inorganic material in some of the lower ash biomass fuels is commonly in this form. It is also possible for inorganic species to be present in very fine particulate form within the organic structure of some of the fuels, and to behave essentially as an inherent component of the fuel.
The extraneous inorganic material, which has been added to the fuel through geological processes or during harvesting, handling and processing of the fuel. Biomass fuels, for instance, are commonly contaminated with soil and other materials, which have become mixed with the fuel during collection, handling and storage [275].
The inherent inorganic materials in biomass fuels can further be categorized as:
water soluble, that is in free ionic form;
organically associated;
precipitated as relatively pure compounds, in crystalline or amorphous forms [276].
Many laboratory characterization techniques for biomass ashes have been adapted from coal studies, e.g., the chemical fractionation technique; this technique is based on the solubility of the fuel subjected to successive leaching with increasing severity (water, ammonium acetate and hydrochloric acid) of the chemical reagents [277–280]. The four fractions obtained include:
water leachable fractions consisting mainly of alkali metal salts, sulfur and chlorine compounds;
buffer solution leachable fractions, usually associated to the organic matrix;
acid leachable fractions, containing mainly carbonates and sulfates;
the residue fraction is mainly silicates and other species not soluble in mineral acids.
The water and acetate-leachable elements are usually considered as those that are readily vaporized, forming the very finest aerosol fraction of the ash generated. The acid-soluble and residue fractions are not considered to be easily vaporized, therefore, they tend to be the major constituents of the coarser fractions of the ash [281].
Vaporization, condensation and coagulation/agglomeration are the main mechanisms involving biomass ash formation; alkali metals and chlorides contained in biomass vaporize to form HCl(g), KCl(g), K2SO4(g), Na2SO4(g), and NaCl(g) [273]. Their condensation constitutes the main contributor to the fine ash fraction. Strand et al. [282] identified two principal routes for condensation of these vapors into particles: a homogeneous nucleation where the vapor condenses into very fine droplets and agglomerates/accumulates into a particle and a heterogeneous condensation where the vapor on the surface of an existing particle entrains in the flue gas.
The inherent and extraneous inorganic materials of coal have different pathways for ash formation. Inherent inorganic materials are close to each other in the char particle; therefore, they become molten and easily coalesce and agglomerate during char burnout. Extraneous minerals mainly form ash through fragmentation, a mechanism that seldom occurs in inherent mineral matters [283]. Conclusions of Wu et al. [283] partially agree [281] that the coarse ash particles result from mechanisms such as coalescence and shedding while fine particles are from vaporization and condensation mechanisms.
Another important characterization technique is the slagging and fouling index, first developed to study the thermal behavior of coal ash, and modified for studying other solid fuels, including biomass. Many authors have developed indices for slagging and fouling; most of these indices are based on the fuel ash content and/or the ash chemical composition. They give an indication of how a fuel is inclined to forming deposits in the gasifier.
5.2. Prediction of Coal-Biomass Ash Behavior
Chemical equilibrium calculation has been used in many studies to predict the behavior of ash at given pressures and temperatures. As stated earlier, because of the lack of co-gasification studies on ash characteristics, almost all the literatures explored here are from co-combustion. We also mentioned earlier that behavior of ash from co-gasification of coal and biomass has to be investigated on a case-by-case basis, given the wide variation in coal fuels as well as biomass fuels. Most of the studies involving co-firing coal and straw have focused on the speciation of alkali metals and the reactions of alkali metal with chlorine to determine the properties of fly ash deposits [284–287]; although equilibrium models have some limitation (no consideration is given to kinetics, gas phase is assumed ideal, perfect mixing prevail that all elements are available for reaction). Significant amounts of K are released in the gas phase as KCl and KOH [284] as well as K2SO4 [284,288–290] have studied reactions involving the reactions of these species with other species present in the gas phase. Zheng et al. [273] observed that the K content in the fly ash is almost the same as that in the fuel ash during combustion and co-combustion involving straw. They concluded that the total K in the fly ash can be predicted when the ash composition of the fuel and straw share are known. Na is likely to be released as NaCl, NaOH, NaSO4 and Na [284] and react with the gas phase compounds. These observations corroborate with the fuel characterization based on the reactive and non-reactive ash-forming matter. This prediction method segregates the ash-forming matter in the biomass fuels into a “reactive” and “nonreactive” fraction. The non-reactive fraction, for instance, silica based mineral matter, is assumed to remain practically unchanged during the thermal conversion process and act as basically inert fly ash. The reactive fraction, typically alkali compounds of various kinds, will be easily converted to new compounds in combustion, it will interact with the gaseous compounds in the flue gases, typically with sulfur and Cl, and with the reactive parts of the other fuels fired in the same furnace [291]. It is interesting to notice that the “reactive” fraction is made of the main compounds of the water and acetate-leachable fraction in chemical fractionation and that the “nonreactive” fraction is essentially the acid soluble and residue. Addition of the non-reactive components to the mixture has been suggested as a way to mitigate deposition during thermochemical conversion of solid fuels. Increasing the amount of Si does decrease the amount of volatile K, but for nearly complete removal of volatile K, both Si and Al have to be added [273]. It can therefore be inferred that using a coal with a high content of Si and Al to co-fire with straw is advantageous in reducing deposits.
5.3. Utilization of Ash from Coal-Biomass Co-Fired Systems
Unlike combustion (and co-combustion) fly ash, gasification (and co-gasification) ash contains a high concentration of unburned carbon and harmful soluble compounds, making it unsuitable for direct use in most combustion ash utilization methods [292,293]. Coal and biomass combustion (and co-combustion) ashes have been used successfully in many applications including fertilizer, row material for fertilizer or soil amendment [294–296], building material or as component in the manufacture of building material [297,298]. The high content of unburned carbon in the gasification ash makes it possible to be reused as fuel. Pels et al. [293] suggests that utilization as a fuel should be envisioned only if the carbon content is larger than 35 wt% or the caloric value is higher than 15 MJ/kg. Exploring possible valorization options of co-gasification ash is capital to increasing the attractiveness of this process. Specific articles discussing usability of co-gasification ashes, highlighting the difference with combustion, gasification and co-combustion ashes are scarce. However, understanding the various applications of combustion, gasification, and co-combustion ashes should provide enough guideline for the usability of co-gasification ash.
5.3.1. Fertilizer and Fertilization
Extraction of biomass from farmland or forest removes nutrients and acid-buffering capacity from the soil. N, phosphorus (P) and K are the main elements present in a complete fertilizer. Nitrogen in the gasification feedstock is generally released as ammonia, amines, and urea in the volatiles during the gasification process and only a negligible amount remains in the ash. Since ash from thermal processes are nearly deprived of nitrogen while P is present in a form which is poorly soluble at soil conditions, utilization of combustion and gasification ashes in all-purpose fertilizer is limited [293]. However, biomass ash can be useful as a soil amendment agent, partially replacing some nutrients extracted and restituting the acid-buffering capacity of the soil [299]. Although ash from pure agricultural residues (farm litter, agricultural wastes) are low in toxic heavy metals, coal and some biomass (MSW, sewage sludge and demolition) ashes usually have significant amounts of heavy metals and, when applied to the soil, can be phytotoxic or increase the concentration of these elements in the plant material to become toxic to animals under continuous long term grazing conditions and transferred to man through the food chain [300].
5.3.2. Cement Manufacture and Civil Engineering Application
Coal fired fly ash has pozzolanic properties; it can react with lime to form a hardened mass. The particle shape, fineness, chemistry and pozzolanic reactivity of fly ash provide a low permeability when used in concrete, improving the long term strength gain, durability and reduced shrinkage [301]. The use of fly ash in concrete is done either as a blend to the cement or by adding it to the mixture when concrete is being prepared. The addition of fly ash to concrete impacts all the major properties of the concrete. As part of the composite concrete mass, fly ash acts both as a fine aggregate and as a cementitious component. It influences the rheological properties of the fresh concrete and the strength, finish, porosity, and durability of the hardened mass as well as the cost and energy consumed in manufacturing the final product [302].
Bottom ash is the easiest for utilization as building material. Bottom ash from fluidized bed combustion or gasification consist for a large part of sand and may replace other kinds of sand in road base and sub-base aggregate, use as structural fill material, and as fine aggregate in asphalt paving. Bottom ashes from grate stokers and entrained-flow gasification can be made into granulate and find its way to road constructions and concrete. Other applications of bottom ash include snow and ice control, as aggregate in lightweight concrete masonry units, and raw feed material for the production of Portland cement [302].
6. Summary and Conclusions
In the preceding, we have explored the current status of coal and biomass co-conversion in thermochemical systems. It has been noted that this process allows for compensating the shortcomings of one fuel by another, since both fuels seems to be complementary in their drawbacks and advantages.
GHG mitigation is one of the most attractive benefits of co-converting coal and biomass. Although lifecycle assessments of co-gasification processes are scarce, lifecycle assessment on co-firing coal and biomass have shown that CO2 emission declines proportionally to the amount of coal offset by biomass, considering biomass as a carbon-neutral source produced sustainably [14]. With further reduction in CH4 and N2O emission, the overall outcome of co-firing is a percentage reduction of global warming potential (CO2 equivalent) higher than the percentage of biomass in the blend.
Entrained flow gasification produces a relatively clean gas compared to fixed bed and fluidized bed gasification. However, size reduction of biomass to the order of hundreds of μm, required in entrained flow systems, is difficult to achieve, requiring preprocessing steps like torrefaction or fast pyrolysis.
Catalytic gasification of coal by AAEM is well documented in the literature [84–87] and given the high content of biomass in these elements; co-gasification of coal and biomass could exhibit some catalytic enhancement depending on the type of coal and biomass.
There is no common agreement regarding the synergistic effect of coal and biomass in the literature. Those authors favoring interaction between coal and biomass have suggested that free radicals formation and hydrogen transfer from biomass to coal could be the key explanation behind this phenomenon. Temperature dependence of the synergy has also been mentioned, with prominent synergy at lower temperature up to an optimum above which no synergy is noticeable [95,116,303,304].
Thermodynamic equilibrium of co-gasification shows that the amount of gases increases as the percentage of biomass in the blend increases, increasing the gross calorific value and coal gas efficiency of the gas. Increasing the reaction temperature favors endothermic reactions; formation of CO and H2 are thus enhanced while the fractions of CO2 and H2O in the gas are reduced. Meanwhile, the cold gas efficiency and the gross calorific value increase up to an optimum temperature above which they begin to decrease. Increasing the pressure has an opposite effect to that of temperature with an increase in CO2, H2O, and CH4 while CO and H2 are reduced. This leads to a decrease of the cold gas efficiency and gross calorific value; however at higher temperatures, the effect of pressure is eclipsed by that of temperature.
Biomass mineral matter is usually rich in AAEM and low in alumino-silicates, whereas the content of alumino-silicates is higher in coal. Because of the low melting temperature of AAEM and high melting temperature of alumino-silicates metals, biomass ash tend to melt at lower temperatures than coal ash. It's therefore important to know the behavior of the ash mixture from coal and biomass in the design of thermal co-conversion systems. The wide variation of mineral matter content and quality observed in biomasses as well as in coals causes the design of co-conversion systems to be regarded at a case-by-case basis. There is a need for models of predicting the formation and characteristics of coal-biomass ash mixtures under different gasification conditions.
The formation, behavior, and use of ash under combustion conditions for coal [88,263,264], biomass [263,265–268] and coal-biomass (co-firing) [101,263,269–273] is abundant whereas very few are available for coal-biomass co-gasification. The thermochemistry in the two environments is not the same, e.g., gasification ashes have a high content of unburned carbon preventing its direct utilization in some applications where combustion ashes are used.
Among all renewable energies, biomass is the only source of concentrated carbon and as such, is the only renewable energy source for replacement of transportation fuels, and therefore, will certainly play an important role in meeting the future energy need of the world. The share of biofuel in the transportation fuel market is likely to grow rapidly in the next decade due to numerous benefits, including sustainability, reduction of GHG emissions, regional development, reduction of rural poverty and energy security. Coal gasification is a proven technology that has been operated successfully at commercial scale for heat, power generation and synthetic fuels; some commercial applications of biomass gasification for heat and power are also available. However, biomass gasification and subsequent production of synthetic fuels (diesel fuel, methanol and dimethylether, etc.) appears to be more complex (tar cracking, syngas cleaning, etc.) and more expensive to compete with fossil-based liquid fuels. As a result, economies of scale are essential to ensure the economic viability of biomass gasification plants [305]. It has been calculated that only large biomass-to-liquid (BtL) plants with synfuel production capacities of at least 1,000,000 t a−1 (input at least 6,000,000 t a−1 dry lignocellulose) would profit from meaningful economies of scale [306]. Biomass, like most existing renewable energy resources is both diffuse, that is, thinly scattered; and sporadic, that is, variable over time. Regular and constant supply of such a huge amount of biomass, coupled with processing and handling difficulties makes a dedicated biomass power and heat plant or biomass to liquid plant nearly infeasible; but this limitation is lifted if coal and biomass are both used as feedstock. It is well known that most renewable energy resources lack the ability to dispatch the power to meet demand at any given time and need to be backed up by storage facilities or facilities that use fossil fuel resources. Although storage ability of biomass energy systems makes it available almost 80% to 100% of the time [307], the association of coal-biomass gives a complete palliation to the diffuse and sporadic issue of dedicated biomass thermochemical energy systems.
