Abstract
We have simulated the effect of changing the end groups in BTP core with five organic units of 1,3-Indandione (IN), 2-thioxothiazolidin-4-one (Rhodanine), propanedinitrile (Malononitrile), (2-(6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene)malononitrile) (CPTCN) and 2-(3-oxo-2,3-dihydroinden-1-ylidene (IC), and two halogenated units of (4F) IC and (4Cl) IC on the optical and photovoltaic properties of the BTP DA’D core molecular unit. Thus modified, seven molecular structures are considered and their optical properties, including HOMO and LUMO energies and absorption spectra are simulated in this paper. On the basis of HOMO and LUMO energies, it is found that two of the seven molecules, BTP-IN and BTP-Rhodanine, can act as donors and the other four, BTP-(4F) IC, BTP-(4Cl) IC, BTP-CPTCN and BTP-IC, as acceptors in designing bulk heterojunction (BHJ) organic solar cells (OSCs). Using these combinations of donors and acceptors in the active layer, eight BHJ OSCs, such as BTP-IN: BTP-(4F) IC, BTP-IN: BTP-(4Cl) IC, BTP-IN: BTP-CPTCN, BTP-IN: BTP-IC, BTP-Rhodanine: BTP-(4F) IC, BTP-Rhodanine: BTP-(4Cl) IC, BTP-Rhodanine: BTP-CPTCN and BTP-Rhodanine: BTP-IC, are designed, and their photovoltaic performance is simulated. The photovoltaic parameters , and FF for all eight BHJ OSCs and their power conversion efficiency (PCE) are simulated. It is found that the BHJ OSC of the BTP-IN: BTP-CPTCN donor–acceptor blend gives the highest PCE (14.73%) and that of BTP-Rhodanine: BTP-(4F) IC gives the lowest PCE (12.07%). These results offer promising prospects for the fabrication of high-efficiency BHJ OSCs with the blend of both donor and acceptor based on the same core structure.
1. Introduction
The conversion of solar energy into electricity using organic solar cells (OSCs) is key to achieving high renewable energy targets and cut down on CO2 emissions emanating from fossil-based energy generation sources [1]. The use of room-temperature-based chemical processes, their lower cost, and efficient manufacturing are the key advantages in advancing the use of OSCs [2]. As organic semiconductors are low dielectric constant materials, the absorption of solar photons in OSCs first generates Frenkel excitons, which are bound excited electron and hole pairs. These excitons need to be dissociated into free electron and hole pairs and move to their respective electrodes to generate a current in OSCs [3]. Accordingly, the power conversion efficiency (PCE) of the OSCs is intricately linked to the efficiency of photon absorption and subsequent generation and dissociation of excitons [4]. In the development of OSCs, the state-of-the-art structure is regarded to be the bulk-heterojunction (BHJ) in which the donor (D) and acceptor (A) organic materials are blended together in the active layer, which is then sandwiched between the electrodes. By blending D and A materials, the overall absorption of solar photons is enhanced due to complementary absorption in both D and A components [5]. Also, the blending of D and A leads to the formation of multiple D–A interfaces within the active layer, which introduces energy offsets due to differences in ionization potential and electron affinities of the donor and acceptor materials and aids in the dissociation of excitons [6]. The dissociated free electron and hole pairs are then separated and transported to their respective electrodes due to the electric field generated between the electrodes by the difference in their work functions [7].
