

Practical Aspects of the Analysis of Thermal Dissociation and Pyrolysis Processes in Terms of Transition State Theory


Abstract
1. Introduction
2. Purpose of the Work
3. Theory
4. Rate Constant by Arrhenius
5. Rate Constant by Eyring
6. Relation Between TST and Free Energy
6.1. Approach to No. 1
6.2. Approach to No. 2
7. Balance of Free Energy of Activation in Terms of Arrhenius Rate Constant
7.1. Calcite Thermolysis
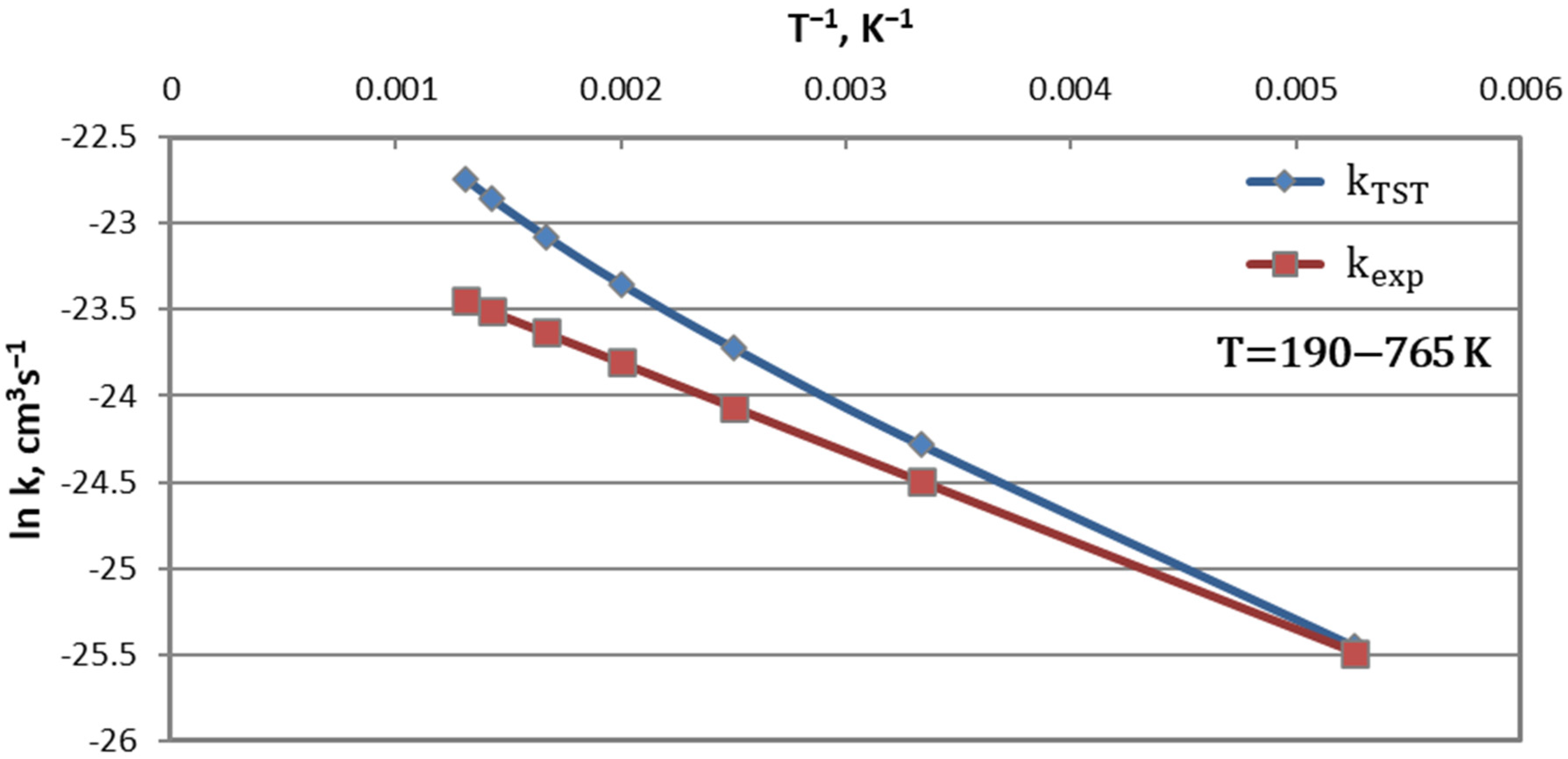
7.2. Methane Thermolysis
- and , (it should be, )
- and , (it should be ).