Acknowledgments
This technical work was performed with partial support National Energy Technology Laboratory under the RES contract DE-FE0004000.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Letcher, T.M. Future Energy: Improved, Sustainable and Clean Options for Our Planet, 1st ed.; Elsevier Ltd.: Oxford, UK, 2008. [Google Scholar]
- U.S. Energy Information Administration (EIA). Annual Energy Outlook 2013 with Projections to 2040; EIA: Washington, DC, USA, 2013. [Google Scholar]
- International Energy Agency (IEA). 21st Century Coal Advanced Technology and Global Energy Solution; IEA Coal Industry Advisory Board (CIAB): Paris, France, 2013. [Google Scholar]
- Balat, M. Influence of coal as an energy source on environmental pollution. Energy Sources Part A Recovery Util. Environ. Eff. 2007, 29, 581–589. [Google Scholar]
- International Energy Agency (IEA). World Energy Outlook 2013; IEA: Paris, France, 2013. [Google Scholar]
- Demirbas, A.; Sahin-Demirbas, A. Global energy sources, energy usage, and future developments. Energy Sources 2004, 26, 191–204. [Google Scholar]
- Balat, M.; Balat, H.; Acici, N. Environmental issues relating to greenhouse carbon dioxide emissions in the world. Energy Explor. Exploit. 2003, 21, 457–473. [Google Scholar]
- Schuck, S. Biomass as an energy source. Int. J. Environ. Stud. 2006, 63, 823–835. [Google Scholar]
- Hoogwijk, M.; Faaij, A.; van den Broek, R.; Berndes, G.; Gielen, D.; Turkenburg, W. Exploration of the ranges of the global potential of biomass for energy. Biomass Bioenergy 2003, 25, 119–133. [Google Scholar]
- Slade, R.; Saunders, R.; Gross, R.; Bauen, A. Energy from Biomass: The Size of the Global Resource; Imperial College Centre for Energy Policy and Technology and UK Energy Research Centre: London, UK, 2011. [Google Scholar]
- International Energy Agency (IEA). Potential Contributions of Bio-Energy to the World's Future Energy Demand; IEA: Paris, France, 2007. [Google Scholar]
- Sven, B.; Martina, J. Carbon dioxide capture and storage—Liability for non-permanence under the UNFCCC. Int. Environ. Agreem. Polit. Law Econ. 2006, 6, 173–186. [Google Scholar]
- Stangeland, A. A model for the CO2 capture potential. Int. J. Greenh. Gas Control 2007, 1, 418–429. [Google Scholar]
- Caputo, J.; Hacker, J. Biomass Cofiring: A Transition to A Low-Carbon Future; Environmental and Energy Study Institute: Washington, DC, USA, 2009. [Google Scholar]
- Gogebakan, Z.; Gogebakan, Y.; Selcuk, N. Co-firing of olive residue with lignite in bubbling FBC. Combust. Sci. Technol. 2008, 180, 854–868. [Google Scholar]
- Forster, P.; Ramaswamy, V.; Artaxo, P.; Berntsen, T.; Betts, R.; Fahey, D.W.; Haywood, J.; Lean, J.; Lowe, D.C.; Myhre, G.; et al. Changes in Atmospheric Constituents and in Radiative Forcing. In Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K.B., Tignor, M., Miller, H.L., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2007. [Google Scholar]
- Mann, M.K.; Spath, P.L. Life cycle assessment of biomass cofiring in a coal-fired power plant. Clean Prod. Process. 2001, 3, 81–91. [Google Scholar]
- Souza-Santos, M.L.D. Solid Fuel Combustion and Gasification: Modeling, Simulation and Equipment Operation; Marcel Dekker, Inc.: New York, NY, USA, 2004. [Google Scholar]
- Knoef, H.A.M. Handbook Biomass Gasification; Biomass Technology Group BTG: Meppel, The Netherlands, 2005. [Google Scholar]
- Henrich, E.; Weirich, F. Pressurized entrained flow gasifiers for biomass. Environ. Eng. Sci. 2004, 21, 53–64. [Google Scholar]
- Vakkilainen, E.; Kuparinen, K.; Heinimö, J. Large Industrial Users of Energy Biomass; IEA Bioenergy Task 40; Lappeenranta University of Technology: Lappeenranta, Finland, 2013. [Google Scholar]
- Ciolkosz, D.; Wallace, R. A review of torrefaction for bioenergy feedstock production. Biofuels Bioprod. Biorefining 2011, 5, 317–329. [Google Scholar]
- Bergman, P.C.A.; Boersma, A.R.; Kiel, J.H.A.; Wilberink, R.W.A.; Bodenstaff, H.; Heere, P.G.T. Torrefaction for Entrained Flow Gasification of Biomass; ECN and TU/e, Report ECN, Petten (NL) ECN-C-05-067; Energy Research Center of the Netherlands (ECN): Petten, The Netherlands, 2003. [Google Scholar]
- Tumuluru, J.S.; Hess, J.R.; Boardman, R.D.; Wright, C.T.; Westover, T.L. Formulation, pretreatment, and densification options to improve biomass specifications for co-firing high percentages with coal. Ind. Biotechnol. 2012, 8, 113–132. [Google Scholar]
- Van der Drift, A.; Boerrigter, H.; Coda, B.; Cieplik, M.K.; Hemmes, K. Entrained Flow Gasification of Biomass: Ash Behaviour, Feeding Issues, and System Analyses; Energy Research Center of the Netherlands (ECN): Petten, The Netherlands, 2004. [Google Scholar]
- Zulfiqar, M.; Moghtaderi, B.; Wall, T.F. Flow properties of biomass and coal blends. Fuel Process. Technol. 2006, 87, 281–288. [Google Scholar]
- Neves, D.; Thunman, H.; Matos, A.; Tarelho, L.; Gómez-Barea, A. Characterization and prediction of biomass pyrolysis products. Prog. Energy Combust. Sci. 2011, 37, 611–630. [Google Scholar]
- Henrich, E.; Dahmen, N.; Raffelt, K.; Stahl, R.; Weirich, F. The Karlsruhe “Bioliq” Process for Biomass Gasification. Proceedings of the 2nd European Summer School on Renewable Motor Fuels, Warsaw, Poland, 29–31 August 2007.
- Dobel, G.; Urbanovich, I.; Volpert, A.; Kampars, V.; Samulis, E. Fast pyrolysis–effect of wood drying on the yield and properties of bio-oil. BioResources 2007, 2, 698–706. [Google Scholar]
- Franco, C.; Pinto, F.; André, R.N.; Tavares, C.; Dias, M.; Gulyurtlu, I.; Cabrita, I. Experience Using INETI Experimental Facilities in Co-Gasification of Coal and Wastes. Proceedings of the Workshop Co-Processing of Different Waste Materials with Coal for Energy, Lisbon, Portugal, 23 November 2001; pp. 238–247.
- Pan, Y.G.; Velo, E.; Roca, X.; Manya, J.J.; Puigjaner, L. Fluidized-bed co-gasification of residual biomass/poor coal blends for fuel gas production. Fuel 2000, 79, 1317–1326. [Google Scholar]
- Aznar, M.P.; Caballero, M.A.; Sancho, J.A.; Frances, E. Plastic waste elimination by co-gasification with coal and biomass in fluidized bed with air in pilot plant. Fuel Process. Technol. 2006, 87, 409–420. [Google Scholar]
- Kumabe, K.; Hanaoka, T.; Fujimoto, S.; Minowa, T.; Sakanishi, K. Co-gasification of woody biomass and coal with air and steam. Fuel 2007, 86, 684–689. [Google Scholar]
- McIlveen-Wright, D.R.; Pinto, F.; Armesto, L.; Caballero, M.A.; Aznar, M.P.; Cabanillas, A.; Huang, Y.; Franco, C.; Gulyurtlu, I.; McMullan, J.T. A comparison of circulating fluidised bed combustion and gasification power plant technologies for processing mixtures of coal, biomass and plastic waste. Fuel Process. Technol. 2006, 87, 793–801. [Google Scholar]
- Andre, R.N.; Pinto, F.; Franco, C.; Dias, M.; Gulyurtlu, I.; Matos, M.A.A.; Cabrita, I. Fluidised bed co-gasification of coal and olive oil industry wastes. Fuel 2005, 84, 1635–1644. [Google Scholar]
- Cascarosa, E.; Gasco, L.; García, G.; Gea, G.; Arauzo, J. Meat and bone meal and coal co-gasification: Environmental advantages. Resour. Conserv. Recycl. 2012, 59, 32–37. [Google Scholar]
- Collot, A.G.; Zhuo, Y.; Dugwell, D.R.; Kandiyoti, R. Co-pyrolysis and co-gasification of coal and biomass in bench-scale fixed-bed and fluidised bed reactors. Fuel 1999, 78, 667–679. [Google Scholar]
- De Jong, W.B.; Andries, J.; Hein, K.R.G. Coal/biomass co-gasification in a pressurised fluidised bed reactor. Renew. Energy 1999, 16, 1110–1113. [Google Scholar]
- McLendon, T.R.; Lui, A.P.; Pineault, R.L.; Beer, S.K.; Richardson, S.W. High-pressure co-gasification of coal and biomass in a fluidized bed. Biomass Bioenergy 2004, 26, 377–388. [Google Scholar]
- Pinto, F.; Franco, C.; Andre, R.N.; Tavares, C.; Dias, M.; Gulyurtlu, I.; Cabrita, I. Effect of experimental conditions on co-gasitication of coal, biomass and plastics wastes with air/steam mixtures in a fluidized bed system. Fuel 2003, 82, 1967–1976. [Google Scholar]
- Pinto, F.; Franco, C.; Lopes, H.; Andre, R.; Gulyurtlu, I.; Cabrita, I. Effect of used edible oils in coal fluidised bed gasification. Fuel 2005, 84, 2236–2247. [Google Scholar]
- Priyadarsan, S.; Annamalai, K.; Sweeten, J.M.; Holtzapple, M.T.; Mukhtar, S. Co-gasification of blended coal with feedlot and chicken litter biomass. Proc. Combust. Inst. 2005, 30, 2973–2980. [Google Scholar]
- Lapuerta, M.; Hernández, J.J.; Pazo, A.; López, J. Gasification and co-gasification of biomass wastes: Effect of the biomass origin and the gasifier operating conditions. Fuel Process. Technol. 2008, 89, 828–837. [Google Scholar]
- Biagini, E.; Lippi, F.; Petarca, L.; Tognotti, L. Devolatilization rate of biomasses and coal-biomass blends: An experimental investigation. Fuel 2002, 81, 1041–1050. [Google Scholar]
- Meesri, C.; Moghtaderi, B. Lack of synergetic effects in the pyrolytic characteristics of woody biomass/coal blends under low and high heating rate regimes. Biomass Bioenergy 2002, 23, 55–66. [Google Scholar]
- Moghtaderi, B.; Meesri, C.; Wall, T.F. Pyrolytic characteristics of blended coal and woody biomass. Fuel 2004, 83, 745–750. [Google Scholar]
- Vamvuka, D.; Pasadakis, N.; Kastanaki, E.; Grammelis, P.; Kakaras, E. Kinetic modeling of coal/agricultural by-product blends. Energy Fuels 2003, 17, 549–558. [Google Scholar]
- Vuthaluru, H.B. Thermal behaviour of coal/biomass blends during co-pyrolysis. Fuel Process. Technol. 2004, 85, 141–155. [Google Scholar]
- Lu, K.-M.; Lee, W.-J.; Chen, W.-H.; Lin, T.-C. Thermogravimetric analysis and kinetics of co-pyrolysis of raw/torrefied wood and coal blends. Appl. Energy 2013, 105, 57–65. [Google Scholar]
- Kirtania, K.; Bhattacharya, S. Pyrolysis kinetics and reactivity of algae–coal blends. Biomass Bioenergy 2013, 55, 291–298. [Google Scholar]
- Li, S.; Chen, X.; Wang, L.; Liu, A.; Yu, G. Co-pyrolysis behaviors of saw dust and Shenfu coal in drop tube furnace and fixed bed reactor. Bioresour. Technol. 2013, 148, 24–29. [Google Scholar]
- Sadhukhan, A.K.; Gupta, P.; Goyal, T.; Saha, R.K. Modelling of pyrolysis of coal-biomass blends using thermogravimetric analysis. Bioresour. Technol. 2008, 99, 8022–8026. [Google Scholar]
- Pan, Y.G.; Velo, E.; Puigjaner, L. Pyrolysis of blends of biomass with poor coals. Fuel 1996, 75, 412–418. [Google Scholar]
- Aboyade, A.O.; Görgens, J.F.; Carrier, M.; Meyer, E.L.; Knoetze, J.H. Thermogravimetric study of the pyrolysis characteristics and kinetics of coal blends with corn and sugarcane residues. Fuel Process. Technol. 2013, 106, 310–320. [Google Scholar]
- Li, S.; Chen, X.; Liu, A.; Wang, L.; Yu, G. Study on co-pyrolysis characteristics of rice straw and Shenfu bituminous coal blends in a fixed bed reactor. Bioresour. Technol. 2014, 155, 252–257. [Google Scholar]
- Gil, M.V.; Casal, D.; Pevida, C.; Pis, J.J.; Rubiera, F. Thermal behaviour and kinetics of coal/biomass blends during co-combustion. Bioresour. Technol. 2010, 101, 5601–5608. [Google Scholar]
- Haykiri-Acma, H.; Yaman, S. Synergy in devolatilization characteristics of lignite and hazelnut shell during co-pyrolysis. Fuel 2007, 86, 373–380. [Google Scholar]
- Suelves, I.; Lázaro, M.J.; Moliner, R. Synergetic effects in the co-pyrolysis of samca coal and a model aliphatic compound studied by analytical pyrolysis. J. Anal. Appl. Pyrolysis 2002, 65, 197–206. [Google Scholar]
