
Solvolysis and Mild Hydrogenolysis of Lignin Pyrolysis Bio-Oils for Bunker Fuel Blends

 ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Lignin Pyrolysis Bio-Oil Production
2.2. Catalyst Synthesis and Characterization
2.3. Hydrogenolysis of Lignin Pyrolysis Bio-Oil
2.4. Characterization of Initial and Hydrotreated Bio-Oils
2.5. Characterization of Spent Catalysts
3. Results
3.1. Characterization of Catalysts
3.2. Mild Hydrogenolysis of Lignin Pyrolysis Bio-Oils
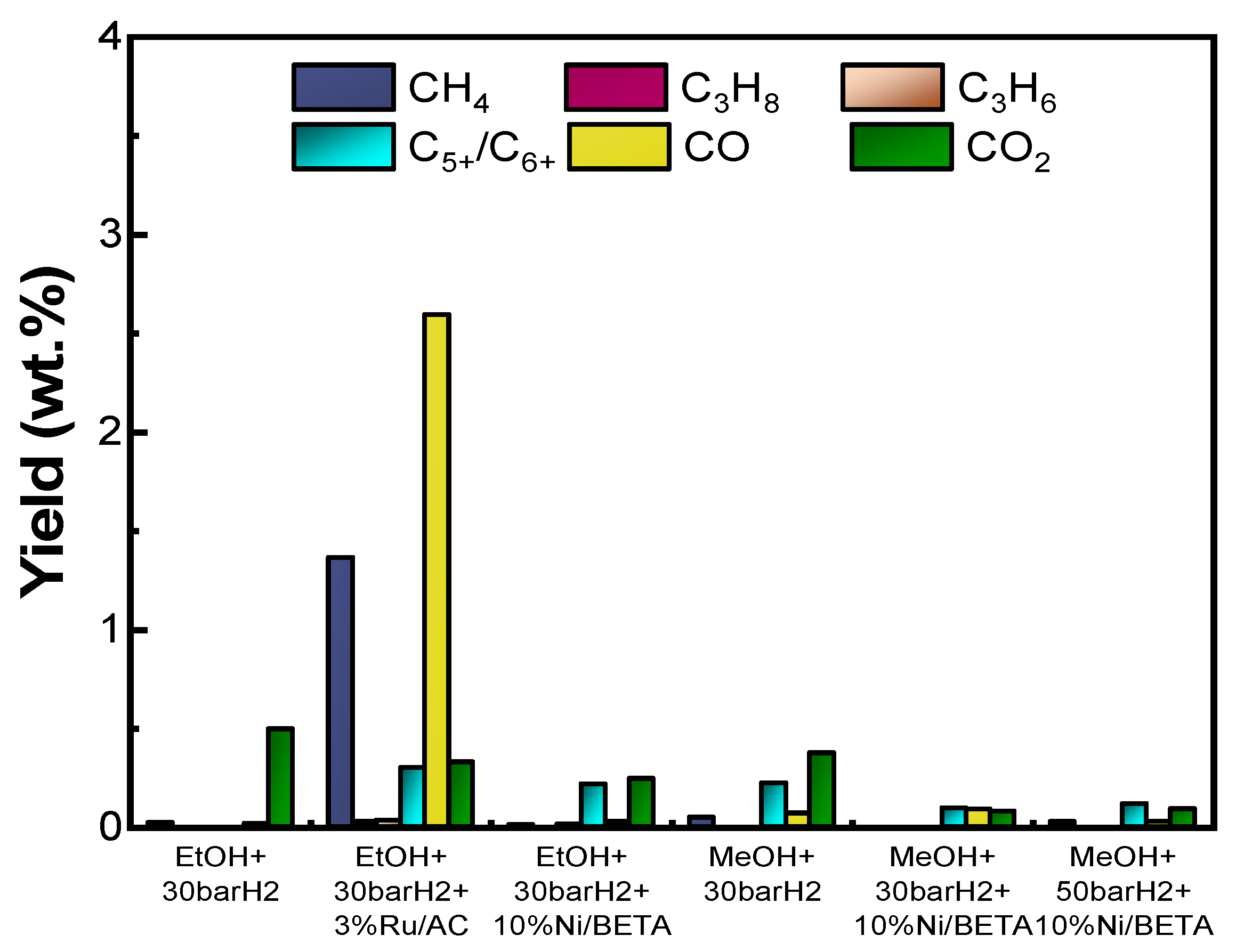
3.2.1. Bio-Oil and Gaseous Product Yields
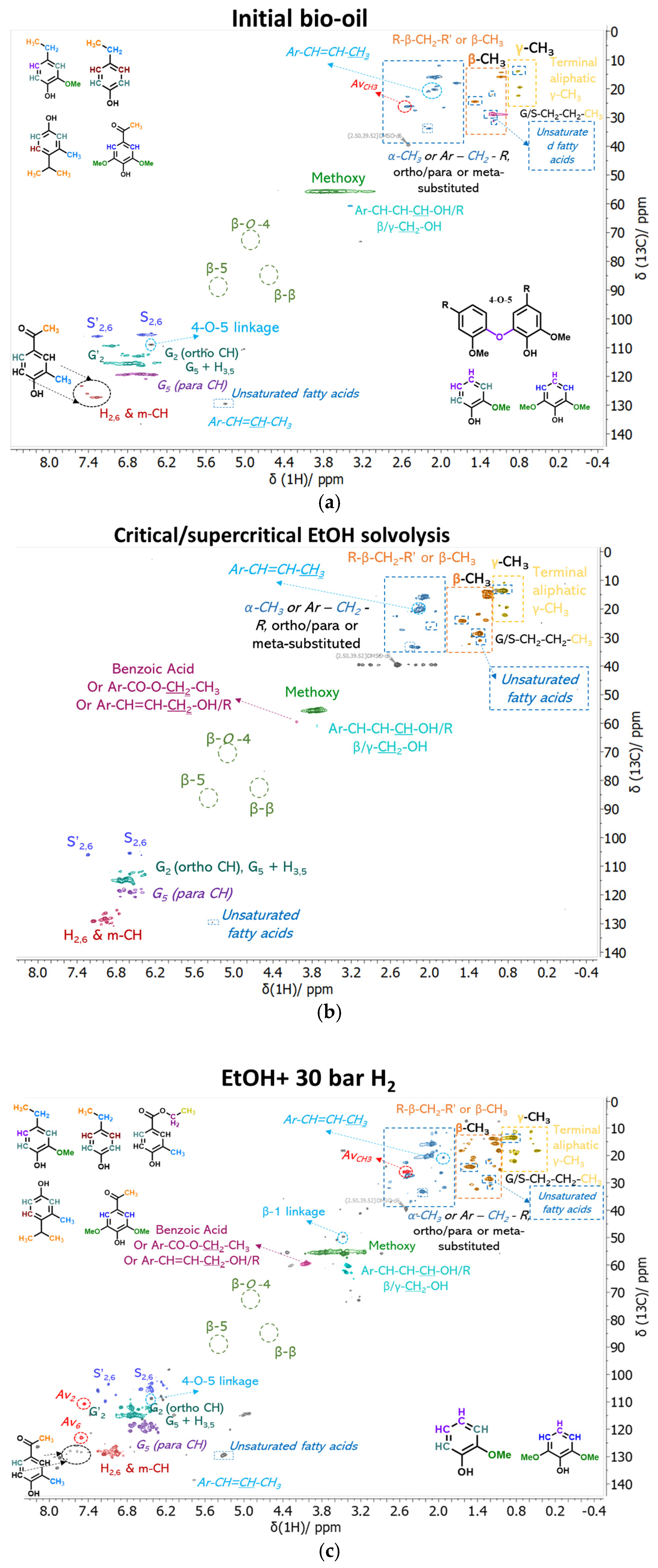
3.2.2. Analysis of Bio-Oil Compositions
3.2.3. Evaluation of Bio-Oil Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Conditions | C 1 (wt.%) | H 1 (wt.%) | N 1 (wt.%) | S 1 (wt.%) | O 2 (wt.%) | HHV 3 (MJ/kg) | Mn (g/mol) | Mw (g/mol) | PDI | Viscosity (cP) |
|---|---|---|---|---|---|---|---|---|---|---|
| Protobind lignin | 59.7 | 6.1 | 0.9 | ~0 | 33.3 | 24.9 | 1746 4 | 2987 4 | 1.7 4 | - |
| Initial bio-oil | 68.9 | 7.1 | 1.2 | ~0 | 22.8 | 30.5 | 245 | 572 | 2.3 | 298,147 |
| EtOH solvolysis | 73.2 | 8.0 | 0.9 | ~0 | 17.9 | 33.5 | 335 | 816 | 2.4 | 1778 |
| EtOH + 30 bar H2 | 72.6 | 7.4 | 1.2 | ~0 | 18.8 | 32.5 | 428 | 1738 | 4.1 | 1500 |
| EtOH + 30 bar H2+ 3% Ru/AC | 63.9 | 7.5 | 0.7 | ~0 | 27.9 | 28.5 | 341 | 832 | 2.4 | 1276 |
| EtOH + 30 bar H2+ 10% Ni/BETA | 65.9 | 7.1 | 0.7 | ~0 | 26.3 | 29.0 | 377 | 1096 | 2.9 | 4439 |
| MeOH + 30 bar H2 | 69.4 | 7.5 | 0.8 | ~0 | 22.3 | 31.1 | 434 | 1247 | 2.9 | 2625 |
| MeOH + 30 bar H2+ 10% Ni/BETA | 66.9 | 7.3 | 0.7 | ~0 | 25.1 | 29.7 | 211 | 744 | 3.5 | 8273 |
| MeOH + 50 bar H2+ 10% Ni/BETA | 62.9 | 6.8 | 0.6 | ~0 | 29.7 | 27.2 | 368 | 982 | 2.7 | 526 |
3.3. Characterization of Char and Spent Catalysts
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hong, J.; Chen, B.; Wang, T.; Zhao, X. A promising technical route for converting lignocellulose to bio-jet fuels based on bioconversion of biomass and coupling of aqueous ethanol: A techno-economic assessment. Fuel 2025, 381, 133670. [Google Scholar] [CrossRef]
- Ashokkumar, V.; Venkatkarthick, R.; Jayashree, S.; Chuetor, S.; Dharmaraj, S.; Kumar, G.; Chen, W.-H.; Ngamcharussrivichai, C. Recent advances in lignocellulosic biomass for biofuels and value-added bioproducts—A critical review. Bioresour. Technol. 2022, 344, 126195. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-J.; Yu, H.-Q. Thermochemical Conversion of Lignocellulosic Biomass into Mass-Producible Fuels: Emerging Technology Progress and Environmental Sustainability Evaluation. ACS Environ. Au 2022, 2, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, P.P.; Matsakas, L.; Rova, U.; Christakopoulos, P. Recent advances in organosolv fractionation: Towards biomass fractionation technology of the future. Bioresour. Technol. 2020, 306, 123189. [Google Scholar] [CrossRef]