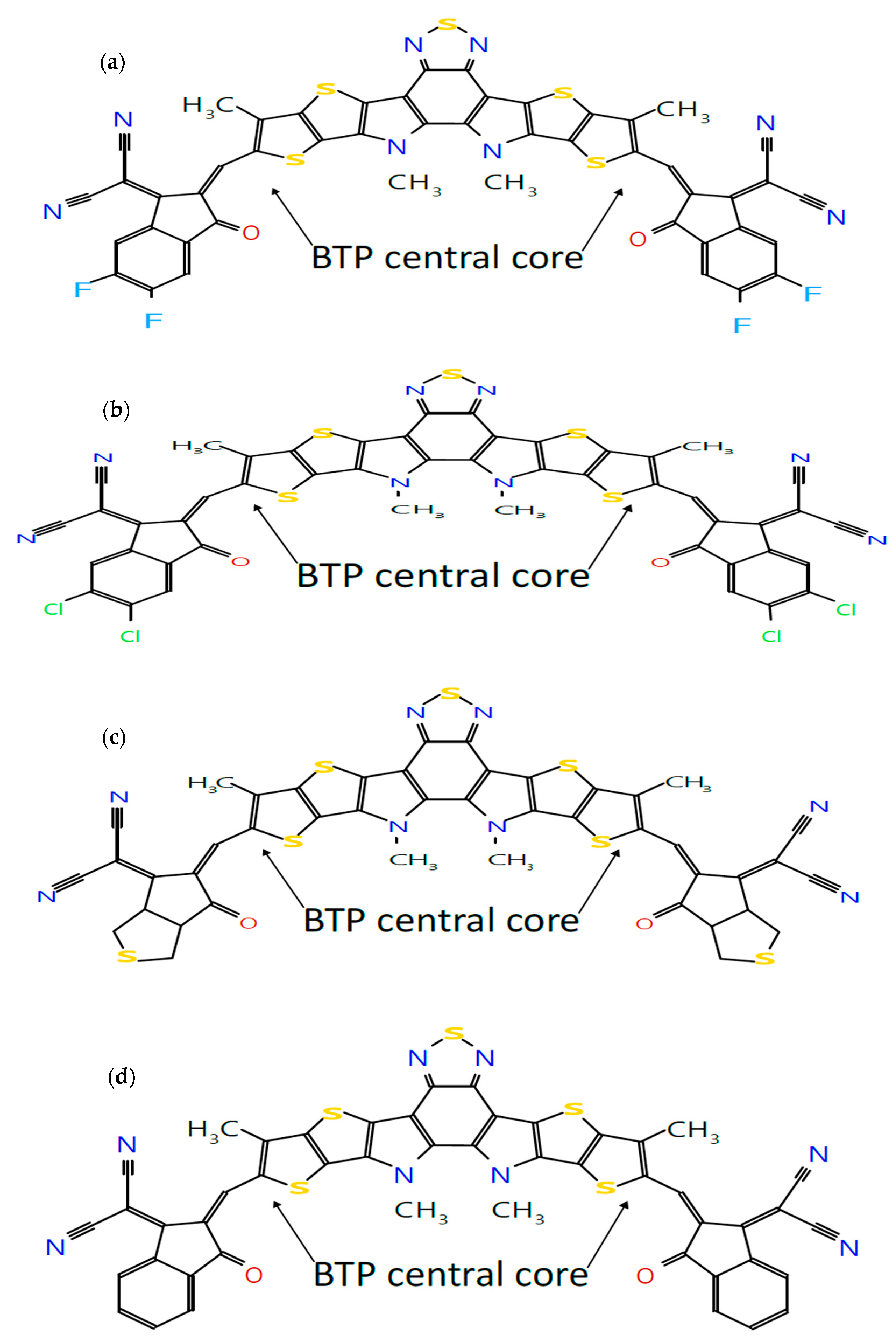
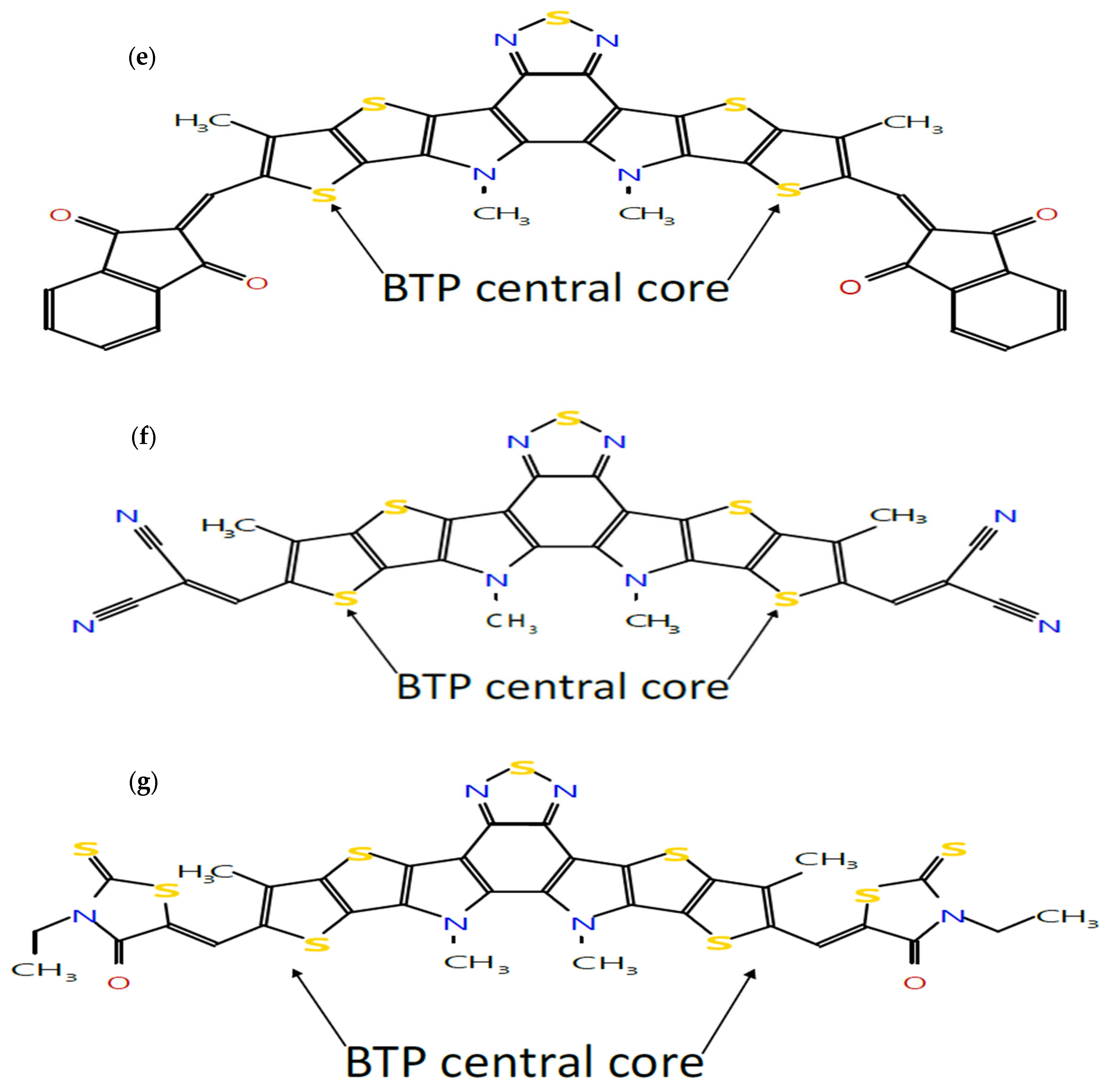
BHJ OSCs with the active layer consisting of a blend of an organic donor material and a fullerene-based organic acceptor material, commonly used PCBM, have been studied extensively and reached a PCE of about 8% [8]. This low PCE is mainly due to the limited light-absorbing capabilities of fullerene-based acceptor materials. Despite these limitations, fullerene-based acceptors have remained the focus of extensive research efforts due to their exceptional charge transport properties [9,10]. For enhancing the absorption, non-fullerene acceptor (NFA) materials, primarily based on small molecules, have now emerged as promising alternatives. These NFA-based OSCs offer a range of advantages, including broad absorption profiles, the tunability of energy levels, high charge carrier mobility, enhanced open-circuit voltage (), and improved stability [11]. The molecular engineering of non-fullerene small-molecule acceptors (SMA) plays a key role in enhancing the performance of organic solar cells. An effective strategy is to introduce