- Zhang, L.; Xu, S.; Zhao, W.; Liu, S. Co-pyrolysis of biomass and coal in a free fall reactor. Fuel 2007, 86, 353–359. [Google Scholar]
- Haykiri-Acma, H.; Yaman, S. Combinations of synergistic interactions and additive behavior during the co-oxidation of chars from lignite and biomass. Fuel Process. Technol. 2008, 89, 176–182. [Google Scholar]
- Sonobe, T.; Worasuwannarak, N.; Pipatmanomai, S. Synergies in co-pyrolysis of thai lignite and corncob. Fuel Process. Technol. 2008, 89, 1371–1378. [Google Scholar]
- Edreis, E.M.A.; Luo, G.; Li, A.; Xu, C.; Yao, H. Synergistic effects and kinetics thermal behaviour of petroleum coke/biomass blends during H2O co-gasification. Energy Convers. Manag. 2014, 79, 355–366. [Google Scholar]
- Krerkkaiwan, S.; Fushimi, C.; Tsutsumi, A.; Kuchonthara, P. Synergetic effect during co-pyrolysis/gasification of biomass and sub-bituminous coal. Fuel Process. Technol. 2013, 115, 11–18. [Google Scholar]
- Park, D.K.; Kim, S.D.; Lee, S.H.; Lee, J.G. Co-pyrolysis characteristics of sawdust and coal blend in TGA and a fixed bed reactor. Bioresour. Technol. 2010, 101, 6151–6156. [Google Scholar]
- Wei, L.G.; Zhang, L.; Xu, S.P. Effects of feedstock on co-pyrolysis of biomass and coal in a free-fall reactor. J. Fuel Chem. Technol. 2011, 39, 728–734. [Google Scholar]
- Blesa, M.J.; Miranda, J.L.; Moliner, R.; Izquierdo, M.T.; Palacios, J.M. Low-temperature co-pyrolysis of a low-rank coal and biomass to prepare smokeless fuel briquettes. J. Anal. Appl. Pyrolysis. 2003, 70, 665–677. [Google Scholar]
- Sjostrom, K.; Chen, G.; Yu, Q.; Brage, C.; Rosen, C. Promoted reactivity of char in co-gasification of biomass and coal: Synergies in the thermochemical process. Fuel 1999, 78, 1189–1194. [Google Scholar]
- Cordero, T.; Rodriguez-Mirasol, J.; Pastrana, J.; Rodriguez, J.J. Improved solid fuels from co-pyrolysis of a high-sulphur content coal and different lignocellulosic wastes. Fuel 2004, 83, 1585–1590. [Google Scholar]
- Haykiri-Acma, H.; Yaman, S. Interaction between biomass and different rank coals during co-pyrolysis. Renew. Energy 2010, 35, 288–292. [Google Scholar]
- Yuan, S.; Dai, Z.-H.; Zhou, Z.-J.; Chen, X.-L.; Yu, G.-S.; Wang, F.-C. Rapid co-pyrolysis of rice straw and a bituminous coal in a high-frequency furnace and gasification of the residual char. Bioresour. Technol. 2012, 109, 188–197. [Google Scholar]
- Xu, C.; Hu, S.; Xiang, J.; Zhang, L.; Sun, L.; Shuai, C.; Chen, Q.; He, L.; Edreis, E.M.A. Interaction and kinetic analysis for coal and biomass co-gasification by TG–FTIR. Bioresour. Technol. 2014, 154, 313–321. [Google Scholar]
- Shafizadeh, F. Themal Conversion of Cellulosic Materials for Fuel and Chemicals. In Wood and Agricultural Residues: Reseach on Use for Feed, Fuels, and Chemicals; Soltes, E.J., Ed.; Academic Press Inc.: New York, NY, USA, 1983. [Google Scholar]
- Zanzi, R.; Sjöström, K.; Björnbom, E. Rapid pyrolysis of agricultural residues at high temperature. Biomass Bioenergy 2002, 23, 357–366. [Google Scholar]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar]
- Kaloustian, J.; Pauli, A.; Pastor, J. Decomposition of bio-polymers of some mediterranean plants during heating. J. Therm. Anal. Calorim. 2000, 61, 13–21. [Google Scholar]
- Miller, R.S.; Bellan, J. A generalized biomass pyrolysis model based on superimposed cellulose, hemicelluloseand liqnin kinetics. Combust. Sci. Technol. 1997, 126, 97–137. [Google Scholar]
- Rao, T.R.; Sharma, A. Pyrolysis rates of biomass materials. Energy 1998, 23, 973–978. [Google Scholar]
- Raveendran, K.; Ganesh, A.; Khilar, K.C. Pyrolysis characteristics of biomass and biomass components. Fuel 1996, 75, 987–998. [Google Scholar]
- Couhert, C.; Commandre, J.-M.; Salvador, S. Is it possible to predict gas yields of any biomass after rapid pyrolysis at high temperature from its composition in cellulose, hemicellulose and lignin? Fuel 2009, 88, 408–417. [Google Scholar]
- Wei, L.; Xu, S.; Zhang, L.; Zhang, H.; Liu, C.; Zhu, H.; Liu, S. Characteristics of fast pyrolysis of biomass in a free fall reactor. Fuel Process. Technol. 2006, 87, 863–871. [Google Scholar]
- Storm, C.; Rudiger, H.; Spliethoff, H.; Hein, K.R.G. Co-pyrolysis of coal/biomass and coal/sewage sludge mixtures. J. Eng. Gas Turbines Power 1999, 121, 55–63. [Google Scholar]
- Kastanaki, E.; Vamvuka, D. A comparative reactivity and kinetic study on the combustion of coal-biomass char blends. Fuel 2006, 85, 1186–1193. [Google Scholar]
- Vamvuka, D.; Kakaras, E.; Kastanaki, E.; Grammelis, P. Pyrolysis characteristics and kinetics of biomass residuals mixtures with lignite. Fuel 2003, 82, 1949–1960. [Google Scholar]
- Radovic, L.R.; Walker, P.L., Jr.; Jenkins, R.G. Catalytic coal gasification: Use of calcium versus potassium. Fuel 1984, 63, 1028–1030. [Google Scholar]
- Walker, P.L., Jr.; Matsumoto, S.; Hanzawa, T.; Muira, T.; Ismail, I.M.K. Catalysis of gasification of coal-derived cokes and chars. Fuel 1983, 62, 140–149. [Google Scholar]
- Freund, H. Kinetics of carbon gasification by CO2. Fuel 1985, 64, 657–660. [Google Scholar]
- McKee, D.W.; Spiro, C.L.; Kosky, P.G.; Lamby, E.J. Eutectic salt catalysts for graphite and coal char gasification. Fuel 1985, 64, 805–809. [Google Scholar]
- Huffman, G.P.; Huggins, F.E.; Shah, N.; Shah, A. Behavior of basic elements during coal combustion. Prog. Energy Combust. Sci. 1990, 16, 243–251. [Google Scholar]
- Huffman, G.P.; Huggins, F.E.; Shoenberger, R.W.; Walker, J.S.; Lytle, F.W.; Greegor, R.B. Investigation of the structural forms of potassium in coke by electron microscopy and X-ray absorption spectroscopy. Fuel 1986, 65, 621–632. [Google Scholar]
- Huggins, F.E.; Huffman, G.P.; Lytle, F.W.; Greegor, R.B. The form of occurrence of chlorine in U.S. Coals: An XAFS investigation. ACS Div. Fuel Chem. Prepr. 1989, 34, 551–558. [Google Scholar]
- Huffman, G.P.; Huggins, F.E. Analysis of the Inorganic Constituents of Low-Rank Coals. In The Chemistry of Low Rank Coals; Schobert, H.H., Ed.; American Chemical Society: Washington, DC, USA, 1984; pp. 159–174. [Google Scholar]
- Lang, R.J.; Neavel, R.C. Behaviour of calcium as a steam gasification catalyst. Fuel 1982, 61, 620–626. [Google Scholar]
- Kreith, F. The CRC Handbook of Mechanical Engineering; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Ren, H.; Zhang, Y.; Fang, Y.; Wang, Y. Co-gasification behavior of meat and bone meal char and coal char. Fuel Process. Technol. 2011, 92, 298–307. [Google Scholar]
- Zhu, W.; Song, W.; Lin, W. Catalytic gasification of char from co-pyrolysis of coal and biomass. Fuel Process. Technol. 2008, 89, 890–896. [Google Scholar]
- Brown, R.C.; Liu, Q.; Norton, G. Catalytic effects observed during the co-gasification of coal and switchgrass. Biomass Bioenergy 2000, 18, 499–506. [Google Scholar]
- McKee, D.W.; Spiro, C.L.; Kosky, P.G.; Lamby, E.J. Catalysis of coal char gasification by alkali metal salts. Fuel 1983, 62, 217–220. [Google Scholar]
- Srivastava, S.K.; Saran, T.; Sinha, J.; Ramachandran, L.V.; Rao, S.K. Influence of alkali on pyrolysis of coals. Fuel 1988, 67, 1683–1684. [Google Scholar]
- Brage, C.; Yu, Q.; Chen, G.; Sjostrom, K. Tar evolution profiles obtained from gasification of biomass and coal. Biomass Bioenergy 2000, 18, 87–91. [Google Scholar]
- Habibi, R.; Kopyscinski, J.; Masnadi, M.S.; Lam, J.; Grace, J.R.; Mims, C.A.; Hill, J.M. Co-gasification of biomass and non-biomass feedstocks: Synergistic and inhibition effects of switchgrass mixed with sub-bituminous coal and fluid coke during CO2 gasification. Energy Fuels 2012, 27, 494–500. [Google Scholar]
- Pedersen, L.S.; Nielsen, H.P.; Kiil, S.; Hansen, L.A.; Dam-Johansen, K.; Kildsig, F.; Christensen, J.; Jespersen, P. Full-scale co-firing of straw and coal. Fuel 1996, 75, 1584–1590. [Google Scholar]
- Coughlin, R.W.; Davoudzadeh, F. Coliquefaction of lignin and bituminous coal. Fuel 1986, 65, 95–106. [Google Scholar]
- Feng, Z.; Zhao, J.; Rockwell, J.; Bailey, D.; Huffman, G. Direct liquefaction of waste plastics and coliquefaction of coal-plastic mixtures. Fuel Process. Technol. 1996, 49, 17–30. [Google Scholar]
- Mastral, A.M.; Murillo, R.; Callén, M.S.; GarcIa, T. Evidence of coal and tire interactions in coal-tire coprocessing for short residence times. Fuel Process. Technol. 2001, 69, 127–140. [Google Scholar]
- Mastral, A.M.; Mayoral, M.C.; Murillo, R.; Callen, M.; Garcia, T.; Tejero, M.P.; Torres, N. Evaluation of synergy in tire rubber-coal coprocessing. Ind. Eng. Chem. Res. 1998, 37, 3545–3550. [Google Scholar]
- Taghiei, M.M.; Feng, Z.; Huggins, F.E.; Huffman, G.P. Coliquefaction of waste plastics with coal. Energy Fuels 1994, 8, 1228–1232. [Google Scholar]
- Kanno, T.; Kimura, M.; Ikenaga, N.; Suzuki, T. Coliquefaction of coal with polyethylene using Fe(CO)5-S as catalyst. Energy Fuels 2000, 14, 612–617. [Google Scholar]
- Howaniec, N.; Smoliński, A. Steam co-gasification of coal and biomass–Synergy in reactivity of fuel blends chars. Int. J. Hydrog. Energy 2013, 38, 16152–16160. [Google Scholar]
- Morgan, T.J.; Kandiyoti, R. Pyrolysis of coals and biomass: Analysis of thermal breakdown and its products. Chem. Rev. 2014, 114, 1547–1607. [Google Scholar]
- Malhotra, R.; McMillen, D.F. Relevance of cleavage of strong bonds in coal liquefaction. Energy Fuels 1993, 7, 227–233. [Google Scholar]
- Vuthaluru, H.B. Investigations into the pyrolytic behaviour of coal/biomass blends using thermogravimetric analysis. Bioresour. Technol. 2004, 92, 187–195. [Google Scholar]
- Idris, S.S.; Rahman, N.A.; Ismail, K.; Alias, A.B.; Rashid, Z.A.; Aris, M.J. Investigation on thermochemical behaviour of low rank malaysian coal, oil palm biomass and their blends during pyrolysis via thermogravimetric analysis (TGA). Bioresour. Technol. 2010, 101, 4584–4592. [Google Scholar]
- Jones, J.M.; Kubacki, M.; Kubica, K.; Ross, A.B.; Williams, A. Devolatilisation characteristics of coal and biomass blends. J. Anal. Appl. Pyrolysis 2005, 74, 502–511. [Google Scholar]
- Hayashi, J.; Mizuta, H.; Kusakabe, K.; Morooka, S. Flash copyrolysis of coal and polyolefin. Energy Fuels 1994, 8, 1353–1359. [Google Scholar]
- Onay, O.; Bayram, E.; Kockar, O.M. Copyrolysis of Seyitömer lignite and safflower seed: Influence of the blending ratio and pyrolysis temperature on product yields and oil characterization. Energy Fuels 2007, 21, 3049–3056. [Google Scholar]
- Gao, C.; Vejahati, F.; Katalambula, H.; Gupta, R. Co-gasification of biomass with coal and oil sand coke in a drop tube furnace. Energy Fuels 2009, 24, 232–240. [Google Scholar]
- Ulloa, C.A.; Gordon, A.L.; García, X.A. Thermogravimetric study of interactions in the pyrolysis of blends of coal with radiata pine sawdust. Fuel Process. Technol. 2009, 90, 583–590. [Google Scholar]
- Kubacki, M.L. Co-Pyrolysis and Co-Combustion of Coal and Biomass. Ph.D. Thesis, University of Leeds, Leeds, UK, 2007. [Google Scholar]
- Chen, G.; Yu, Q.; Brage, C.; Sjostrom, K. Co-Gasification of Birch Wood and Daw Mill Coal in Pressurized Fluidized Bed Gasifier: The Investigation into the Synergies in the Process. Proceedings of the 1st World Conference on Biomass for Energy and Industry, Sevilla, Spain, 5–9 June 2000; pp. 1545–1548.