- Tofani, G.; Jasiukaitytė-Grojzdek, E.; Grilc, M.; Likozar, B. Organosolv biorefinery: Resource-based process optimisation, pilot technology scale-up and economics. Green Chem. 2024, 26, 186–201. [Google Scholar] [CrossRef]
- Margellou, A.G.; Psochia, E.A.; Torofias, S.A.; Pappa, C.P.; Triantafyllidis, K.S. Isolation of Highly Crystalline Cellulose via Combined Pretreatment/Fractionation and Extraction Procedures within a Biorefinery Concept. ACS Sustain. Resour. Manag. 2024, 1, 1432–1443. [Google Scholar] [CrossRef]
- Kammoun, M.; Margellou, A.; Toteva, V.B.; Aladjadjiyan, A.; Sousa, A.F.; Luis, S.V.; Garcia-Verdugo, E.; Triantafyllidis, K.S.; Richel, A. The key role of pretreatment for the one-step and multi-step conversions of European lignocellulosic materials into furan compounds. RSC Adv. 2023, 13, 21587–21612. [Google Scholar] [CrossRef]
- Lu, X.; Gu, X. A review on lignin-based epoxy resins: Lignin effects on their synthesis and properties. Int. J. Biol. Macromol. 2023, 229, 778–790. [Google Scholar] [CrossRef]
- Pappa, C.P.; Torofias, S.; Triantafyllidis, K.S. Sub-Micro Organosolv Lignin as Bio-Based Epoxy Polymer Component: A Sustainable Curing Agent and Additive. ChemSusChem 2023, 16, e202300076. [Google Scholar] [CrossRef]
- Gioia, C.; Colonna, M.; Tagami, A.; Medina, L.; Sevastyanova, O.; Berglund, L.A.; Lawoko, M. Lignin-Based Epoxy Resins: Unravelling the Relationship between Structure and Material Properties. Biomacromolecules 2020, 21, 1920–1928. [Google Scholar] [CrossRef]
- Pang, B.; Yang, S.; Fang, W.; Yuan, T.-Q.; Argyropoulos, D.S.; Sun, R.-C. Structure-property relationships for technical lignins for the production of lignin-phenol-formaldehyde resins. Ind. Crops Prod. 2017, 108, 316–326. [Google Scholar] [CrossRef]
- Gaudenzi, E.; Cardone, F.; Lu, X.; Canestrari, F. The use of lignin for sustainable asphalt pavements: A literature review. Constr. Build. Mater. 2023, 362, 129773. [Google Scholar] [CrossRef]
- Domínguez-Robles, J.; Cárcamo-Martínez, Á.; Stewart, S.A.; Donnelly, R.F.; Larrañeta, E.; Borrega, M. Lignin for pharmaceutical and biomedical applications—Could this become a reality? Sustain. Chem. Pharm. 2020, 18, 100320. [Google Scholar] [CrossRef]
- Alqahtani, M.S.; Alqahtani, A.; Al-Thabit, A.; Roni, M.; Syed, R. Novel lignin nanoparticles for oral drug delivery. J. Mater. Chem. B 2019, 7, 4461–4473. [Google Scholar] [CrossRef]