functional groups into SMA end groups to tune the electronic and morphological properties of non-fullerene SMAs [12]. The development of small molecule NFA-based BHJ OSCs has heralded a significant breakthrough and achieved a record efficiency of more than 15% [13]. It has recently been reported [14] that an NFA with a core unit (D) connected with two end groups (A) in the form of acceptor–donor–acceptor (A-D-A) appears to be one of the most successful design strategies to achieve high PCE in OSCs. This classification as A-D-A-type molecules is derived from their electron–donor central backbone flanked on both sides by electron-withdrawing groups. Following [14], this concept has been implemented in this paper to design seven NF molecular structures with one central core unit of dithienothiophen[3,2-b]-pyrrolobenzothiadiazole (BTP) [15] (D) and seven end groups (A). The seven end groups have been selected as follows: The fluorinated (4F) IC is selected to form Y6, a well-known NFA, and the chlorinated (4Cl) IC, previously reported as Y7, is selected as another halogenated end group [14]. The other five end groups are rhodanine, IC, IN, CPCTN and malonitrile which have been used in [16], where the effect of different end groups on the calamitic-type small molecules NFAs with a structural configuration of A-π-D-π-A has been studied. Thus formed, the seven NF molecular structures with the central core unit of BTP are shown in Figure 1. Thus, we have considered seven end groups on the central core unit of BTP as discussed below. It is found that such designs of donor and acceptor molecules enhance the charge carrier mobility and minimize the typical batch-to-batch variations in molecular weight, polydispersity, and purity often associated with polymer-based donors and acceptors used in the active layer of OSCs [17]. The molecular configuration of the BTP core involves the insertion of an electron-deficient (A′) moiety into the middle of the central D conjugated building block, forming a fused DA’D backbone [18,19]. The development and use of such A-DA’D-A molecules has been found to enhance PCE exceeding 18% [18,20].
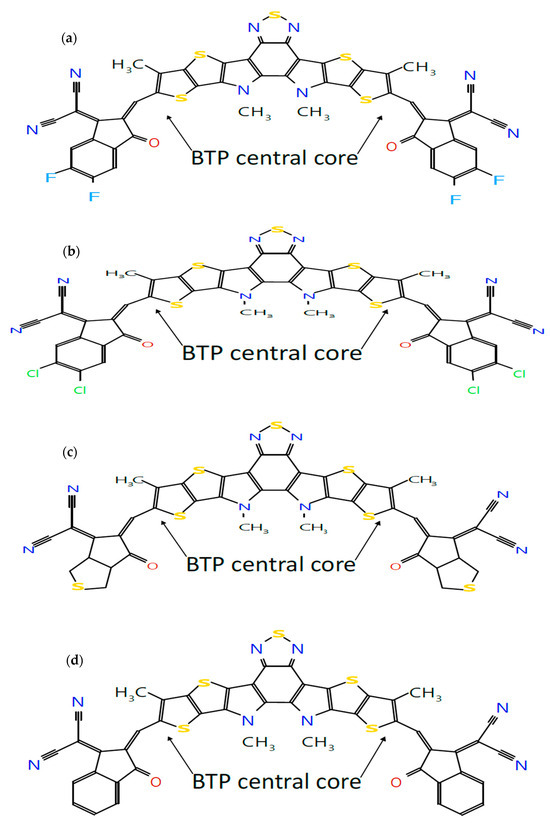
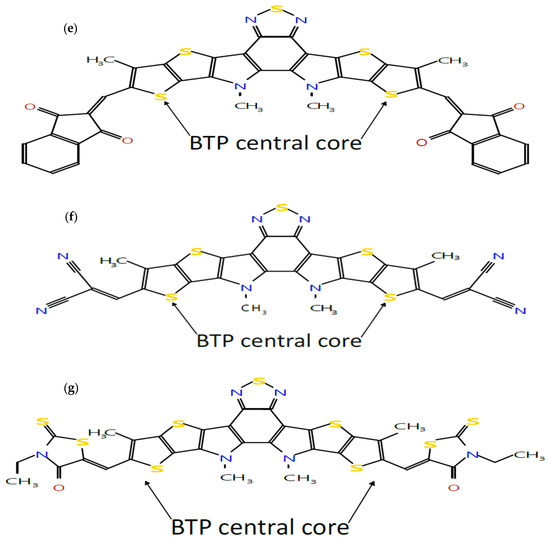
Figure 1.