- Squir, K.R.; Solomon, P.R.; Carangelo, R.M.; DiTaranto, M.B. Tar evolution from coal and model polymers: 2. The effects of aromatic ring sizes and donatable hydrogens. Fuel 1986, 65, 833–843. [Google Scholar]
- Ahmaruzzaman, M.; Sharma, D.K. Coprocessing of petroleum vacuum residue with plastics, coal, and biomass and its synergistic effects. Energy Fuels 2007, 21, 891–897. [Google Scholar]
- Miura, K.; Mae, K.; Asaoka, S.; Yoshimura, T.; Hashimoto, K. A new coal flash pyrolysis method utilizing effective radical transfer from solvent to coal. Energy Fuels 1991, 5, 340–346. [Google Scholar]
- Miura, K.; Mae, K.; Sakurada, K.; Hashimoto, K. Flash pyrolysis of coal swollen by tetralin vapor. Energy Fuels 1993, 7, 434–435. [Google Scholar]
- Miura, K.; Mae, K.; Yoshimura, T.; Masuda, K.; Hashimoto, K. Mechanism of radical transfer during the flash pyrolysis of solvent-swollen coal. Energy Fuels 1991, 5, 803–808. [Google Scholar]
- Hayashi, J.; Kawakami, T.; Kusakabe, K.; Morooka, S. Physical and chemical modification of low-rank coals with alkyl chains and the roles of incorporated groups in pyrolysis. Energy Fuels 1993, 7, 1118–1122. [Google Scholar]
- Curran, G.P.; Struck, R.T.; Gorin, E. Mechanism of hydrogen-transfer process to coal and coal extract. Ind. Eng. Chem. Process Des. Dev. 1967, 6, 166–173. [Google Scholar]
- Neavel, R.C. Liquefaction of coal in hydrogen-donor and non-donor vehicles. Fuel 1976, 55, 237–242. [Google Scholar]
- Vernon, L.W. Free radical chemistry of coal liquefaction: Role of molecular hydrogen. Fuel 1980, 59, 102–106. [Google Scholar]
- Li, X.; Hu, H.; Jin, L.; Hu, S.; Wu, B. Approach for promoting liquid yield in direct liquefaction of Shenhua coal. Fuel Process. Technol 2008, 89, 1090–1095. [Google Scholar]
- McMillen, D.F.; Malhotra, R.; Chang, S.; Ogier, W.C.; Nigenda, S.E.; Fleming, R.H. Mechanisms of hydrogen transfer and bond scission of strongly bonded coal structures in donor-solvent systems. Fuel 1987, 66, 1611–1620. [Google Scholar]
- Wei, X.; Ogata, E.; Zong, Z.; Zhou, S.; Qin, Z.; Liu, J.; Shen, K.; Li, H. Advances in the study of hydrogen transfer to model compounds for coal liquefaction. Fuel Process. Technol. 2000, 62, 103–107. [Google Scholar]
- Miura, K.; Mae, K.; Sakurada, K.; Hashimoto, K. Flash pyrolysis of coal following thermal pretreatment at low temperature. Energy Fuels 1992, 6, 16–21. [Google Scholar]
- Mae, K.; Hoshika, N.; Hashimoto, K.; Miura, K. A new coal flash pyrolysis method suppressing crosslinking through the swelling of coal by pyridine vapor. Energy Fuels 1994, 8, 868–873. [Google Scholar]
- Awan, I.A.; Mahmood, T.; Nisar, J. Flash pyrolysis of Lakhara lignite utilizing effective radical transfer. J. Chin. Chem. Soc. 2004, 51, 291–296. [Google Scholar]
- Mae, K.; Inoue, S.; Miura, K. Flash pyrolysis of coal modified through liquid phase oxidation and solvent swelling. Energy Fuels 1996, 10, 364–370. [Google Scholar]
- Milne, T.A.; Evans, R.J.; Abatzoglou, N. Biomass Gasifier “Tars”: heir Nature, Formation, and Conversion; NREL/TP-57025357; National Renewable Energy Laboratory: Golden, CO, USA, 1998; p. 204. [Google Scholar]
- Pinto, F.; Lopes, H.; Andre, R.N.; Gulyurtlu, I.; Cabrita, I. Effect of catalysts in the quality of syngas and by-products obtained by co-gasification of coal and wastes. 1. Tars and nitrogen compounds abatement. Fuel 2007, 86, 2052–2063. [Google Scholar]
- Mettler, M.S.; Vlachos, D.G.; Dauenhauer, P.J. Top ten fundamental challenges of biomass pyrolysis for biofuels. Energy Environ. Sci. 2012, 5, 7797–7809. [Google Scholar]
- Bahng, M.-K.; Mukarakate, C.; Robichaud, D.J.; Nimlos, M.R. Current technologies for analysis of biomass thermochemical processing: A review. Anal. Chim. Acta 2009, 651, 117–138. [Google Scholar]
- Lee, C.W.; Scaroni, A.W.; Jenkins, R.G. Effect of pressure on the devolatilization and swelling behaviour of a softening coal during rapid heating. Fuel 1991, 70, 957–965. [Google Scholar]
- Sun, C.L.; Xiong, Y.Q.; Liu, Q.X.; Zhang, M.Y. Thermogravimetric study of the pyrolysis of two chinese coals under pressure. Fuel 1997, 76, 639–644. [Google Scholar]
- Seebauer, V.; Petek, J.; Staudinger, G. Effects of particle size, heating rate and pressure on measurement of pyrolysis kinetics by thermogravimetric analysis. Fuel 1997, 76, 1277–1282. [Google Scholar]
- Gibbins, J.R.; Gonenc, Z.S.; Kandiyoti, R. Pyrolysis and hydropyrolysis of coal: Comparison of product distributions from a wire-mesh and a hot-rod reactor. Fuel 1991, 70, 621–626. [Google Scholar]
- Shan, G. A Comprehensive Model for Coal Devolatilisation. Ph.D. Thesis, The University of Newcastle, Newcastle, Australia, 2000. [Google Scholar]
- Wall, T.F.; Liu, G.; Wu, H.; Roberts, D.G.; Benfell, K.E.; Gupta, S.; Lucas, J.A.; Harris, D.J. The effects of pressure on coal reactions during pulverised coal combustion and gasification. Prog. Energy Combust. Sci. 2002, 28, 405–433. [Google Scholar]
- Pindoria, R.V.; Lim, J.-Y.; Hawkes, J.E.; Lazaro, M.-J.; Herod, A.A.; Kandiyoti, R. Structural characterization of biomass pyrolysis tars/oils from eucalyptus wood waste: Effect of H2 pressure and sample configuration. Fuel 1997, 76, 1013–1023. [Google Scholar]
- Yu, J.; Lucas, J.A.; Wall, T.F. Formation of the structure of chars during devolatilization of pulverized coal and its thermoproperties: A review. Prog. Energy Combust. Sci. 2007, 33, 135–170. [Google Scholar]
- Suuberg, E.M. Rapid Pyrolysis and Hydropyrolysis of Coal. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 1978. [Google Scholar]
- Lee, C.W.; Jenkins, R.G.; Schobert, H.H. Structure and reactivity of char from elevated pressure pyrolysis of Illinois No. 6 bituminous coal. Energy Fuels 1992, 6, 40–47. [Google Scholar]
- Sha, X.-Z.; Chen, Y.-G.; Cao, J.; Yang, Y.-M.; Ren, D.-Q. Effects of operating pressure on coal gasification. Fuel 1990, 69, 656–659. [Google Scholar]
- Mühlen, H.J.; van Heek, K.H.; Jüntgen, H. Influence of pretreatment temperature and pressure on the char reactivity during hydrogasification. Fuel 1986, 65, 591–593. [Google Scholar]
- Cetin, E.; Moghtaderi, B.; Gupta, R.; Wall, T.F. Influence of pyrolysis conditions on the structure and gasification reactivity of biomass chars. Fuel 2004, 83, 2139–2150. [Google Scholar]
- Cetin, E.; Gupta, R.; Moghtaderi, B. Effect of pyrolysis pressure and heating rate on radiata pine char structure and apparent gasification reactivity. Fuel 2005, 84, 1328–1334. [Google Scholar]
- Pindoria, R.V.; Megaritis, A.; Herod, A.A.; Kandiyoti, R. A two-stage fixed-bed reactor for direct hydrotreatment of volatiles from the hydropyrolysis of biomass: Effect of catalyst temperature, pressure and catalyst ageing time on product characteristics. Fuel 1998, 77, 1715–1726. [Google Scholar]
- Kessler, F.; Knechtle, N.; Frischknecht, R. Heizenergie aus Heizöl, Erdgas oder Holz? Swiss Federal Office of Environment (BUWAL): Berne, Switzerland, 2000; p. 163, in German. [Google Scholar]
- Burchill, P.; Welch, L.S. Variation of nitrogen content and functionality with rank for some UK bituminous coals. Fuel 1989, 68, 100–104. [Google Scholar]
- Wallace, S.; Bartle, K.D.; Perry, D.L. Quantification of nitrogen functional groups in coal and coal derived products. Fuel 1989, 68, 1450–1455. [Google Scholar]
- Li, C.-Z. Conversion of Coal-N and Coal-S during Pyrolysis, Gasification and Combustion. In Advances in the Science of Victorian Brown Coal; Elsevier Ltd.: Oxford, UK, 2004. [Google Scholar]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar]
- Kambara, S.; Takarada, T.; Yamamoto, Y.; Kato, K. Relation between functional forms of coal nitrogen and formation of nitrogen oxide (NOx) precursors during rapid pyrolysis. Energy Fuels 1993, 7, 1013–1020. [Google Scholar]
- Hansson, K.-M.; Samuelsson, J.; Tullin, C.; Åmand, L.-E. Formation of HNCO, HCN, and NH3 from the pyrolysis of bark and nitrogen-containing model compounds. Combust. Flame 2004, 137, 265–277. [Google Scholar]
- Tian, F.-J.; Yu, J.; McKenzie, L.J.; Hayashi, J.-I.; Li, C.-Z. Conversion of fuel-N into HCN and NH3 during the pyrolysis and gasification in steam: A comparative study of coal and biomass. Energy Fuels 2007, 21, 517–521. [Google Scholar]
- Dey, P.M.; Harborne, J.B. Plant Biochemistry; Academic Press: San Diego, CA, USA, 1997; p. 554. [Google Scholar]
- Straka, F.; Jenicek, P.; Zabranska, J.; Dohanyos, M.; Kuncarova, M. Anaerobic Fermentation of Biomass and Wastes with Respect to Sulfur and Nitrogen Contents in Treated Materials. Proceedings of the Eleventh International Waste Management and Landfill Symposium, Environmental Sanitary Engineering Centre, Sardinia, Italy, 1–5 October 2007.