- Abu-Omar, M.M.; Barta, K.; Beckham, G.T.; Luterbacher, J.S.; Ralph, J.; Rinaldi, R.; Román-Leshkov, Y.; Samec, J.S.M.; Sels, B.F.; Wang, F. Guidelines for performing lignin-first biorefining. Energy Environ. Sci. 2021, 14, 262–292. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Schutyser, W.; Sels, B.F. Lignin-first biomass fractionation: The advent of active stabilisation strategies. Energy Environ. Sci. 2017, 10, 1551–1557. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bossche, G.; Vangeel, T.; Van Aelst, K.; Sels, B. Reductive catalytic fractionation: State of the art of the lignin-first biorefinery. Curr. Opin. Biotechnol. 2019, 56, 193–201. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Ma, R.; Song, G. Catalytic Hydrogenolysis of Lignin into Serviceable Products. Acc. Chem. Res. 2025, 58, 529–542. [Google Scholar] [CrossRef]
- Margellou, A.; Triantafyllidis, K.S. Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts 2019, 9, 43. [Google Scholar] [CrossRef]
- Liu, X.; Bouxin, F.P.; Fan, J.; Budarin, V.L.; Hu, C.; Clark, J.H. Recent Advances in the Catalytic Depolymerization of Lignin towards Phenolic Chemicals: A Review. ChemSusChem 2020, 13, 4296–4317. [Google Scholar] [CrossRef]
- Charisteidis, I.; Lazaridis, P.; Fotopoulos, A.; Pachatouridou, E.; Matsakas, L.; Rova, U.; Christakopoulos, P.; Triantafyllidis, K. Catalytic Fast Pyrolysis of Lignin Isolated by Hybrid Organosolv—Steam Explosion Pretreatment of Hardwood and Softwood Biomass for the Production of Phenolics and Aromatics. Catalysts 2019, 9, 935. [Google Scholar] [CrossRef]
- Margellou, A.G.; Lazaridis, P.A.; Charisteidis, I.D.; Nitsos, C.K.; Pappa, C.P.; Fotopoulos, A.P.; Van den Bosch, S.; Sels, B.F.; Triantafyllidis, K.S. Catalytic fast pyrolysis of beech wood lignin isolated by different biomass (pre)treatment processes: Organosolv, hydrothermal and enzymatic hydrolysis. Appl. Catal. A-Gen. 2021, 623, 118298. [Google Scholar] [CrossRef]
- Soldatos, P.; Margellou, A.; Pappa, C.; Torofias, S.; Matsakas, L.; Rova, U.; Christakopoulos, P.; Triantafyllidis, K. Conversion of beechwood organosolv lignin via fast pyrolysis and in situ catalytic upgrading towards aromatic and phenolic-rich bio-oil. Sustain. Chem. Environ. 2024, 6, 100107. [Google Scholar] [CrossRef]
- Pappa, C.; Feghali, E.; Vanbroekhoven, K.; Triantafyllidis, K.S. Recent advances in epoxy resins and composites derived from lignin and related bio-oils. Curr. Opin. Green Sustain. Chem. 2022, 38, 100687. [Google Scholar] [CrossRef]
- Vithanage, A.E.; Chowdhury, E.; Alejo, L.D.; Pomeroy, P.C.; DeSisto, W.J.; Frederick, B.G.; Gramlich, W.M. Renewably sourced phenolic resins from lignin bio-oil. J. Appl. Polym. Sci. 2017, 134, 44827. [Google Scholar] [CrossRef]