8. Discussion
9. Conclusions
- In order to determine thermodynamic activation functions, it has been established that the isokinetic temperature is the most reasonable for comparative purposes, both from a thermodynamic and kinetic point of view. The proof is based on deduction, as the central point for the solid phase approaches the equilibrium temperature. The experiment supports the possibility of such a substitution in the terms of mathematical formalism.
- The suggestion made here to use as the central point allows TST to be used when the equilibrium temperature is unknown. However, this is only possible when analysing an elementary reaction or a separated fragments of a complex reaction pathway. Some guidance can be provided by Equation (41), which is a mathematical inversion of Eyring’s concept, assuming equal kinetic rates: Arrhenius and Eyring.
- The fundamental initial condition of TST, expressed as (see Equation (13)), is expanded to encompass an additional term that represents processes of a physical nature. In essence, a variation of the Gibbs free energy as a function of the reaction pathway is proposed, integrating the relationship of phenomenological thermodynamics with elements of kinetics, with activation thermodynamics serving as the unifying element (Figure 2).
- In the context of calcite thermolysis, the term in Equation (53) represents the nucleation of the product (CaO). It has been demonstrated that a maximum nucleation rate is guaranteed when the activation energy . In the vicinity of the centre point, when , there is a pronounced increase in activation energy and an inhibition of the structural transformation process.
- In contrast to calcite thermolysis, in methane thermolysis, this additional term depends on the main factors in which the reaction takes place, which is ultimately expressed in terms of the activation energy characteristic of the system. The characteristics of the free energy changes of the accompanying processes, i.e., processes of a physical nature, are related to the formation of a solid phase in the form of a carbon deposit. In the context of non-catalytic reactions/processes, the observed effects can be classified as either endothermic (low E) or exothermic. The occurrence of these effects can be attributed, for instance, to the specific manner in which the reaction/process is conducted (2). The concept is illustrated by way of the bypass reactor, which is presented as a model (very high E).
- A general free energy of activation equation for the thermolysis reaction/process has been proposed, for the temperature range This equation is equivalent to that given in Equation (76), in which the activation energy is expressed in and a pre-exponential factor in . In order to achieve equilibrium, Equation (77) is employed, incorporating an additional term that is intended to signify the impact of concomitant physical processes.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Nomenclature
| pre-exponential factor, s–1 | |
| exponents | |
| B | ratio of Boltzmann to Planck constant, |
| intercept in Equations.(51) and (52) | |
| the thermodynamic limit acc. [42], | |
| activation energy, | |
| kinetic function of conversion degree argument, | |
| ∆G, ∆H, ∆S | thermodynamic functions; accordingly free energy , enthalpy , and entropy , respectively |
| excess free energy, | |
| Planck constant, | |
| rate constant, | |
| Boltzmann constant, | |
| equilibrium constant | |
| molecularity of the reaction, or | |
| Avogadro constant, | |
| pressure, | |
| partial pressure, Pa | |
| universal gas constant, , | |
| heating rate, , | |
| reaction rate, , | |
| supersaturation, | |
| time, | |
| lifetime of transition-state acc. [60] | |
| absolute temperature, | |
| volume, | |
| mol fraction, , | |
| constant, , Equations (62)–(64) | |
| conversion degree, | |
| vibrational and/or oscillation frequency, s–1, (in [60] is defined as decomposition frequency of TS) | |
| stoichiometric ratio | |
| specific surface energy, , | |