- Jiachun, Z.; Masutani, S.M.; Ishimura, D.M.; Turn, S.Q.; Kinoshita, C.M. Release of Fuel-Bound Nitrogen in Biomass during High Temperature Pyrolysis and Gasification. Proceedings of the 32nd Intersociety Energy Conversion Engineering Conference (IECEC), Honolulu, HI, USA, 27 July–1 August 1997; Volume 3, pp. 1785–1790.
- De Jong, W. Nitrogen Compounds in Pressurised Fluidised Bed Gasification of Biomass and Fossil Fuels. Ph.D. Thesis, Technische Universiteit Delft, Delft, The Netherlands, 2005. [Google Scholar]
- Blair, D.W.; Wendt, J.O.L.; Bartok, W. Evolution of nitrogen and other species during controlled pyrolysis of coal. Symp. Int. Combust. 1977, 16, 475–489. [Google Scholar]
- Slaughter, D.M.; Overmoe, B.J.; Pershing, D.W. Inert pyrolysis of stoker-coal fines. Fuel 1988, 67, 482–489. [Google Scholar]
- Solomon, P.R.; Colket, M.B. Evolution of fuel nitrogen in coal devolatilization. Fuel 1978, 57, 749–755. [Google Scholar]
- Pohl, J.H.; Sarofim, A.F. Devolatilization and oxidation of coal nitrogen. Symp. Int. Combust. 1977, 16, 491–501. [Google Scholar]
- Cai, H.Y.; Güell, A.J.; Dugwell, D.R.; Kandiyoti, R. Heteroatom distribution in pyrolysis products as a function of heating rate and pressure. Fuel 1993, 72, 321–327. [Google Scholar]
- Basiuk, V.A. Pyrolysis of valine and leucine at 500 °C: Identification of less-volatile products using gas chromatography-Fourier transform infrared spectroscopy-mass spectrometry. J. Anal. Appl. Pyrolysis 1998, 47, 127–143. [Google Scholar]
- Chiavari, G.; Galletti, G.C. Pyrolysis-gas chromatography/mass spectrometry of amino acids. J. Anal. Appl. Pyrolysis 1992, 24, 123–137. [Google Scholar]
- Ratcliff, M.A.; Medley, E.E.; Simmonds, P.G. Pyrolysis of amino acids. Mechanistic considerations. J. Org. Chem. 1974, 39, 1481–1490. [Google Scholar]
- Ren, Q.; Zhao, C. NOx and N2O precursors from biomass pyrolysis: Nitrogen transformation from amino acid. Environ. Sci. Technol. 2012, 46, 4236–4240. [Google Scholar]
- Ren, Q.; Zhao, C.; Chen, X.; Duan, L.; Li, Y.; Ma, C. NOx and N2O precursors (NH3 and HCN) from biomass pyrolysis: Co-pyrolysis of amino acids and cellulose, hemicellulose and lignin. Proc. Combust. Inst. 2011, 33, 1715–1722. [Google Scholar]
- Simmonds, P.G.; Medley, E.E.; Ratcliff, M.A.; Shulman, G.P. Thermal decomposition of aliphatic monoaminomonocarboxylic acids. Anal. Chem. 1972, 44, 2060–2066. [Google Scholar]
- Ren, Q.; Zhao, C.; Wu, X.; Liang, C.; Chen, X.; Shen, J.; Tang, G.; Wang, Z. Effect of mineral matter on the formation of NOx precursors during biomass pyrolysis. J. Anal. Appl. Pyrolysis 2009, 85, 447–453. [Google Scholar]
- Ren, Q.; Zhao, C.; Wu, X.; Liang, C.; Chen, X.; Shen, J.; Wang, Z. Catalytic effects of Fe, Al and Si on the formation of NOx precursors and HCl during straw pyrolysis. J. Therm. Anal. Calorim. 2010, 99, 301–306. [Google Scholar]
- Ren, Q.; Zhao, C.; Wu, X.; Liang, C.; Chen, X.; Shen, J.; Wang, Z. Formation of NOx precursors during wheat straw pyrolysis and gasification with O2 and CO2. Fuel 2010, 89, 1064–1069. [Google Scholar]
- Becidan, M.; Skreiberg, Ø.; Hustad, J.E. NOx and N2O precursors (NH3 and HCN) in pyrolysis of biomass residues. Energy Fuels 2007, 21, 1173–1180. [Google Scholar]
- Kleemann, M.; Elsener, M.; Koebel, M.; Wokaun, A. Hydrolysis of isocyanic acid on SCR catalysts. Ind. Eng. Chem. Res. 2000, 39, 4120–4126. [Google Scholar]
- Ogawa, M.; Yoshida, N. Nitrous oxide emission from the burning of agricultural residue. Atmos. Environ. 2005, 39, 3421–3429. [Google Scholar]
- De Soete, G. Nitrous oxide from combustion and industry: Chemistry, emissions and control. Oil Gas Sci. Technol. 1993, 48, 413–451. [Google Scholar]
- De Soete, G.G. Nitrous oxide formation and destruction by industrial NO abatement techniques including SCR. Oil Gas Sci. Technol. 1990, 45, 663–682. [Google Scholar]
- Kilpinen, P.; Hupa, M. Homogeneous N2O chemistry at fluidized bed combustion conditions: A kinetic modeling study. Combust. Flame 1991, 85, 94–104. [Google Scholar]
- Kramlich, J.C.; Cole, J.A.; McCarthy, J.M.; Lanier, W.S.; McSorley, J.A. Mechanisms of nitrous oxide formation in coal flames. Combust. Flame 1989, 77, 375–384. [Google Scholar]
- Schäfer, S.; Bonn, B. Hydrolysis of HCN as an important step in nitrogen oxide formation in fluidised combustion. Part II: Heterogeneous reactions involving limestone. Fuel 2002, 81, 1641–1646. [Google Scholar]
- Wojtowicz, M.A.; Bassilakis, R.; Serio, M.A. Ammonia and hydrogen cyanide release during coal pyrolysis at low heating rates. ACS Div. Fuel Chem. Prepr. 2001, 46, 133–137. [Google Scholar]
- Samuelsson, J.I. Conversion of Nitrogen in a Fixed Burning Biofuel Bed. Ph.D. Thesis, Chalmers University of Techonolgy Göteborg, Göteborg, Sweden, 2006. [Google Scholar]
- Tian, F.-J.; Yu, J.-L.; McKenzie, L.J.; Hayashi, J.-I.; Chiba, T.; Li, C.-Z. Formation of NOx precursors during the pyrolysis of coal and biomass. Part VII. Pyrolysis and gasification of cane trash with steam. Fuel 2005, 84, 371–376. [Google Scholar]
- Xie, K.-C.; Lin, J.-Y.; Li, W.-Y.; Chang, L.-P.; Feng, J.; Zhao, W. Formation of HCN and NH3 during coal macerals pyrolysis and gasification with CO2. Fuel 2005, 84, 271–277. [Google Scholar]
- Nelson, P.F.; Buckley, A.N.; Kelly, M.D. Functional forms of nitrogen in coals and the release of coal nitrogen as NOx precursors (HCN and NH3). Symp. Int. Combust. 1992, 24, 1259–1267. [Google Scholar]
- Jiachun, Z.; Masutani, S.M.; Ishimura, D.M.; Turn, S.Q.; Kinoshita, C.M. Simulation of Fuel-Bound Nitrogen Evolution in Biomass Gasification. Proceedings of the 32nd Intersociety Energy Conversion Engineering Conference (IECEC), Honolulu, HI, USA, 27 July–1 August 1997; Volume 3, pp. 1791–1796.
- Abbas, T.; Costa, M.; Costen, P.; Godoy, S.; Lockwood, F.C.; Ou, J.J.; Romo-Millares, C.; Zhou, J. NOx formation and reduction mechanisms in pulverized coal flames. Fuel 1994, 73, 1423–1436. [Google Scholar]
- Bassilakis, R.; Zhao, Y.; Solomon, P.R.; Serio, M.A. Sulfur and nitrogen evolution in the argonne coals. Experiment and modeling. Energy Fuels 1993, 7, 710–720. [Google Scholar]
- Solomon, P.R.; Hamblen, D.G.; Carangelo, R.M.; Krause, J.L. Coal thermal decomposition in an entrained flow reactor. Proc. Combust. Inst. 1982, 19, 1139–1149. [Google Scholar]
- Zhang, H.; Fletcher, T.H. Nitrogen transformations during secondary coal pyrolysis. Energy Fuels 2001, 15, 1512–1522. [Google Scholar]
- Glarborg, P.; Jensen, A.D.; Johnsson, J.E. Fuel nitrogen conversion in solid fuel fired systems. Prog. Energy Combust. Sci. 2003, 29, 89–113. [Google Scholar]
- Leppälahti, J. Formation of NH3 and HCN in slow-heating-rate inert pyrolysis of peat, coal and bark. Fuel 1995, 74, 1363–1368. [Google Scholar]
- Li, C.Z.; Nelson, P.F.; Ledesma, E.B.; Mackie, J.C. An experimental study of the release of nitrogen from coals pyrolyzed in fluidized-bed reactors. Proc. Combust. Inst. 1996, 26, 3205–3211. [Google Scholar]
- Yuan, S.; Zhou, Z.; Li, J.; Chen, X.; Wang, F. HCN and NH3 released from biomass and soybean cake under rapid pyrolysis. Energy Fuels 2010, 24, 6166–6171. [Google Scholar]
- Abelha, P.; Gulyurtlu, I.; Cabrita, I. Release of nitrogen precursors from coal and biomass residues in a bubbling fluidized bed. Energy Fuels 2007, 22, 363–371. [Google Scholar]
- Furimsky, E.; Ohtsuka, Y. Formation of nitrogen-containing compounds during slow pyrolysis and oxidation of petroleum coke. Energy Fuels 1997, 11, 1073–1080. [Google Scholar]
- Axworthy, A.E.; Dayan, V.H.; Martin, G.B. Reactions of fuel-nitrogen compounds under conditions of inert pyrolysis. Fuel 1978, 57, 29–35. [Google Scholar]
- Lifshitz, A.; Tamburu, C.; Suslensky, A. Isomerization and decomposition of pyrrole at elevated temperatures: Studies with a single-pulse shock tube. J. Phys. Chem. 1989, 93, 5802–5808. [Google Scholar]
- Mackie, J.C.; Colket, M.B.; Nelson, P.F.; Esler, M. Shock tube pyrolysis of pyrrole and kinetic modeling. Int. J. Chem. Kinet. 1991, 23, 733–760. [Google Scholar]
- Patterson, J.M.; Tsamasfyros, A.; Smith, W.T. Pyrolysis of pyrrole. J. Heterocycl. Chem. 1968, 5, 727–729. [Google Scholar]
- Wang, B.; Zhang, R. Quantum Chemistry Study on the Pyrolysis Mechanism of Coal-Related Model Compounds. In Rate Constant Calculation for Thermal Reactions: Methods and Applications; DaCosta, H., Fan, M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Yuan, S.; Zhou, Z.; Li, J.; Wang, F. Nitrogen conversion during rapid pyrolysis of coal and petroleum coke in a high-frequency furnace. Appl. Energy 2012, 92, 854–859. [Google Scholar]
- Li, C.; Tan, L.L. Formation of NOx and SOx precursors during the pyrolysis of coal and biomass. Part III. Further discussion on the formation of HCN and NH3 during pyrolysis. Fuel 2000, 79, 1899–1906. [Google Scholar]
- Tan, L.L.; Li, C.-Z. Formation of NOx and SOx precursors during the pyrolysis of coal and biomass. Part I. Effects of reactor configuration on the determined yields of HCN and NH3 during pyrolysis. Fuel 2000, 79, 1883–1889. [Google Scholar]
- Tan, L.L.; Li, C. Formation of NOx and SOx precursors during the pyrolysis of coal and biomass. Part II. Effects of experimental conditions on the yields of NOx and SOx precursors from the pyrolysis of a Victorian brown coal. Fuel 2000, 79, 1891–1897. [Google Scholar]
- Xu, W.C.; Kumagai, M. Nitrogen evolution during rapid hydropyrolysis of coal. Fuel 2002, 81, 2325–2334. [Google Scholar]
- Aho, M.J.; Hämäläinen, J.P.; Tummavuori, J.L. Importance of solid fuel properties to nitrogen oxide formation through HCN and NH3 in small particle combustion. Combust. Flame 1993, 95, 22–30. [Google Scholar]
- Nelson, P.F.; Li, C.; Ledesma, E. Formation of HNCO from the rapid pyrolysis of coals. Energy Fuels 1996, 10, 264–265. [Google Scholar]
- Yuan, S.; Chen, X.; Li, W.; Liu, H.; Wang, F. Nitrogen conversion under rapid pyrolysis of two types of aquatic biomass and corresponding blends with coal. Bioresour. Technol. 2012, 102, 10124–10130. [Google Scholar]
- Yuan, S.; Zhou, Z.; Li, J.; Chen, X.; Wang, F. HCN and NH3 (NOx precursors) released under rapid pyrolysis of biomass/coal blends. J. Anal. Appl. Pyrolysis 2011, 92, 463–469. [Google Scholar]
- Di Nola, G.; de Jong, W.; Spliethoff, H. The fate of main gaseous and nitrogen species during fast heating rate devolatilization of coal and secondary fuels using a heated wire mesh reactor. Fuel Process. Technol. 2009, 90, 388–395. [Google Scholar]
- Di Nola, G.; de Jong, W.; Spliethoff, H. TG-FTIR characterization of coal and biomass single fuels and blends under slow heating rate conditions: Partitioning of the fuel-bound nitrogen. Fuel Process. Technol. 2010, 91, 103–115. [Google Scholar]
- World Bank Group, United Nations Environment Programme, United Nations Industrial Development Organization. Nitrogen Oxides: Pollution Prevention and Control. In Pollution Prevention and Abatement Handbook, 1998: Toward Cleaner Production; The World Bank Group: Washington, DC, USA, 1999; pp. 245–249. [Google Scholar]
- Konttinen, J.; Kallio, S.; Hupa, M.; Winter, F. NO formation tendency characterization for solid fuels in fluidized beds. Fuel 2013, 108, 238–246. [Google Scholar]
- De Kok, L.J. Sulfur Nutrition and Assimilation in Higher Plants: Regulatory Agricultural and Environmental Aspects; SPB Academic Publishing: The Hague, The Netherlands, 1993. [Google Scholar]
- Hawkesford, M.J.; de Kok, L.J. Managing sulphur metabolism in plants. Plant Cell Environ. 2006, 29, 382–395. [Google Scholar]
- Rennenberg, H.; Brunold, C.H.; de Kok, L.J.; Stulen, I. Sulfur Nutrition and Sulfur Assimilation in Higher Plants: Fundamental Environmental and Agricultural Aspects; SPB Academic Publishing: The Hague, The Netherlands, 1990. [Google Scholar]
- Calkins, W.H. The chemistry of sulfur in coal—A historical perspective. Prepr. Am. Chem. Soc. Fuel Chem. Div. 1993, 32, 358–368. [Google Scholar]
- Chou, C.L. Geologic Factors Affecting the Abundance Distribution Speciation of Sulfur in Coals. Proceedings of the 30th International Geological Congress Geology of Fossil Fuels—Coal, Beijing, China, 4–14 August 1997; Volume 18, Part B. pp. 47–57.