- Agbo, P.; Mali, A.; Deng, D.; Zhang, L. Bio-Oil-Based Epoxy Resins from Thermochemical Processing of Sustainable Resources: A Short Review. J. Compos. Sci. 2023, 7, 374. [Google Scholar] [CrossRef]
- Chaouch, M.; Diouf, P.N.; Laghdir, A.; Yin, S. Bio-oil from whole-tree feedstock in resol-type phenolic resins. J. Appl. Polym. Sci. 2014, 131, 40014. [Google Scholar] [CrossRef]
- Saidi, M.; Moradi, P. Chapter 3—Conversion of lignin-derived bio-oil to bio-jet fuel. In Sustainable Alternatives for Aviation Fuels; Yousuf, A., Gonzalez-Fernandez, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 49–68. [Google Scholar]
- Kong, X.; Liu, C.; Fan, Y.; Li, M.; Xiao, R. Enhanced Upgrading of Crude Lignin Bio-Oil to Jet Fuel Precursors with a Moderate Organic Acid-Modified Copper Catalyst. ACS Sustain. Chem. Eng. 2023, 11, 7454–7465. [Google Scholar] [CrossRef]
- Zormpa, F.F.; Margellou, A.G.; Karakoulia, S.A.; Delli, E.; Triantafyllidis, K.S. Hydrodeoxygenation of lignin bio-oil model compounds and surrogate mixtures over zeolite supported nickel catalysts. Catal. Today 2024, 433, 114654. [Google Scholar] [CrossRef]
- Margellou, A.G.; Zormpa, F.F.; Karfaridis, D.; Karakoulia, S.A.; Triantafyllidis, K.S. Hydrodeoxygenation of Phenolic Compounds and Lignin Bio-Oil Surrogate Mixture over Ni/BEA Zeolite Catalyst and Investigation of Its Deactivation. Catalysts 2025, 15, 48. [Google Scholar] [CrossRef]
- Luo, Z.; Liu, C.; Radu, A.; de Waard, D.F.; Wang, Y.; Behaghel de Bueren, J.T.; Kouris, P.D.; Boot, M.D.; Xiao, J.; Zhang, H.; et al. Carbon–carbon bond cleavage for a lignin refinery. Nat. Chem. Eng. 2024, 1, 61–72. [Google Scholar] [CrossRef]
- Duan, H.; Dong, J.; Gu, X.; Peng, Y.-K.; Chen, W.; Issariyakul, T.; Myers, W.K.; Li, M.-J.; Yi, N.; Kilpatrick, A.F.R.; et al. Hydrodeoxygenation of water-insoluble bio-oil to alkanes using a highly dispersed Pd–Mo catalyst. Nat. Commun. 2017, 8, 591. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.-P.; Zhou, Q.-J.; Zhao, Y.-P.; Wu, Y.-F.; Liu, F.-J.; Zhong, M.; Li, J.; Liang, J.; Cao, J.-P. Selective hydrodeoxygenation of guaiacol and bio-oil to cycloalkanes over Ni-based catalysts supported on altered Hβ zeolite. Mol. Catal. 2025, 576, 114932. [Google Scholar] [CrossRef]
- Kumar, A.; Khani, Y.; Ko, C.H.; Jae, J.; Banerjee, A.; Bhaskar, T.; Park, Y.-K. Production of Valuable Aromatics from the Hydrodeoxygenation of Guaiacol and Real Bio-Oil over Ni–Nb/HZSM-5 under Supercritical Ethanol. ACS Sustain. Chem. Eng. 2024, 12, 10786–10804. [Google Scholar] [CrossRef]