| when acc. [39] | |
| transmission coefficient in Equations (22), (26) and (28), assumed | |
| apparent contact angle between the embryophase and the solid phase, o | |
| Subscripts | |
| acc. to | forwards, backwards |
| 1, 2, n | refers to Figure 7 |
| c | refers to carbon |
| equilibrium | |
| experiment | |
| refers to harmonic mean | |
| isokinetic | |
| equilibrium for forwards | |
| equilibrium for backwards | |
| melting temperature | |
| max | maximum of rate reaction/process |
| nucleation | |
| nucleation inhibition | |
| reaction | |
| products | |
| substrates | |
| transition state | |
| transition state theory | |
| refers to conversion degree | |
| Superscripts | |
| thermodynamic activation functions | |
| standard condition |
Abbreviations
Appendix A
References
- Błażejowski, J.; Szychlínski, J.; Windorpska, K. Thermal Reactions of Lead(IV) Chloride Complexes in the Solid State. Part IV. Thermolysis of Alkali Metal Hexachloroplumbates. Thermochim. Acta 1981, 46, 147–166. [Google Scholar] [CrossRef]
- Xu, R.; Meisner, J.; Chang, A.M.; Thompson, K.C.; Martínez, T.J. First Principles Reaction Discovery: From the Schrodinger Equation to Experimental Prediction for Methane Pyrolysis. Chem. Sci. 2023, 14, 7447–7464. [Google Scholar] [CrossRef]
- Mianowski, A.; Radko, T.; Bigda, R. Isokinetic and Compensation Temperature in the Analysis of Thermal Dissociation of the Solid Phase under Dynamic Conditions. Energies 2023, 16, 5692. [Google Scholar] [CrossRef]
- Mianowski, A.; Radko, T.; Bigda, R. Elements of Transition-State Theory in Relation to the Thermal Dissociation of Selected Solid Compounds. Energies 2024, 17, 2669. [Google Scholar] [CrossRef]
- Mianowski, A.; Szul, M.; Radko, T.; Sobolewski, A.; Iluk, T. Literature Review on Thermodynamic and Kinetic Limitations of Thermal Decomposition of Methane. Energies 2024, 17, 5007. [Google Scholar] [CrossRef]
- Mianowski, A.; Bigda, R. Use of Kinetic Parameters from Thermal Analysis for Balancing Free Energy of Activation Based on Calcite Decomposition. Energies 2025, 18, 570. [Google Scholar] [CrossRef]
- Šimon, P.; Dubaj, T.; Cibulková, Z. An Alternative to the Concept of Variable Activation Energy. J. Therm. Anal. Calorim. 2024, 149, 11507–11516. [Google Scholar] [CrossRef]
- Šimon, P.; Dubaj, T.; Cibulková, Z. Frequent Flaws Encountered in the Manuscripts of Kinetic Papers. J. Therm. Anal. Calorim. 2022, 147, 10083–10088. [Google Scholar] [CrossRef] [PubMed]
- Holba, P.; Šesták, J. Kinetics with Regard to the Equilibrium of Processes Studied by Non-Isothermal Techniques. Z. Für Phys. Chem. 1972, 80, 1–20. [Google Scholar] [CrossRef]
- Šesták, J.; Šatava, V.; Wendlandt, W.W. The Study of Heterogeneous Processes by Thermal Analysis. Thermochim. Acta 1973, 7, 333–556. [Google Scholar] [CrossRef]
- Błażejowski, J. Evaluation of Kinetic Constants for the Solid State Reactions under Linear Temperature Increase Conditions. Thermochim. Acta 1981, 48, 109–124. [Google Scholar] [CrossRef]
- Błażejowski, J. Remarks on the Description of Reaction Kinetics under Non-Isothermal Conditions. Thermochim. Acta 1984, 76, 359–372. [Google Scholar] [CrossRef]
- Koga, N.; Favergeon, L.; Kodani, S. Impact of Atmospheric Water Vapor on the Thermal Decomposition of Calcium Hydroxide: A Universal Kinetic Approach to a Physico-Geometrical Consecutive Reaction in Solid–Gas Systems under Different Partial Pressures of Product Gas. Phys. Chem. Chem. Phys. 2019, 21, 11615–11632. [Google Scholar] [CrossRef]
- Fukuda, M.; Favergeon, L.; Koga, N. Universal Kinetic Description for Thermal Decomposition of Copper(II) Hydroxide over Different Water Vapor Pressures. J. Phys. Chem. C 2019, 123, 20903–20915. [Google Scholar] [CrossRef]
- Kodani, S.; Iwasaki, S.; Favergeon, L.; Koga, N. Revealing the Effect of Water Vapor Pressure on the Kinetics of Thermal Decomposition of Magnesium Hydroxide. Phys. Chem. Chem. Phys. 2020, 22, 13637–13649. [Google Scholar] [CrossRef]