- Khalil, R.A. Thermal Conversion of Biomass with Emphasis on Product Distribution, Reaction Kinetics and Sulfur Abatement. Ph.D. Tesis, Norwegian University of Science and Technology (NTNU), Trondheim, Norway, 2009. [Google Scholar]
- Knudsen, J.N. Volatilization of Inorganic Matter during Combustion of Annual Biomass. Ph.D. Thesis, Techical University of Denmark, Lyngby, Danmark, 2004. [Google Scholar]
- Knudsen, J.N.; Jensen, P.A.; Lin, W.; Frandsen, F.J.; Dam-Johansen, K. Sulfur transformations during thermal conversion of herbaceous biomass. Energy Fuels 2004, 18, 810–819. [Google Scholar]
- Dayton, D.C.; French, R.J.; Milne, T.A. Direct observation of alkali vapor release during biomass combustion and gasification. 1. Application of molecular beam/mass spectrometry to switchgrass combustion. Energy Fuels 1995, 9, 855–865. [Google Scholar]
- Dayton, D.C.; Jenkins, B.M.; Turn, S.Q.; Bakker, R.R.; Williams, R.B.; Belle-Oudry, D.; Hill, L.M. Release of inorganic constituents from leached biomass during thermal conversion. Energy Fuels 1999, 13, 860–870. [Google Scholar]
- Duong, D.N.B.; Tillman, D.A. Chlorine Issues with Biomass Cofiring in Pulverized Coal Boilers: Sources, Reactions, and Consequences—A Literature Review. Proceedings of the 34th International Technical Conference on Coal Utilization & Fuel Systems, Clearwater, FL, USA, 31 May–4 June 2009.
- Miles, T.R.; Baxter, L.L.; Bryers, R.W.; Jenkins, B.M.; Oden, L.L. Alkali Deposits Found in Biomass Power Plants a Preliminary Investigation of Their Extent and Nature; National Renewable Energy Laboratory: Golden, CO, USA, 1996; p. 481. [Google Scholar]
- Jenkins, B.M.; Bakker, R.R.; Wei, J.B. On the properties of washed straw. Biomass Bioenergy 1996, 10, 177–200. [Google Scholar]
- Baxter, L.L.; Miles, T.R.; Jenkins, B.M.; Milne, T.; Dayton, D.; Bryers, R.W.; Oden, L.L. The behavior of inorganic material in biomass-fired power boilers: Field and laboratory experiences. Fuel Process. Technol. 1998, 54, 47–78. [Google Scholar]
- Lewin, C.J.; Reimann, B.E. Silicon and plant growth. Annu. Rev. Plant Physiol. 1969, 20, 289–304. [Google Scholar]
- Yoshida, S. The Physiology of Silicon in Rice; Food and Fertilizer Technology Center: Taipei, Taiwan, 1975. [Google Scholar]
- Marschner, H. Mineral Nutrients in Higher Plants, 2nd ed.; Academic Press: London, UK, 2002. [Google Scholar]
- Zevenhoven-Onderwater, M. Ash-Forming Matter in Biomass Fuels. Ph.D. Thesis, Åbo Akademi University, Turku, Finland, 2001. [Google Scholar]
- Björkman, E.; Strömberg, B. Release of chlorine from biomass at pyrolysis and gasification conditions. Energy Fuels 1997, 11, 1026–1032. [Google Scholar]
- Knudsen, J.N.; Jensen, P.A.; Dam-Johansen, K. Transformation and release to the gas phase of Cl, K, and S during combustion of annual biomass. Energy Fuels 2004, 18, 1385–1399. [Google Scholar]
- Zhao, J.; Hu, X.; Gao, J. Study on the variations of organic sulfur in coal by pyrolysis. Coal Convers. 1993, 16, 77–81. [Google Scholar]
- Calkins, W.H. Determination of organic sulfur-containing structures in coal by flash pyrolysis experiments. Prepr. Am. Chem. Soc. Fuel Chem. Div. 1985, 30, 450–465. [Google Scholar]
- Cullis, C.F.; Norris, A.C. The pyrolysis of organic compounds under conditions of carbon formation. Carbon 1972, 10, 525–537. [Google Scholar]
- Winkler, J.K.; Karow, W.; Rademacher, P. Gas-phase pyrolysis of heterocyclic compounds, part 1 and 2: Flow pyrolysis and annulation reactions of some sulfur heterocycles: Thiophene, benzo[b]thiophene, and dibenzothiophene. A product-oriented study. J. Anal. Appl. Pyrolysis 2002, 62, 123–141. [Google Scholar]
- Ur Rahman Memon, H.; Williams, A.; Williams, P.T. Shock tube pyrolysis of thiophene. Int. J. Energy Res. 2003, 27, 225–239. [Google Scholar]
- Huang, C.; Zhang, J.Y.; Chen, J.; Zheng, C.G. Quantum chemistry study on the pyrolysis of thiophene functionalities in coal. Coal Convers. 2005, 28, 33–35. [Google Scholar]
- Ibarra, J.V.; Palacios, J.M.; Gracia, M.; Gancedo, J.R. Influence of weathering on the sulphur removal from coal by pyrolysis. Fuel Process. Technol. 1989, 21, 63–73. [Google Scholar]
- Cleyle, P.J.; Caley, W.F.; Stewart, L.; Whiteway, S.G. Decomposition of pyrite and trapping of sulphur in a coal matrix during pyrolysis of coal. Fuel 1984, 63, 1579–1582. [Google Scholar]
- Gryglewicz, G.; Jasieńko, S. The behaviour of sulphur forms during pyrolysis of low-rank coal. Fuel 1992, 71, 1225–1229. [Google Scholar]
- Gryglewicz, G. Sulfur transformations during pyrolysis of a high sulfur polish coking coal. Fuel 1995, 74, 356–361. [Google Scholar]
- Telfer, M.A.; Zhang, D.K. Investigation of sulfur retention and the effect of inorganic matter during pyrolysis of south australian low-rank coals. Energy Fuels 1998, 12, 1135–1141. [Google Scholar]
- Yrjas, K.P.; Zevenhoven, C.A.P.; Hupa, M.M. Hydrogen sulfide capture by limestone and dolomite at elevated pressure. 1. Sorbent performance. Ind. Eng. Chem. Res. 1996, 35, 176–183. [Google Scholar]
- Liu, Y.; Morrison, J.L.; Scaroni, A.W. Sulfur capture capacity of limestones in combustion gases: Effect of thermally induced cracking. Prepr. Am. Chem. Soc. Fuel Chem. Div. 1994, 39, 228–232. [Google Scholar]
- Takkinen, S.; Hyppänen, T.; Saastamoinen, J.; Pikkarainen, T. Experimental and modeling study of sulfur capture by limestone in selected conditions of air-fired and oxy-fuel circulating fluidized-bed boilers. Energy Fuels 2011, 25, 2968–2979. [Google Scholar]
- Pisupati, S.V.; Morrison, J.L.; Romans, D.E.; Miller, B.G.; Scaroni, A.W. Importance of Calcium Carbonate Content on the Sulfur Capture Performance of Naturally-Occurring Sorbents in a 30 MW(e) Circulating Fluidized-Bed Cogeneration Plant. Proceedings of the 12th International Conference on Fluidized-Bed Combustion, San Diego, CA, USA, 9–13 May 1993; pp. 1069–1078.
- Pisupati, S.V.; Bhalla, S. Influence of calcium content of biomass-based materials on simultaneous nox and SO2 reduction. Environ. Sci. Technol. 2008, 42, 2509–2514. [Google Scholar]
- Lu, Q.-G.; Li, Z.-W.; Na, Y.-J.; Bao, S.-L.; Sun, Y.-K.; He, J. Low SO2 emission from CFB co-firing MSW and bituminous. J. Environ. Sci. 2004, 16, 821–824. [Google Scholar]
- Kazagic, A.; Smajevic, I. Synergy effects of co-firing wooden biomass with bosnian coal. Energy 2009, 34, 699–707. [Google Scholar]
- Suárez-Ruiz, I.; Crelling, J.C. Applied Coal Petrology: The Role of Petrology in Coal Utilization; Elsevier Ltd.: Burlington, MA, USA, 2008. [Google Scholar]
- Biedermann, F.; Obernberger, I. Ash Related Problems during Biomass Combustion and Possibilities for A Sustainable Ash Utilization. Proceedings of the International Conference on World Renewable Energy Congress (WREC), Aberdeen, Scotland, UK, 22––27 May 2005; p. 8.