- Li, B.-S.; Feng, B.-X.; Wu, K.-Y.; Yang, T.-H. Hydrodeoxygenation of lignin derived bio-oil into aromatic hydrocarbons over Ni-Cu-Ru/HZSM-5 catalyst. J. Fuel Chem. Technol. 2023, 51, 358–365. [Google Scholar] [CrossRef]
- Li, L.; Huang, Z.; Shu, F.; Gao, Y.; Long, J. Hydrodeoxygenation of heavy lignin bio-oil to oxygenated fuel catalyzed by CuxNiy/MgO. Fuel 2024, 357, 129805. [Google Scholar] [CrossRef]
- Shu, R.; Li, R.; Lin, B.; Wang, C.; Cheng, Z.; Chen, Y. A review on the catalytic hydrodeoxygenation of lignin-derived phenolic compounds and the conversion of raw lignin to hydrocarbon liquid fuels. Biomass Bioenergy 2020, 132, 105432. [Google Scholar] [CrossRef]
- Ismail, O.; Hamid, A.; Ali, L.; Shittu, T.; Kuttiyathil, M.S.; Iqbal, M.Z.; Khaleel, A.; Altarawneh, M. Selective formation of fuel BXT compounds from catalytic hydrodeoxygenation of waste biomass over Ni-decorated beta-zeolite. Bioresour. Technol. Rep. 2023, 24, 101616. [Google Scholar] [CrossRef]
- International Maritime Organization. Initial IMO strategy on reduction of GHG emissions from ships. In Resolution MEPC; International Maritime Organization: London, UK, 2018; Volume 304. [Google Scholar]
- Bullermann, J.; Meyer, N.-C.; Krafft, A.; Wirz, F. Comparison of fuel properties of alternative drop-in fuels with standard marine diesel and the effects of their blends. Fuel 2024, 357, 129937. [Google Scholar] [CrossRef]
- Hansson, J.; Månsson, S.; Brynolf, S.; Grahn, M. Alternative marine fuels: Prospects based on multi-criteria decision analysis involving Swedish stakeholders. Biomass Bioenergy 2019, 126, 159–173. [Google Scholar] [CrossRef]
- Zhang, Z.; E, J.; Deng, Y.; Pham, M.; Zuo, W.; Peng, Q.; Yin, Z. Effects of fatty acid methyl esters proportion on combustion and emission characteristics of a biodiesel fueled marine diesel engine. Energy Convers. Manag. 2018, 159, 244–253. [Google Scholar] [CrossRef]
- Masum, F.H.; Zaimes, G.G.; Tan, E.C.D.; Li, S.; Dutta, A.; Ramasamy, K.K.; Hawkins, T.R. Comparing Life-Cycle Emissions of Biofuels for Marine Applications: Hydrothermal Liquefaction of Wet Wastes, Pyrolysis of Wood, Fischer–Tropsch Synthesis of Landfill Gas, and Solvolysis of Wood. Environ. Sci. Technol. 2023, 57, 12701–12712. [Google Scholar] [CrossRef] [PubMed]
- Funke, A.; Tomasi Morgano, M.; Dahmen, N.; Leibold, H. Experimental comparison of two bench scale units for fast and intermediate pyrolysis. J. Anal. Appl. Pyrolysis 2017, 124, 504–514. [Google Scholar] [CrossRef]
- Lazaridis, P.A.; Karakoulia, S.A.; Teodorescu, C.; Apostol, N.; Macovei, D.; Panteli, A.; Delimitis, A.; Coman, S.M.; Parvulescu, V.I.; Triantafyllidis, K.S. High hexitols selectivity in cellulose hydrolytic hydrogenation over platinum (Pt) vs. ruthenium (Ru) catalysts supported on micro/mesoporous carbon. Appl. Catal. B-Environ. 2017, 214, 1–14. [Google Scholar] [CrossRef]