Molecular structures of seven A-DA’D-A-type donor and acceptor molecules: (a) BTP-(4F) IC, (b) BTP-(4Cl) IC, (c) BTP-CPTCN, (d) BTP-IC, (e) BTP-IN, (f) BTP-Malononitrile and (g) BTP-Rhodanine, sketched using the Molsketch software (version 0.7.2).
Since the initial breakthrough in achieving high PCE with the use of A-DA’D-A-type acceptor materials, there has been a notable gap in the literature in this direction. Furthermore, there is a distinct absence of studies exploring all-small molecule-based OSCs that utilize derivatives of the same central core unit of a molecule. Therefore, the primary focus of this paper is to delve into a comprehensive study of the quantum chemical, optoelectronic and photovoltaic properties of the seven derivatives of the BTP central core unit shown in Figure 1. As described above, these seven derivatives emerge from the strategic substitution of seven end groups, including, 1,3-Indandione (IN), propanedinitrile (Malononitrile), 2-thioxothiazolidin-4-one (Rhodanine), (4F) IC, (4Cl) IC, (2-(6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene)malononitrile) (CPTCN), and 2-(3-oxo-2,3-dihydroinden-1- ylidene (IC), onto the central core unit of BTP. By analyzing the absorption spectra of these seven derivatives, we have found that four of them can be used as acceptors and three as donors in the BHJ blend of OSCs. Such modifications in the molecular structure may therefore be regarded as very innovative and economical ways of synthesizing donor and acceptor molecules for BHJ OSCs.
A donor (D) molecule usually has energetically higher frontier molecular orbitals, i.e., highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), in comparison with those of acceptor (A) materials. An efficient acceptor molecule is expected to have a high electron-accepting ability, appropriate HOMO and LUMO energy levels, and high absorption [21,22]. The selected units in this research are efficient electron-withdrawing groups with extended conjugation, which improves the overall absorption and charge transport properties [16]. Such a modification in molecules by combining a core and end groups to form a non-fullerene-based acceptor Y7 and used in the fabrication of BHJ OSCs with the two donors, PTQ10 and PM6, has been recently reported [14]. It may be noted that the acceptor Y7 is the same as one of the acceptors BTP-(4Cl) IC considered in this paper.
However, despite the remarkable PCE enhancement achieved by A-DA’D-A-type molecules, the open-circuit voltage () of such OSCs is still found to be relatively low [23,24]. Achieving higher necessitates a careful selection of donor and acceptor materials with appropriate energies of their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) because the energy difference between the LUMO of acceptor and HOMO of donor at the interface governs the attainable [25]. Also, the energy offsets between donor and acceptor materials at the interface is a crucial inherent property that affects the dissociation of excitons leading to charge separation and recombination and, hence, influences the achievable and [26]. In addition to these material-dependent properties that impose constraints on , various processing-related parameters further contribute to limiting . These include the purity of materials and the morphology of the active layer, as well as the quality of the interface between donor and acceptor materials, and the thermal processing conditions during device fabrication [27]. The effect of the interplay of material and processing parameters on necessitates careful consideration in optimizing the inherent material properties prior to the fabrication of OSCs.
Following the procedures published in our earlier work [28,29] and using self-consistent field (SCF) Density Functional Theory (DFT) through the ORCA software (version 5.1), the ground state structure and energy are first optimized, and then, using Time-Dependent (TD) DFT through ORCA, the excited state energies and the absorption spectra in the UV-VIS region are calculated for all seven molecules.
Finally, by selecting the suitable donors and acceptors, we have designed eight BHJ OSCs whose photovoltaic performance is simulated and analyzed. The exploration of distinctive donor and acceptor configurations as presented here unlocks new venues in the field of organic photovoltaic in developing A-DA’D-A-type donor and acceptor molecules for fabricating more efficient and chemically economical BHJ OSCs.