- Baxter, L.L. Ash deposition during biomass and coal combustion: A mechanistic approach. Biomass Bioenergy 1993, 4, 85–102. [Google Scholar]
- Rushdi, A.; Sharma, A.; Gupta, R. An experimental study of the effect of coal blending on ash deposition. Fuel 2004, 83, 495–506. [Google Scholar]
- Coda, B.; Cieplik, M.K.; de Wild, P.J.; Kiel, J.H.A. Slagging behavior of wood ash under entrained-flow gasification conditions. Energy Fuels 2007, 21, 3644–3652. [Google Scholar]
- Obernberger, I.; Biedermann, F.; Widmann, W.; Riedl, R. Concentrations of inorganic elements in biomass fuels and recovery in the different ash fractions. Biomass Bioenergy 1997, 12, 211–224. [Google Scholar]
- Turn, S.Q.; Kinoshita, C.M.; Ishimura, D.M. Removal of inorganic constituents of biomass feedstocks by mechanical dewatering and leaching. Biomass Bioenergy 1997, 12, 241–252. [Google Scholar]
- Vamvuka, D.; Zografos, D.; Alevizos, G. Control methods for mitigating biomass ash-related problems in fluidized beds. Bioresour. Technol. 2008, 99, 3534–3544. [Google Scholar]
- Pettersson, A.; Zevenhoven, M.; Steenari, B.-M.; Åmand, L.-E. Application of chemical fractionation methods for characterisation of biofuels, waste derived fuels and CFB co-combustion fly ashes. Fuel 2008, 87, 3183–3193. [Google Scholar]
- Chao, C.Y.H.; Kwong, P.C.W.; Wang, J.H.; Cheung, C.W.; Kendall, G. Co-firing coal with rice husk and bamboo and the impact on particulate matters and associated polycyclic aromatic hydrocarbon emissions. Bioresour. Technol. 2008, 99, 83–93. [Google Scholar]
- Jianjun, D.; Shahab, S.; John, R.G.; Xiaotao, B.; Lim, C.J.; Staffan, M. Overview and some issues related to co-firing biomass and coal. Can. J. Chem. Eng. 2008, 86, 367–386. [Google Scholar]
- Doshi, V.; Vuthaluru, H.B.; Korbee, R.; Kiel, J.H.A. Development of a modeling approach to predict ash formation during co-firing of coal and biomass. Fuel Process. Technol. 2009, 90, 1148–1156. [Google Scholar]
- Zheng, Y.; Jensen, P.A.; Jensen, A.D.; Sander, B.; Junker, H. Ash transformation during co-firing coal and straw. Fuel 2007, 86, 1008–1020. [Google Scholar]
- Higman, C.; Burgt, M.V.D. Gasification; Elsevier: New York, NY, USA, 2008. [Google Scholar]
- Livingston, W.R. Biomass ash Characteristics and Behaviour in Combustion, Gasification and Pyrolysis Systems; Doosan Babcock Energy Limited: West Sussex, UK, 2007. [Google Scholar]
- Marschner, H. Mineral Nutrition of Higher Plants; Academic Press Inc.: San Diego, CA, USA, 1986. [Google Scholar]
- Jenkins, B.M.; Baxter, L.L.; Miles, T.R. Combustion properties of biomass. Fuel Process. Technol. 1998, 54, 17–46. [Google Scholar]
- Benson, S.A.; Holm, P.L. Comparison of inorganics in three low-rank coals. Ind. Eng. Chem. Prod. Res. Dev. 2002, 24, 145–149. [Google Scholar]
- Zevenhoven, M.; Skrifvars, B.-J.; Yrjas, P.; Hupa, M.; Nuutinen, L.; Laitinen, R. Searching for Improved Characterisation of Ash Forming Matter in Biomass. Proceedings of the 16th International Conference on Fluidized Bed Combustion,, Reno, NV, USA, 13–16 May 2001.
- Zevenhoven-Onderwater, M.; Blomquist, J.P.; Skrifvars, B.J.; Backman, R.; Hupa, M. The prediction of behaviour of ashes from five different solid fuels in fluidised bed combustion. Fuel 2000, 79, 1353–1361. [Google Scholar]
- Van Loo, S.; Koppejan, J. Handbook of Biomass Combustion and Co-Firing; Earthscan: London, UK, 2008. [Google Scholar]
- Strand, M.; Bohgard, M.; Swietlicki, E.; Gharibi, A.; Sanati, M. Laboratory and field test of a sampling method for characterization of combustion aerosols at high temperatures. Aerosol Sci. Technol. 2004, 38, 757–765. [Google Scholar]
- Wu, H.; Wall, T.; Liu, G.; Bryant, G. Ash liberation from included minerals during combustion of pulverized coal: The relationship with char structure and burnout. Energy Fuels 1999, 13, 1197–1202. [Google Scholar]
- Wei, X.; Lopez, C.; von Puttkamer, T.; Schnell, U.; Unterberger, S.; Hein, K.R.G. Assessment of chlorine-alkali-mineral interactions during co-combustion of coal and straw. Energy Fuels 2002, 16, 1095–1108. [Google Scholar]
- Dayton, D.C.; Belle-Oudry, D.; Nordin, A. Effect of coal minerals on chlorine and alkali metals released during biomass/coal cofiring. Energy Fuels 1999, 13, 1203–1211. [Google Scholar]
- Andersen, K.H.; Frandsen, F.J.; Hansen, P.F.B.; Wieck-Hansen, K.; Rasmussen, I.; Overgaard, P.; Dam-Johansen, K. Deposit formation in a 150 MWe utility PF-boiler during co-combustion of coal and straw. Energy Fuels 2000, 14, 765–780. [Google Scholar]