- Reyhanitash, E.; Tymchyshyn, M.; Yuan, Z.; Albion, K.; van Rossum, G.; Xu, C. Hydrotreatment of fast pyrolysis oil: Effects of esterification pre-treatment of the oil using alcohol at a small loading. Fuel 2016, 179, 45–51. [Google Scholar] [CrossRef]
- Nielsen, J.B.; Jensen, A.; Schandel, C.B.; Felby, C.; Jensen, A.D. Solvent consumption in non-catalytic alcohol solvolysis of biorefinery lignin. Sustain. Energy Fuels 2017, 1, 2006–2015. [Google Scholar] [CrossRef]
- Shafaghat, H.; Lee, I.-G.; Jae, J.; Jung, S.-C.; Park, Y.-K. Pd/C catalyzed transfer hydrogenation of pyrolysis oil using 2-propanol as hydrogen source. Chem. Eng. J. 2019, 377, 119986. [Google Scholar] [CrossRef]
- Mateus, M.M.; Bordado, J.M.; Galhano dos Santos, R. Estimation of higher heating value (HHV) of bio-oils from thermochemical liquefaction by linear correlation. Fuel 2021, 302, 121149. [Google Scholar] [CrossRef]
- Kouris, P.D.; van Osch, D.J.G.P.; Cremers, G.J.W.; Boot, M.D.; Hensen, E.J.M. Mild thermolytic solvolysis of technical lignins in polar organic solvents to a crude lignin oil. Sustain. Energy Fuels 2020, 4, 6212–6226. [Google Scholar] [CrossRef]
- Huang, X.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Catalytic Depolymerization of Lignin in Supercritical Ethanol. ChemSusChem 2014, 7, 2276–2288. [Google Scholar] [CrossRef]
- Kim, Y.; Shim, J.; Choi, J.-W.; Jin Suh, D.; Park, Y.-K.; Lee, U.; Choi, J.; Ha, J.-M. Continuous-flow production of petroleum-replacing fuels from highly viscous Kraft lignin pyrolysis oil using its hydrocracked oil as a solvent. Energy Convers. Manag. 2020, 213, 112728. [Google Scholar] [CrossRef]
- Zhang, L.; Luo, Y.; Wijayapala, R.; Walters, K.B. Alcohol Stabilization of Low Water Content Pyrolysis Oil during High Temperature Treatment. Energy Fuels 2017, 31, 13666–13674. [Google Scholar] [CrossRef]
- de Waard, D.F.; Kouris, P.D.; Boot, M.D.; Hensen, E.J.M. Mixed Cu–Mn Oxide Catalysts for Solvolysis of Technical Lignin. ACS Sustain. Chem. Eng. 2025, 13, 3269–3279. [Google Scholar] [CrossRef] [PubMed]
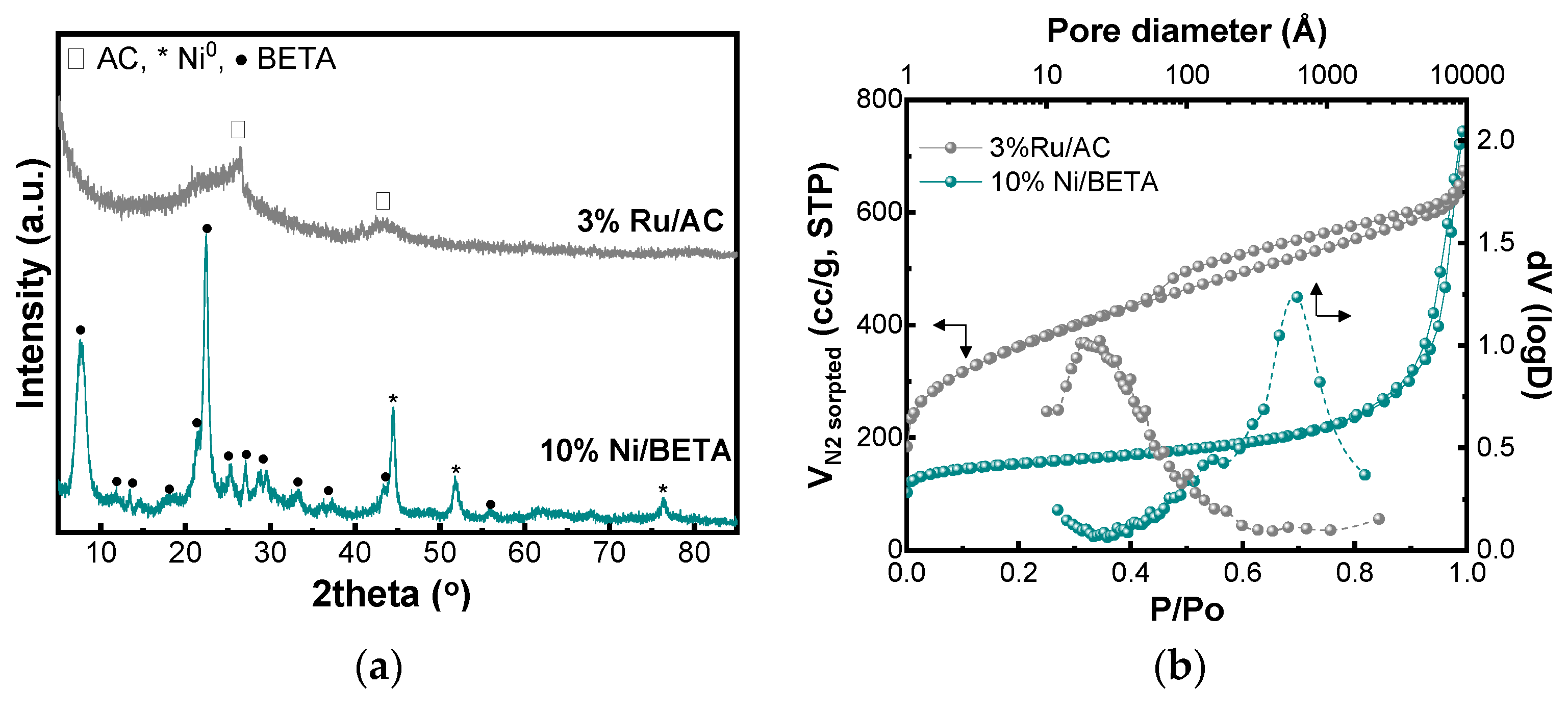
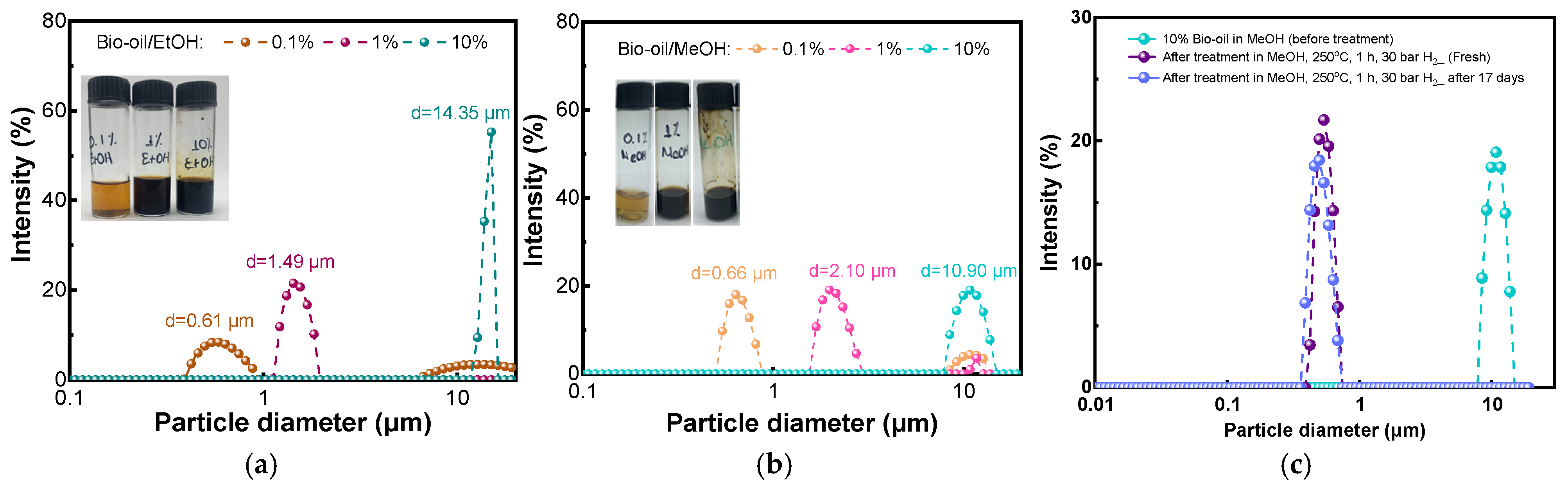


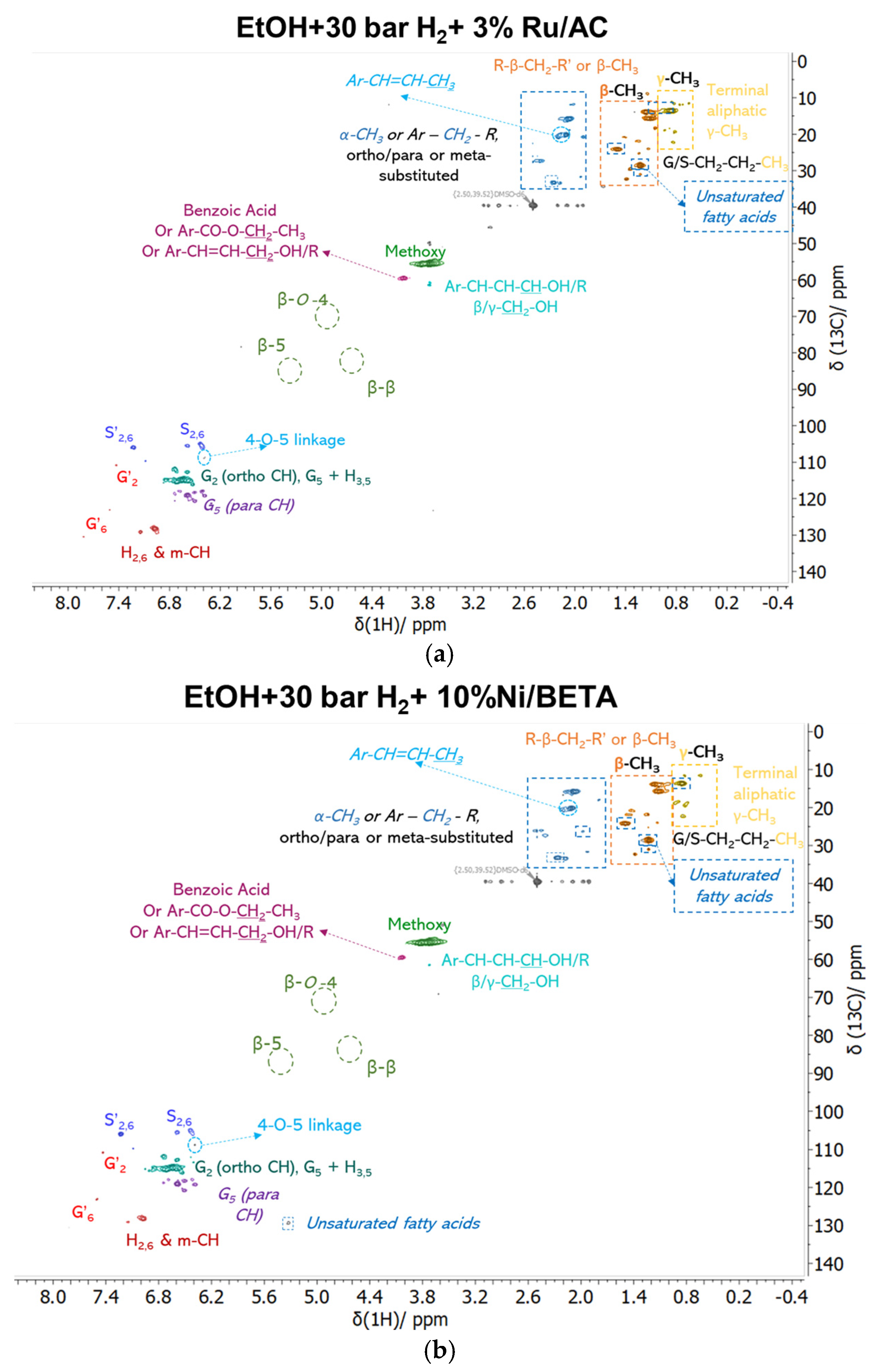
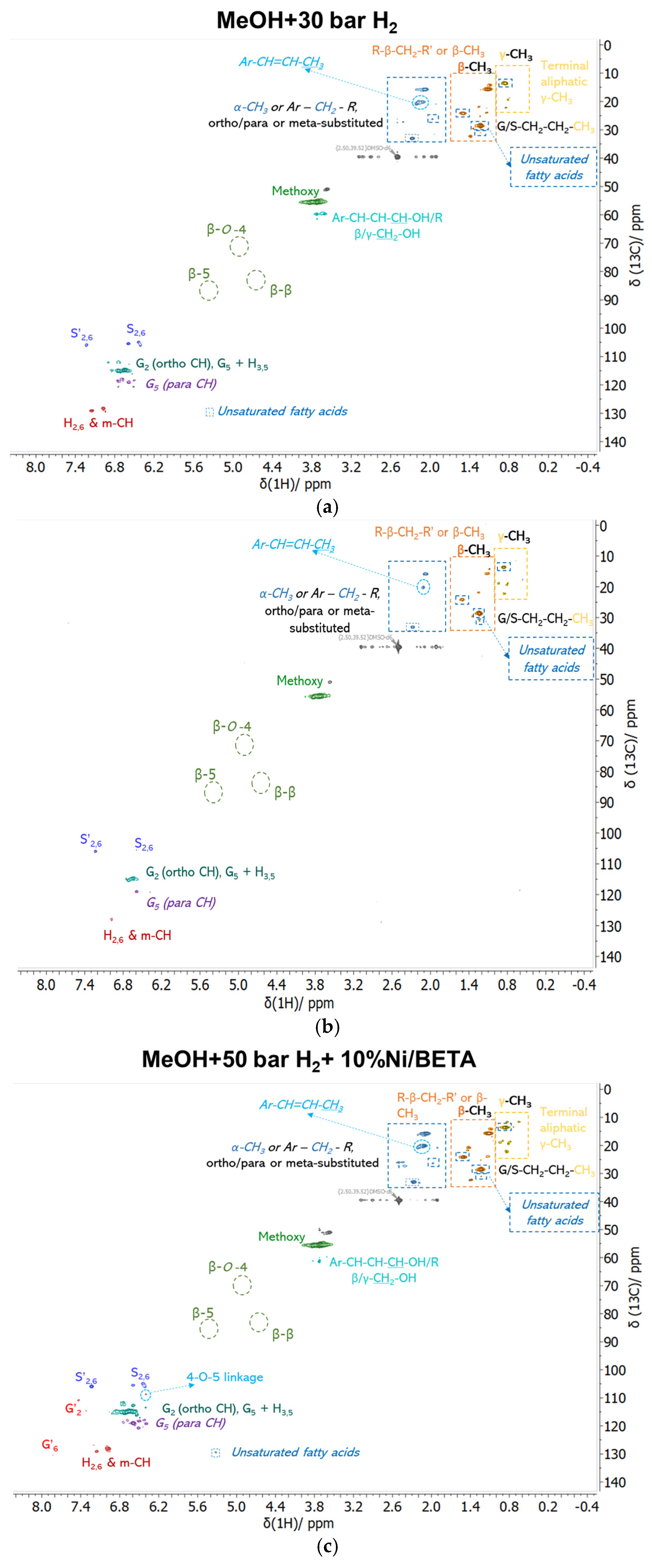


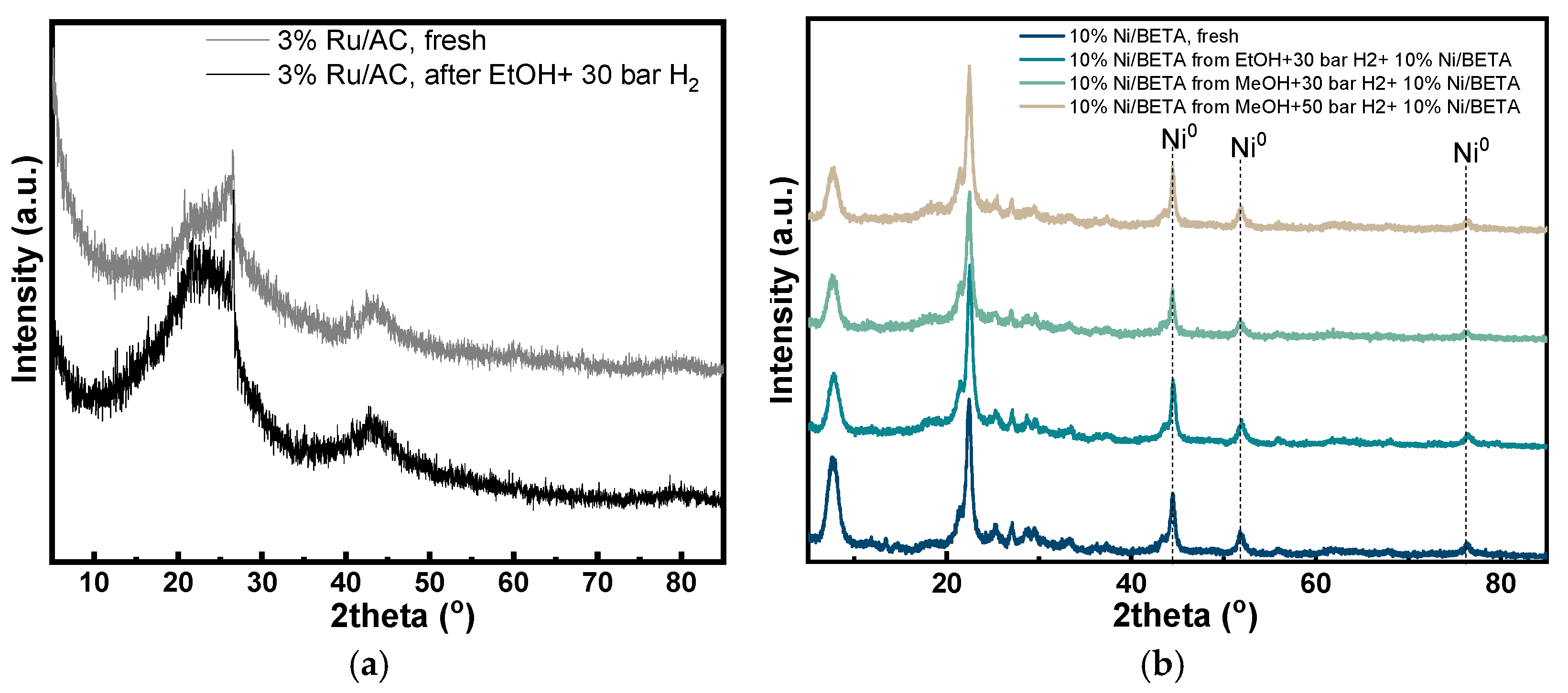
| Catalysts | Dcrystallite (nm) | SBET (m2/g) | Smicro (m2/g) | Smeso (m2/g) | Vtotal (cm3/g) | Vmicro (cm3/g) | Dpore, BJH (nm) |
|---|---|---|---|---|---|---|---|
| 3%Ru/AC | <3 | 1263 | 494 | 769 | 0.826 | 0.232 | 2.4 |
| 10%Ni/BETA | 14 | 562 | 371 | 191 | 1.151 | 0.154 | 62 |