- Iwasaki, S.; Sakai, Y.; Kikuchi, S.; Koga, N. Thermal Decomposition of Perlite Concrete under Different Water Vapor Pressures. J. Therm. Anal. Calorim. 2022, 147, 6309–6322. [Google Scholar] [CrossRef]
- Sakai, Y.; Koga, N. Kinetics of Component Reactions in Calcium Looping Appeared during the Multistep Thermal Decomposition of Portland Cement under Various Atmospheric Conditions. Chem. Eng. J. 2022, 428, 131197. [Google Scholar] [CrossRef]
- Koga, N.; Sakai, Y.; Fukuda, M.; Hara, D.; Tanaka, Y.; Favergeon, L. Universal Kinetics of the Thermal Decomposition of Synthetic Smithsonite over Different Atmospheric Conditions. J. Phys. Chem. C 2021, 125, 1384–1402. [Google Scholar] [CrossRef]
- Hotta, M.; Tone, T.; Favergeon, L.; Koga, N. Kinetic Parameterization of the Effects of Atmospheric and Self-Generated Carbon Dioxide on the Thermal Decomposition of Calcium Carbonate. J. Phys. Chem. C 2022, 126, 7880–7895. [Google Scholar] [CrossRef]
- Hotta, M.; Favergeon, L.; Koga, N. Kinetic Insight into the Effect of Self-Generated CO2 on the Thermal Decomposition of Inorganic Carbonates in an Inert Gas Atmosphere: ZnCO3 and CaCO3. J. Phys. Chem. C 2023, 127, 13065–13080. [Google Scholar] [CrossRef]
- Sakai, Y.; Iwasaki, S.; Kikuchi, S.; Koga, N. Influence of Atmospheric CO2 on the Thermal Decomposition of Perlite Concrete. J. Therm. Anal. Calorim. 2022, 147, 5801–5813. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Favergeon, L.; Koga, N. Thermal Dehydration of Lithium Sulfate Monohydrate Revisited with Universal Kinetic Description over Different Temperatures and Atmospheric Water Vapor Pressures. J. Phys. Chem. C 2020, 124, 11960–11976. [Google Scholar] [CrossRef]
- Iwasaki, S.; Zushi, Y.; Koga, N. An Advanced Kinetic Approach to the Multistep Thermal Dehydration of Calcium Sulfate Dihydrate under Different Heating and Water Vapor Conditions: Kinetic Deconvolution and Universal Isoconversional Analyses. Phys. Chem. Chem. Phys. 2022, 24, 9492–9508. [Google Scholar] [CrossRef] [PubMed]
- Zushi, Y.; Iwasaki, S.; Koga, N. Effect of Atmospheric Water Vapor on Independent-Parallel Thermal Dehydration of a Compacted Composite of an Inorganic Hydrate: Sodium Carbonate Monohydrate Grains Comprising Crystalline Particles and a Matrix. Phys. Chem. Chem. Phys. 2022, 24, 29827–29840. [Google Scholar] [CrossRef] [PubMed]
- Hotta, M.; Zushi, Y.; Iwasaki, S.; Fukunaga, S.; Koga, N. Efflorescence Kinetics of Sodium Carbonate Decahydrate: A Universal Description as a Function of Temperature, Degree of Reaction, and Water Vapor Pressure. Phys. Chem. Chem. Phys. 2023, 25, 27114–27130. [Google Scholar] [CrossRef]
- Hotta, M.; Koga, N. Extended Kinetic Approach to Reversible Thermal Decomposition of Solids: A Universal Description Considering the Effect of the Gaseous Product and the Kinetic Compensation Effect. Thermochim. Acta 2024, 733, 179699. [Google Scholar] [CrossRef]
- Mianowski, A.; Radko, T.; Siudyga, T. Kinetic Compensation Effect of Isoconversional Methods. Reac Kinet. Mech. Cat. 2021, 132, 37–58. [Google Scholar] [CrossRef]
- Lyon, R.E. A Physical Basis for Kinetic Compensation. J. Phys. Chem. A 2023, 127, 2399–2406. [Google Scholar] [CrossRef]
- Koga, N.; Vyazovkin, S.; Burnham, A.K.; Favergeon, L.; Muravyev, N.V.; Pérez-Maqueda, L.A.; Saggese, C.; Sánchez-Jiménez, P.E. ICTAC Kinetics Committee Recommendations for Analysis of Thermal Decomposition Kinetics. Thermochim. Acta 2023, 719, 179384. [Google Scholar] [CrossRef]
- Krug, R.R. Detection of the Compensation Effect(θ Rule). Ind. Eng. Chem. Fund. 1980, 19, 50–59. [Google Scholar] [CrossRef]
- Griessen, R.; Boelsma, C.; Schreuders, H.; Broedersz, C.P.; Gremaud, R.; Dam, B. Single Quality Factor for Enthalpy-Entropy Compensation, Isoequilibrium and Isokinetic Relationships. ChemPhysChem 2020, 21, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Griessen, R.; Dam, B. Simple Accurate Verification of Enthalpy-Entropy Compensation and Isoequilibrium Relationship–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/34213060/ (accessed on 6 April 2025).