2. Computation and Results
2.1. Computational Procedure
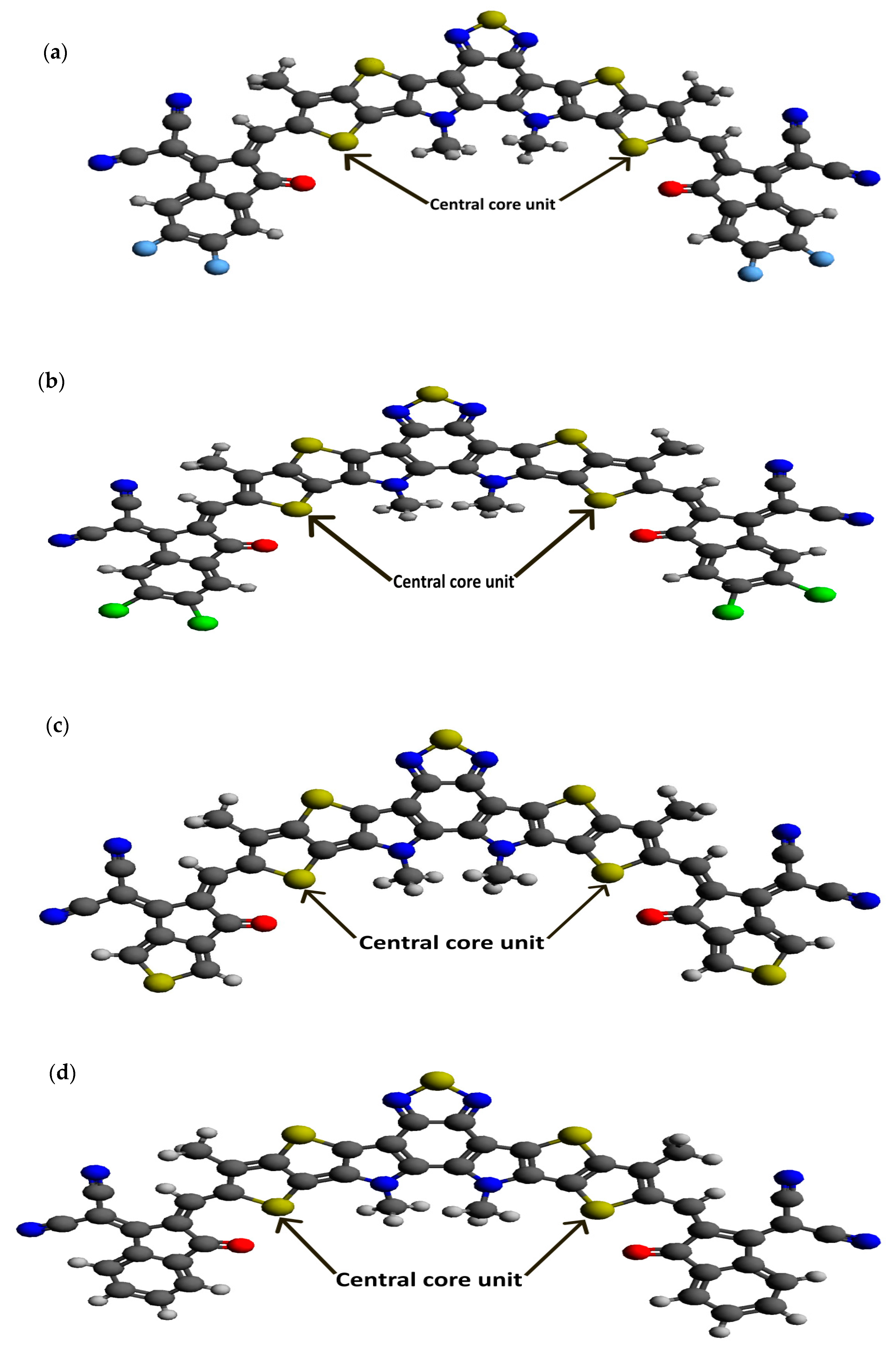
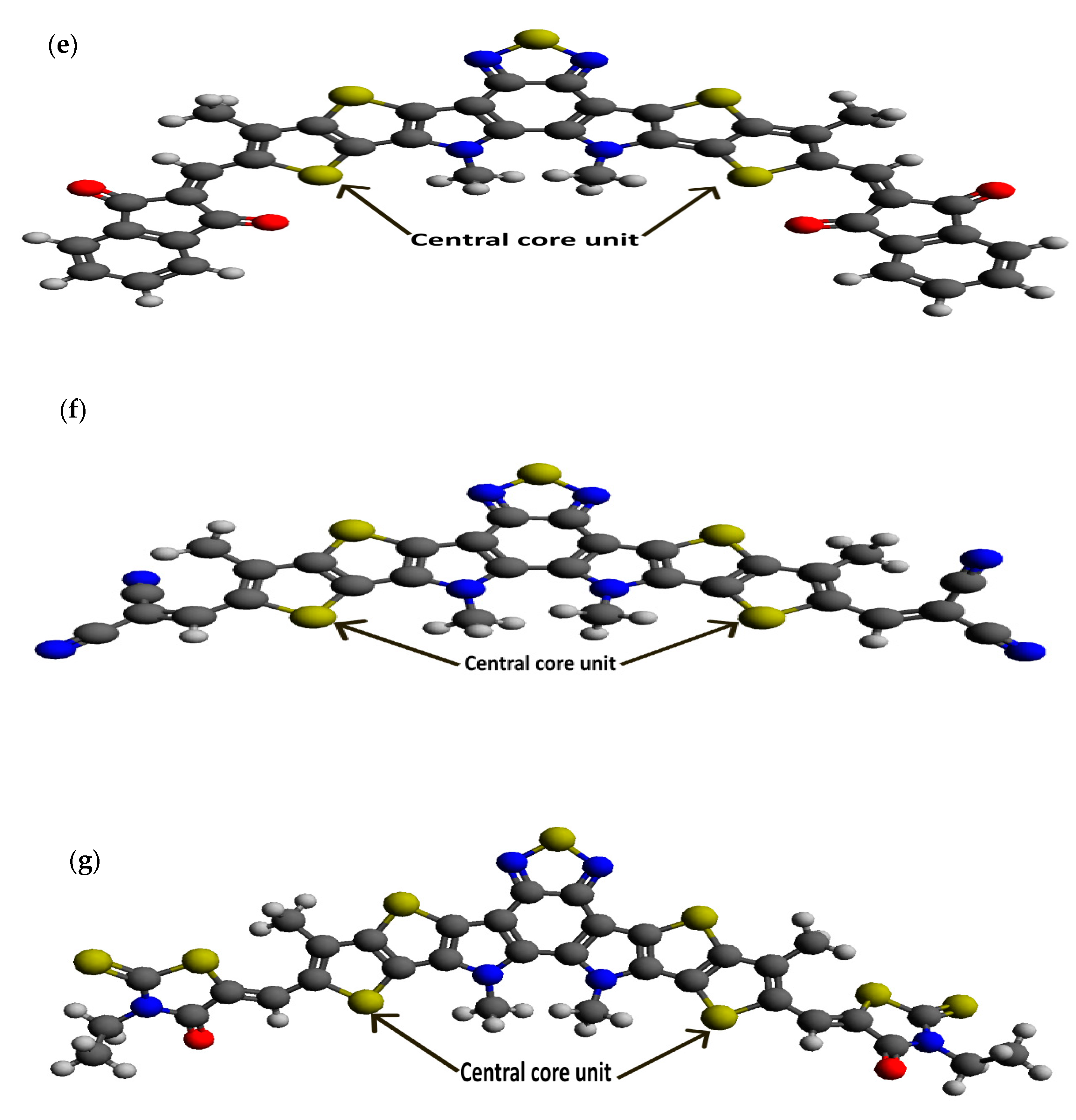
Following our recent simulation work [28,29], the molecular structures are initially designed using Avogadro software (version 1.98.1). The seven distinct molecules, namely (a) BTP-(4F) IC, (b) BTP-(4Cl) IC, (c) BTP-CPTCN, (d) BTP-IC, (e) BTP-IN, (f) BTP-Malononitrile, and (g) BTP-Rhodanine, are subjected to a comprehensive simulation process to attain the optimized ground state equilibrium structures, as shown in Figure 2. The optimization process uses the Avogadro software and starts with the initial molecular geometries shown in Figure 1 for each of the seven derivatives. It may be noted that the initial structures of the seven molecules shown in Figure 1, created using Molsketch software, may visually differ significantly from their respective optimized structures shown in Figure 2, generated through the Avogadro software. The main difference is that Figure 1 shows the 2D sketches of the seven initial molecular structures, whereas Figure 2 illustrates their 3D optimized structures obtained using the Avogadro software.