| Compound | Category | Structure | Relative Abundance (%) |
|---|---|---|---|
| Initial bio-oil | |||
| Ethanone, 1-(4-hydroxy-3,5-dimethoxyphenyl)- | OxyPH | 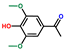 | 12.1 |
| 2-Methoxy-4-vinylphenol | OxyPH | 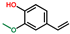 | 8.1 |
| Benzofuran, 2,3-dihydro- | FUR |  | 4.6 |
| Phenol, 2,6-dimethoxy- | OxyPH |  | 4.4 |
| Phenol, 2-methoxy-4-(1-propenyl)-, (E)- | OxyPH | 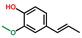 | 3.9 |
| Critical/supercritical EtOH solvolysis | |||
| Phenol, 4-ethyl-2-methoxy- | OxyPH | 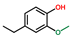 | 5.9 |
| Eicosane | ALI | 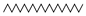 | 5.6 |
| Phenol, p-tert-butyl- | PH | 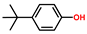 | 4.7 |
| Phenol, 2,6-dimethoxy- | OxyPH | 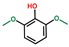 | 3.7 |
| Phenol, 2-methoxy- | OxyPH | 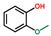 | 3.7 |
| EtOH + 30 bar H2 | |||
| Phenol, 4-ethyl-2-methoxy- | OxyPH | 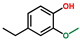 | 9.1 |
| Ethanone, 1-(4-hydroxy-3,5-dimethoxyphenyl)- | OxyPH | 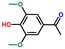 | 7.1 |
| Phenol, 2,6-dimethoxy- | OxyPH | 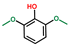 | 6.8 |
| Hexadecanoic acid, ethyl ester | EST | 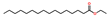 | 6.1 |
| Phenol, 4-ethyl- | PH |  | 5.3 |
| EtOH + 30 bar H2+ 3%Ru/AC | |||
| Phenol, 4-ethyl-2-methoxy- | OxyPH |  | 9.2 |
| Ethanone, 1-(4-hydroxy-3,5-dimethoxyphenyl)- | OxyPH | 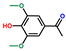 | 6.4 |
| Phenol, 4-ethyl- | PH | 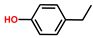 | 5.8 |
| Ethanone, 1-(2,6-dihydroxy-4-methoxyphenyl)- | OxyPH | 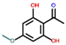 | 4.8 |
| Benzoic acid, 4-hydroxy-3-methoxy- | OxyPH | 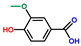 | 4.4 |
| EtOH + 30 bar H2+ 10% Ni/BETA | |||
| Ethanone, 1-(4-hydroxy-3,5-dimethoxyphenyl)- | OxyPH |  | 10.6 |
| Phenol, 4-ethyl-2-methoxy- | OxyPH |  | 8.8 |
| Phenol, 2-methoxy-4-(1-propenyl)-, (E)- | OxyPH | 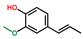 | 5.1 |
| Phenol, 2,6-dimethoxy- | OxyPH | 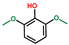 | 5.0 |
| Phenol, 4-ethyl- | PH |  | 4.9 |
| Compound | Category | Structure | Relative Abundance (%) |
|---|---|---|---|
| MeOH + 30 bar H2 | |||
| Phenol, 4-ethyl-2-methoxy- | OxyPH | 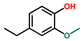 | 10.1 |
| Phenol, 2-methoxy- | OxyPH | 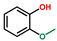 | 8.8 |
| Phenol, 2,6-dimethoxy- | OxyPH |  | 7.6 |
| Ethanone, 1-(4-hydroxy-3,5-dimethoxyphenyl)- | OxyPH | 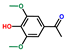 | 4.3 |
| Phenol, 2-methoxy-4-propyl- | OxyPH | 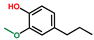 | 3.7 |