- Blevins, L.G.; Cauley, T.H. Fine particulate formation during switchgrass/coal cofiring. J. Eng. Gas Turbines Power 2005, 127, 457–463. [Google Scholar]
- Aho, M.; Silvennoinen, J. Preventing chlorine deposition on heat transfer surfaces with aluminium-silicon rich biomass residue and additive. Fuel 2004, 83, 1299–1305. [Google Scholar]
- Kyi, S.; Chadwick, B.L. Screening of potential mineral additives for use as fouling preventatives in Victorian brown coal combustion. Fuel 1999, 78, 845–855. [Google Scholar]
- Raask, E. Mineral Impurities in Coal Combustion : Behavior, Problems, and Remedial Measures; Hemisphere Publishing Corporation: Washington, DC, USA, 1985. [Google Scholar]
- Hupa, M. Interaction of fuels in co-firing in FBC. Fuel 2005, 84, 1312–1319. [Google Scholar]
- Gomez-Barea, A.; Fernandez-Pereira, C.; Vilches, L.F.; Leiva, C.; Carupoy, M.; Ollero, P. Advanced Utilisation Options for Biomass Gasification Fly Ash. Proceedings of the 15th European Biomass Conference & Exhibition, Berlin, Germany, 7–11 May 2007.
- Pels, J.R.; de Nie, D.S.; Kiel, J.H.A. Utilization of Ashes from Biomass Combustion and Gasification. Proceedings of the 14th European Biomass Conference & Exhibition, Paris, France, 17–21 October 2005.
- Mozaffari, M.; Russelle, M.P.; Rosen, C.J.; Nater, E.A. Nutrient supply and neutralizing value of alfalfa stem gasification ash. Soil Sci. Soc. Am. J. 2002, 66, 171–178. [Google Scholar]
- Patel, B.; McQuigg, K.; Toerne, R.F. Gasification Based Biomass Co-Firing Phase I; Annual Technical Report; Nexant, Inc.: San Francisco, CA, USA; October; 2000 October; 2001.
- Mozaffari, M.; Rosen, C.J.; Russelle, M.P.; Nater, E.A. Chemical characterization of ash from gasification of alfalfa stems: Implications for ash management. J. Environ. Qual. 2000, 29, 963–972. [Google Scholar]
- Pels, J.R. A House Built of Biomass Ash; Newsletter for BioEnergy Network of Excellence: Birmingham, UK; July; 2005; Volume 1. [Google Scholar]
- Forth, J.; Zoorob, S.E. Non-Traditional Binders for Construction Materials. Proceedings of the IABSE Henderson Colloquium, Cambridge, UK, 10–12 July 2006.
- Emilsson, S.; Bergström, J. From Extraction of Forest Fuels to Ash Recycling; Swedish Forest Agency: Jönköping, Sweden, 2006; p. 48. [Google Scholar]
- Francis, C.W.; Davis, E.C.; Goyert, J.C. Plant uptake of trace elements from coal gasification ashes. J. Environ. Qual. 1985, 14, 561–569. [Google Scholar]
- Sear, L.K.A. Properties and Use of Coal Fly Ash a Valuable Industrial By-Product; Thomas Telford Publishing: London, UK, 2001. [Google Scholar]
- Duplessi, H.L. The Utilization of Gasification Ash in the Cement and Concrete. Master's Thesis, University of Pretoria, Pretoria, South Africa, 2006. [Google Scholar]
- Hernaìndez, J.J.; Aranda-Almansa, G.; Serrano, C. Co-gasification of biomass wastes and coal-coke blends in an entrained flow gasifier: An experimental study. Energy Fuels 2010, 24, 2479–2488. [Google Scholar]
- Kajitani, S.; Zhang, Y.; Umemoto, S.; Ashizawa, M.; Hara, S. Co-gasification reactivity of coal and woody biomass in high-temperature gasification. Energy Fuels 2009, 24, 145–151. [Google Scholar]
- Vertes, A.A.; Qureshi, N.; Blaschek, H.P.; Yukawa, H. Biomass to Biofuels: Strategies for Global Industries; John Wiley & Sons Ltd.: West Sussex, UK, 2010. [Google Scholar]
- Henrich, E.; Dahmen, N.; Dinjus, E. Cost estimate for biosynfuel production via biosyncrude gasification. Biofuels Bioprod. Biorefining 2009, 3, 28–41. [Google Scholar]
- Neuhoff, K. Large scale deployment of renewables for electricity generation. Oxf. Rev. Electron. Policy 2005, 21, 88–110. [Google Scholar]
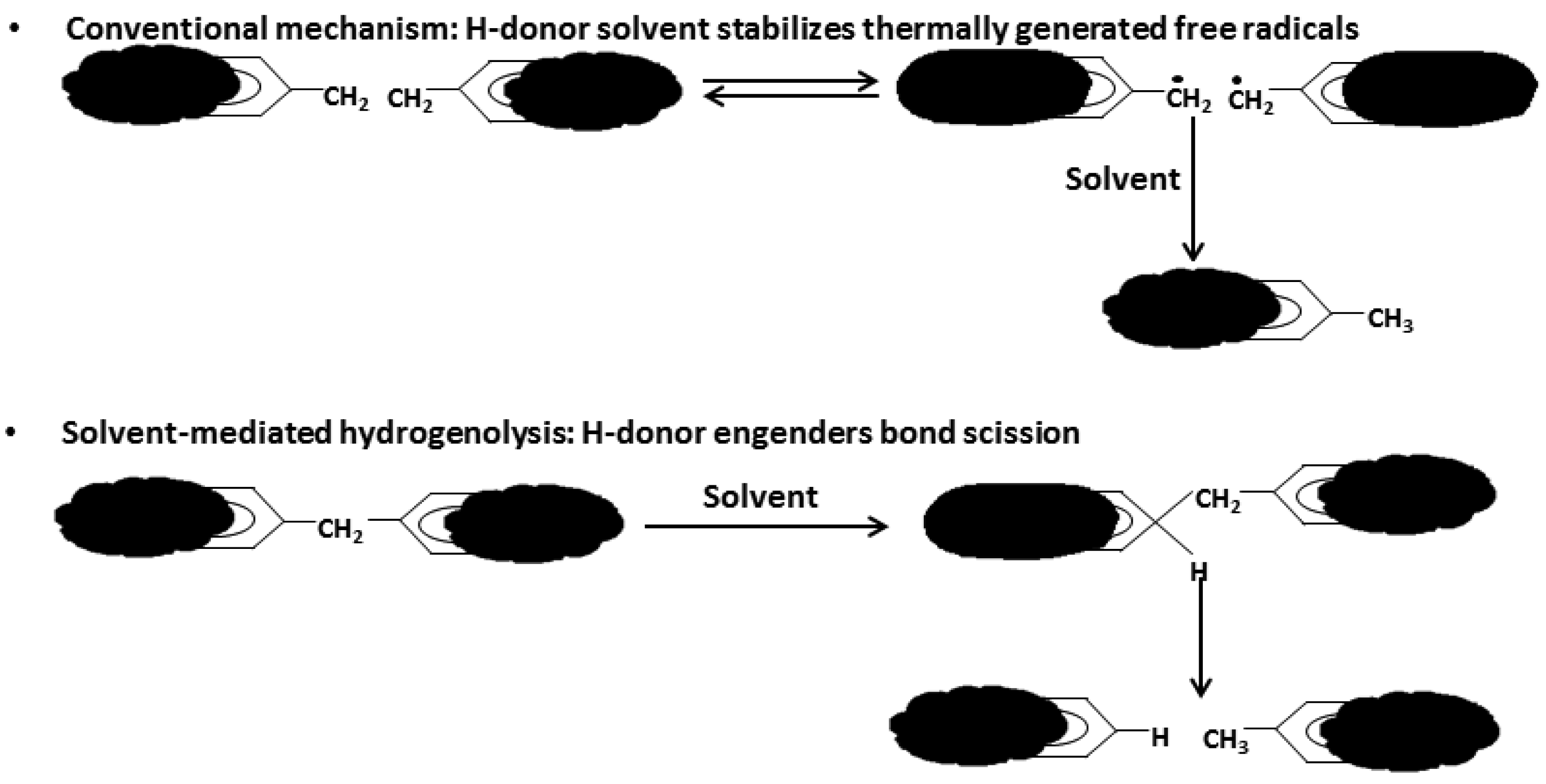
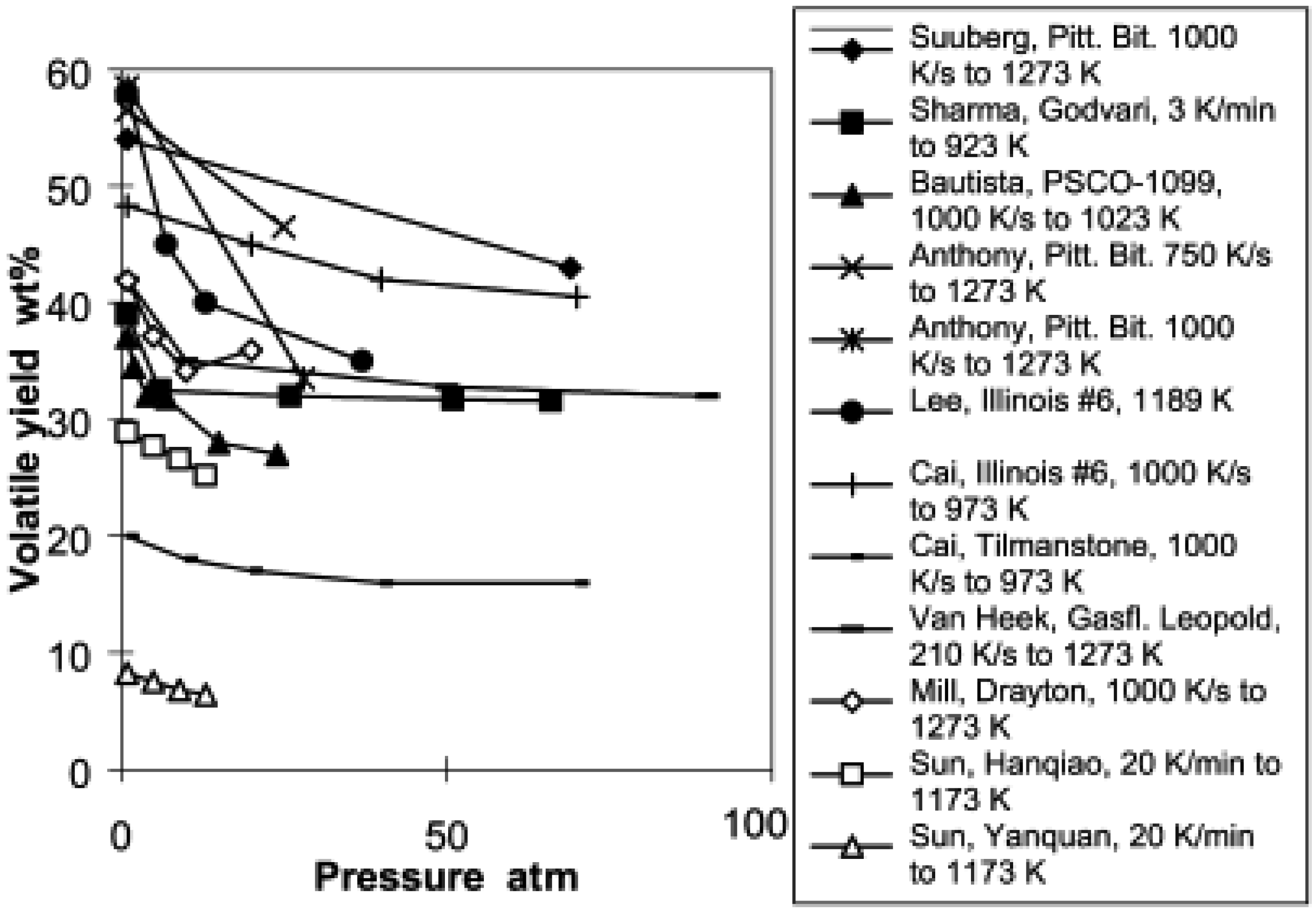

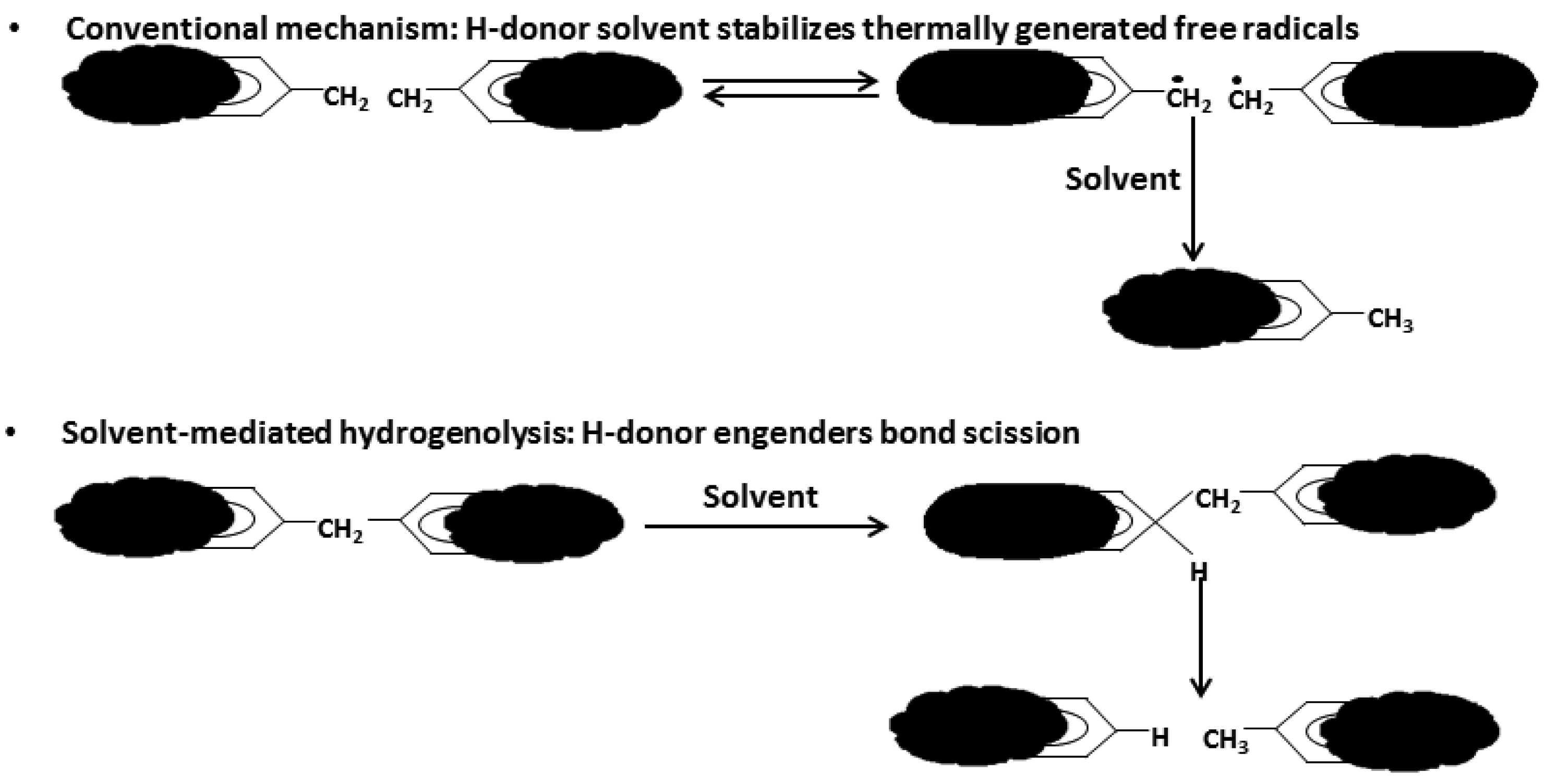{kind=link}
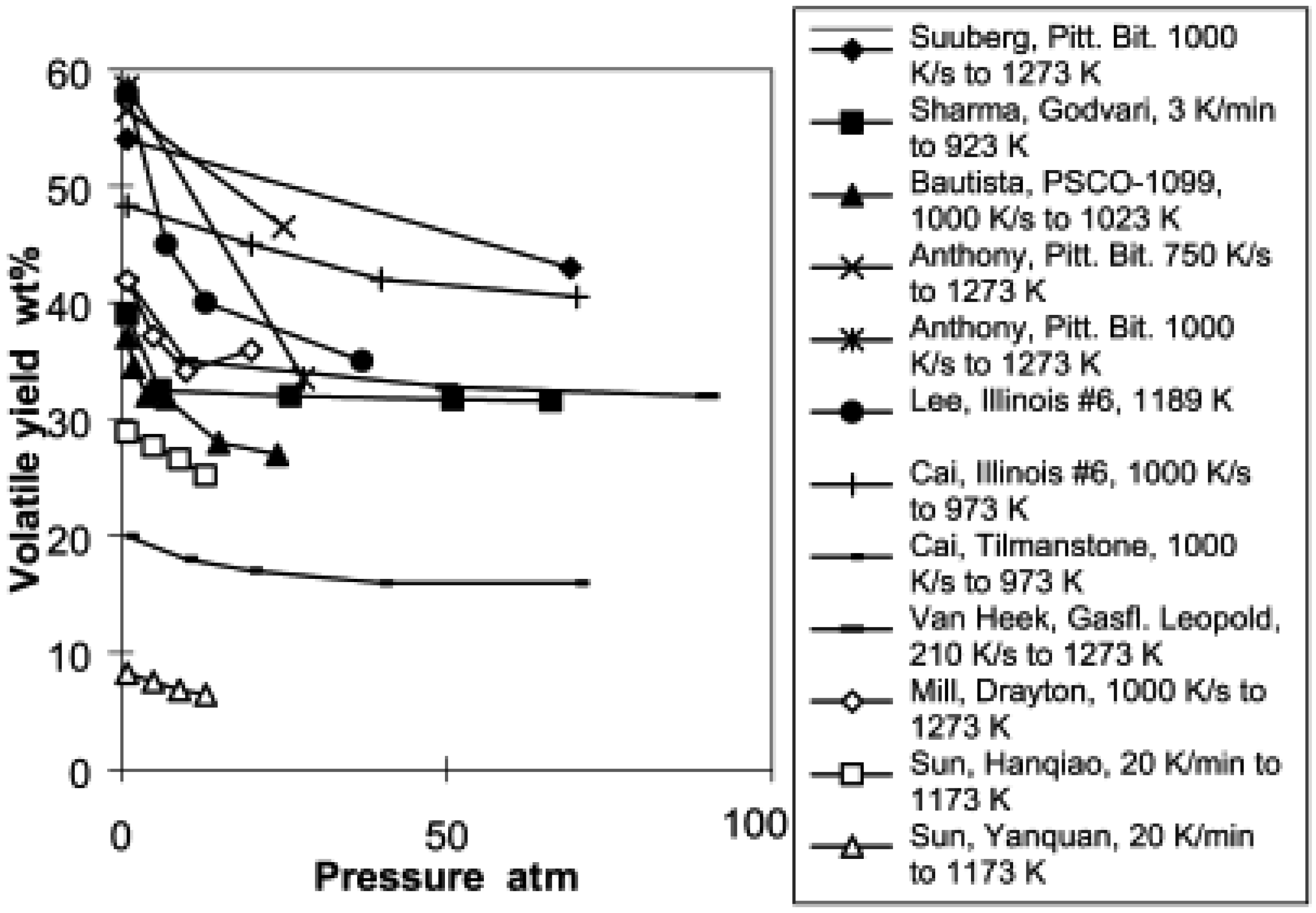{kind=link}
{kind=link}
{kind=link}
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tchapda, A.H.; Pisupati, S.V. A Review of Thermal Co-Conversion of Coal and Biomass/Waste. Energies 2014, 7, 1098-1148. https://doi.org/10.3390/en7031098
Tchapda AH, Pisupati SV. A Review of Thermal Co-Conversion of Coal and Biomass/Waste. Energies. 2014; 7(3):1098-1148. https://doi.org/10.3390/en7031098
Chicago/Turabian StyleTchapda, Aime Hilaire, and Sarma V. Pisupati. 2014. "A Review of Thermal Co-Conversion of Coal and Biomass/Waste" Energies 7, no. 3: 1098-1148. https://doi.org/10.3390/en7031098
APA StyleTchapda, A. H., & Pisupati, S. V. (2014). A Review of Thermal Co-Conversion of Coal and Biomass/Waste. Energies, 7(3), 1098-1148. https://doi.org/10.3390/en7031098