- Arrhenius, S. Über Die Reaktionsgeschwindigkeit Bei Der Inversion von Rohrzucker Durch Säuren. Z. Für Phys. Chem. 1889, 4U, 226–248. [Google Scholar] [CrossRef]
- Logan, S.R. The Origin and Status of the Arrhenius Equation. J. Chem. Educ. 1982, 59, 279. [Google Scholar] [CrossRef]
- Laidler, K.J. The Development of the Arrhenius Equation. J. Chem. Educ. 1984, 61, 494. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N. Determination of Pre-Exponential Factors and of the Mathematical Functions f(α) or G(α) That Describe the Reaction Mechanism in a Model-Free Way. Thermochim. Acta 2013, 564, 59–69. [Google Scholar] [CrossRef]
- Mianowski, A.; Sciążko, M.; Radko, T. Vyazovkin’s Isoconversional Method as a Universal Approach. Thermochim. Acta 2021, 696, 178822. [Google Scholar] [CrossRef]
- Nakamura, K.; Takayanagi, T.; Sato, S. A Modified Arrhenius Equation. Chem. Phys. Lett. 1989, 160, 295–298. [Google Scholar] [CrossRef]
- Aquilanti, V.; Mundim, K.C.; Elango, M.; Kleijn, S.; Kasai, T. Temperature Dependence of Chemical and Biophysical Rate Processes: Phenomenological Approach to Deviations from Arrhenius Law. Chem. Phys. Lett. 2010, 498, 209–213. [Google Scholar] [CrossRef]
- Silva, V.H.C.; Aquilanti, V.; De Oliveira, H.C.B.; Mundim, K.C. Uniform Description of Non-Arrhenius Temperature Dependence of Reaction Rates, and a Heuristic Criterion for Quantum Tunneling vs Classical Non-Extensive Distribution. Chem. Phys. Lett. 2013, 590, 201–207. [Google Scholar] [CrossRef]
- Carvalho-Silva, V.H.; Aquilanti, V.; De Oliveira, H.C.B.; Mundim, K.C. Deformed Transition-State Theory: Deviation from Arrhenius Behavior and Application to Bimolecular Hydrogen Transfer Reaction Rates in the Tunneling Regime. J. Comput. Chem. 2017, 38, 178–188. [Google Scholar] [CrossRef]
- Carvalho-Silva, V.H.; Coutinho, N.D.; Aquilanti, V. Temperature Dependence of Rate Processes Beyond Arrhenius and Eyring: Activation and Transitivity. Front. Chem. 2019, 7, 380. [Google Scholar] [CrossRef]
- Carvalho-Silva, V.H.; Coutinho, N.D.; Aquilanti, V. From the Kinetic Theory of Gases to the Kinetics of Rate Processes: On the Verge of the Thermodynamic and Kinetic Limits. Molecules 2020, 25, 2098. [Google Scholar] [CrossRef] [PubMed]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Eyring, H. The Theory of Absolute Reaction Rates. Trans. Faraday Soc. 1938, 34, 41. [Google Scholar] [CrossRef]
- Wigner, E. The Transition State Method. Trans. Faraday Soc. 1938, 34, 29. [Google Scholar] [CrossRef]
- Henderson, D. My Friend, Henry Eyring. J. Phys. Chem. 1983, 87, 2638–2656. [Google Scholar] [CrossRef]
- Vyazovkin, S. Misinterpretation of Thermodynamic Parameters Evaluated from Activation Energy and Preexponential Factor Determined in Thermal Analysis Experiments. Thermo 2024, 4, 373–381. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. On the Introduction of Thermodynamic Variables into Reaction Kinetics. Trans. Faraday Soc. 1937, 33, 448. [Google Scholar] [CrossRef]
- Cai, J.; Li, Z. First Principle-Based Rate Equation Theory for the Calcination Kinetics of CaCO3 in Calcium Looping. Energy Fuels 2024, 38, 8145–8156. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current Status of Transition-State Theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Shannon, R.D. Activated Complex Theory Applied to the Thermal Decomposition of Solids. Trans. Faraday Soc. 1964, 60, 1902. [Google Scholar] [CrossRef]