| MeOH + 30 bar H2+ 10%Ni/BETA | |||
| Ethanone, 1-(4-hydroxy-3,5-dimethoxyphenyl)- | OxyPH | 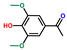 | 11.6 |
| Phenol, 4-ethyl-2-methoxy- | OxyPH | 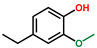 | 6.6 |
| Phenol, 2-methoxy-4-(1-propenyl)-, (E)- | OxyPH | 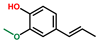 | 4.8 |
| Phenol, 2,6-dimethoxy- | OxyPH |  | 4.4 |
| Phenol, 4-ethyl- | PH | 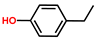 | 4.4 |
| MeOH + 50 bar H2+ 10%Ni/BETA | |||
| Ethanone, 1-(4-hydroxy-3,5-dimethoxyphenyl)- | OxyPH | 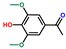 | 9.3 |
| Phenol, 4-ethyl-2-methoxy- | OxyPH | 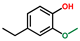 | 8.4 |
| Phenol, 4-ethyl- | PH |  | 5.2 |
| Phenol, 2-methoxy-4-(1-propenyl)-, (E) | OxyPH | 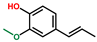 | 4.8 |
| Phenol, 2,6-dimethoxy- | OxyPH | 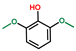 | 4.1 |
| Solid | Reaction Conditions | DXRD (nm) | C (wt.%) | H (wt.%) | N (wt.%) | S (wt.%) | O (wt.%) |
|---|---|---|---|---|---|---|---|
| Char | EtOH | - | 85.4 | 4.8 | 0.8 | 0.0 | 9.0 |
| Char | EtOH + 30 bar H2 | - | 70.7 | 4.8 | 2.3 | 0.0 | 22.1 |
| Catalyst | EtOH + 30 bar H2+ 3% Ru/AC | - | 4.1 | 1.9 | 0.4 | 0.0 | 93.6 |
| Catalyst | EtOH + 30 bar H2+ 10% Ni/BETA | 15 | 13.4 | 1.9 | 0.7 | 0.0 | 84.0 |
| Char | MeOH + 50 bar H2 | - | 77.7 | 5.8 | 1.2 | 0.0 | 15.3 |
| Catalyst | MeOH + 30 bar H2+ 10% Ni/BETA | 15 | 35.1 | 3.5 | 1.2 | 0.1 | 60.0 |
| Catalyst | MeOH + 50 bar H2+ 10% Ni/BETA | 16 | 27.8 | 3.2 | 1.1 | 0.1 | 67.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margellou, A.G.; Langschwager, F.; Pappa, C.P.; Araujo, A.C.C.; Funke, A.; Triantafyllidis, K.S. Solvolysis and Mild Hydrogenolysis of Lignin Pyrolysis Bio-Oils for Bunker Fuel Blends. Energies 2025, 18, 3683. https://doi.org/10.3390/en18143683
Margellou AG, Langschwager F, Pappa CP, Araujo ACC, Funke A, Triantafyllidis KS. Solvolysis and Mild Hydrogenolysis of Lignin Pyrolysis Bio-Oils for Bunker Fuel Blends. Energies. 2025; 18(14):3683. https://doi.org/10.3390/en18143683
Chicago/Turabian StyleMargellou, Antigoni G., Fanny Langschwager, Christina P. Pappa, Ana C. C. Araujo, Axel Funke, and Konstantinos S. Triantafyllidis. 2025. "Solvolysis and Mild Hydrogenolysis of Lignin Pyrolysis Bio-Oils for Bunker Fuel Blends" Energies 18, no. 14: 3683. https://doi.org/10.3390/en18143683
APA StyleMargellou, A. G., Langschwager, F., Pappa, C. P., Araujo, A. C. C., Funke, A., & Triantafyllidis, K. S. (2025). Solvolysis and Mild Hydrogenolysis of Lignin Pyrolysis Bio-Oils for Bunker Fuel Blends. Energies, 18(14), 3683. https://doi.org/10.3390/en18143683