- Garrett, B.C. Perspective on “The Transition State Method”. In Theoretical Chemistry Accounts; Cramer, C.J., Truhlar, D.G., Eds.; Springer: Berlin/Heidelberg, Germany, 2000; pp. 200–204. ISBN 978-3-540-67867-0. [Google Scholar]
- Conner, W. A General Explanation for the Compensation Effect: The Relationship between $Delta;S? And Activation Energy. J. Catal. 1982, 78, 238–246. [Google Scholar] [CrossRef]
- Laidler, K.J.; King, M.C. Development of Transition-State Theory. J. Phys. Chem. 1983, 87, 2657–2664. [Google Scholar] [CrossRef]
- Hänggi, P.; Talkner, P.; Borkovec, M. Reaction-Rate Theory: Fifty Years after Kramers. Rev. Mod. Phys. 1990, 62, 251–341. [Google Scholar] [CrossRef]
- Topaler, M.; Makri, N. Quantum Rates for a Double Well Coupled to a Dissipative Bath: Accurate Path Integral Results and Comparison with Approximate Theories. J. Chem. Phys. 1994, 101, 7500–7519. [Google Scholar] [CrossRef]
- Yelon, A.; Movaghar, B.; Crandall, R.S. Multi-Excitation Entropy: Its Role in Thermodynamics and Kinetics. Rep. Prog. Phys. 2006, 69, 1145–1194. [Google Scholar] [CrossRef]
- Yelon, A.; Sacher, E.; Linert, W. Multi-Excitation Entropy, Entropy–Enthalpy Relations, and Their Impact on Catalysis. Catal. Lett. 2011, 141, 954–957. [Google Scholar] [CrossRef]
- Yin, C.; Du, J. The Power-Law Reaction Rate Coefficient for an Elementary Bimolecular Reaction. Phys. A Stat. Mech. Its Appl. 2014, 395, 416–424. [Google Scholar] [CrossRef]
- Sanjeev, R.; Padmavathi, A.D.; Jagannadham, V. The ‘Yard Stick’ to Interpret the Entropy of Activation in Chemical Kinetics: A Physical-Organic Chemistry Exercise. World J. Chem. Educ. 2018, 6, 78–81. [Google Scholar] [CrossRef]
- Sapunov, V.N.; Saveljev, E.A.; Voronov, M.S.; Valtiner, M.; Linert, W. The Basic Theorem of Temperature-Dependent Processes. Thermo 2021, 1, 45–60. [Google Scholar] [CrossRef]
- Vyazovkin, S. Determining Preexponential Factor in Model-Free Kinetic Methods: How and Why? Molecules 2021, 26, 3077. [Google Scholar] [CrossRef] [PubMed]
- Galukhin, A.; Nikolaev, I.; Nosov, R.; Islamov, D.; Vyazovkin, S. Solvent-Induced Changes in the Reactivity of Tricyanate Esters Undergoing Thermal Polymerization. Polym. Chem. 2021, 12, 6179–6187. [Google Scholar] [CrossRef]
- Brill, T.B.; Gongwer, P.E.; Williams, G.K. Thermal Decomposition of Energetic Materials. 66. Kinetic Compensation Effects in HMX, RDX, and NTO. J. Phys. Chem. 1994, 98, 12242–12247. [Google Scholar] [CrossRef]
- Linert, W. The Isokinetic Relationship. X. Experimental Examples to the Relationship between Isokinetic Temperatures and Active Heat Bath Frequencies. Chem. Phys. 1989, 129, 381–393. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Linert, W. Thermally Induced Reactions of Solids: Isokinetic Relationships of Non-Isothermal Systems. Int. Rev. Phys. Chem. 1995, 14, 355–369. [Google Scholar] [CrossRef]
- Skrdla, P.; Simon, P. Isoconversional Methods of Thermal Analysis Are Incompatible with Turnbull-Fisher Kinetics: Case Study of Crystallization of Piperine from the Melt. ChemRxiv 2025. [Google Scholar] [CrossRef]
- Vyazovkin, S. Isoconversional Kinetics of Thermally Stimulated Processes; Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-14174-9. [Google Scholar]
- Alvares, C.M.S.; Deffrennes, G.; Pisch, A.; Jakse, N. Thermodynamics and Structural Properties of CaO: A Molecular Dynamics Simulation Study. J. Chem. Phys. 2020, 152, 084503. [Google Scholar] [CrossRef] [PubMed]
- Mianowski, A.; Baraniec-Mazurek, I.; Bigda, R. Some Remarks on Equilibrium State in Dynamic Condition. J. Therm. Anal. Calorim. 2012, 107, 1155–1165. [Google Scholar] [CrossRef]
- Steinberg, M. Production of Hydrogen and Methanol from Natural Gas with Reduced CO2 Emission. Int. J. Hydrogen Energy 1998, 23, 419–425. [Google Scholar] [CrossRef]
- Paxman, D.; Trottier, S.; Flynn, M.R.; Kostiuk, L.; Secanell, M. Experimental and Numerical Analysis of a Methane Thermal Decomposition Reactor. Int. J. Hydrogen Energy 2017, 42, 25166–25184. [Google Scholar] [CrossRef]
- Grabke, H.-J. Die Kinetik der Entkohlung und Aufkohlung von γ-Eisen in Methan-Wasserstoff-Gemischen. Ber. Bunsenges. Phys. Chem. 1965, 69, 409–414. [Google Scholar] [CrossRef]
- Kang, D.; Rahimi, N.; Gordon, M.J.; Metiu, H.; McFarland, E.W. Catalytic Methane Pyrolysis in Molten MnCl2-KCl. Appl. Catal. B Environ. 2019, 254, 659–666. [Google Scholar] [CrossRef]
- Msheik, M.; Rodat, S.; Abanades, S. Experimental Comparison of Solar Methane Pyrolysis in Gas-Phase and Molten-Tin Bubbling Tubular Reactors. Energy 2022, 260, 124943. [Google Scholar] [CrossRef]
- Sorcar, S.; Rosen, B.A. Methane Pyrolysis Using a Multiphase Molten Metal Reactor. ACS Catal. 2023, 13, 10161–10166. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Sadykov, A.R.; Melnikov, I.N.; Fomenkov, I.V.; Fershtat, L.L.; Pivkina, A.N.; Muravyev, N.V. Too Fast and Too Furious: Thermoanalytical and Quantum Chemical Study of the Thermal Stability of 4,4′-Dinitro-3,3′-Diazenofuroxan. Phys. Chem. Chem. Phys. 2024, 26, 29541–29551. [Google Scholar] [CrossRef]
- Lei, J.; Yang, L.; Wang, Y.; Zhao, D. Pyrolysis Kinetics, Mechanism and Thermodynamics of Peanut Shell Based on Gaussian Function Deconvolution. Heliyon 2025, 11, e42800. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 116.4 | 30.6 | 6.417 | 113.63 |
| 147.5 | 40.8 | 91.220 | 108.10 |
| 292.9 | 80.3 | 419.623 | 111.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mianowski, A.; Szul, M. Practical Aspects of the Analysis of Thermal Dissociation and Pyrolysis Processes in Terms of Transition State Theory. Energies 2025, 18, 2619. https://doi.org/10.3390/en18102619
Mianowski A, Szul M. Practical Aspects of the Analysis of Thermal Dissociation and Pyrolysis Processes in Terms of Transition State Theory. Energies. 2025; 18(10):2619. https://doi.org/10.3390/en18102619
Chicago/Turabian StyleMianowski, Andrzej, and Mateusz Szul. 2025. "Practical Aspects of the Analysis of Thermal Dissociation and Pyrolysis Processes in Terms of Transition State Theory" Energies 18, no. 10: 2619. https://doi.org/10.3390/en18102619
APA StyleMianowski, A., & Szul, M. (2025). Practical Aspects of the Analysis of Thermal Dissociation and Pyrolysis Processes in Terms of Transition State Theory. Energies, 18(10), 2619. https://doi.org/10.3390/en18102619