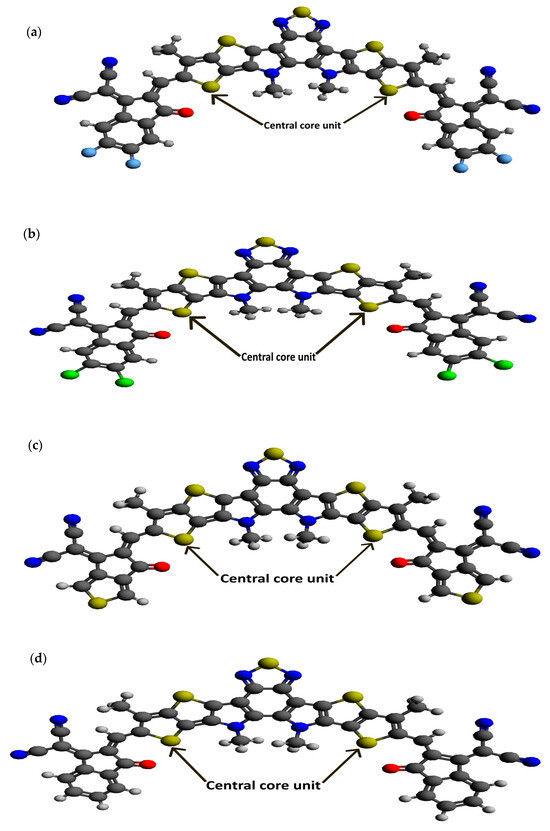
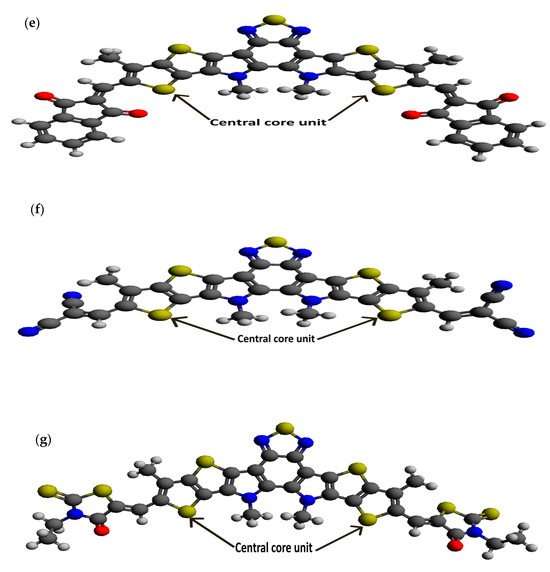
Figure 2.
Optimized ground state structures of the seven molecules: (a) BTP-(4F) IC, (b) BTP-(4Cl) IC, (c) BTP-CPTCN, (d) BTP-IC, (e) BTP-IN, (f) BTP-Malononitrile and (g) BTP-Rhodanine.
The Universal Force Field and the steepest descent method are employed for energy minimization, aiming to obtain improved geometries for subsequent iterations. To calculate the ground state equilibrium structures, the Kohn–Sham Density Functional Theory (DFT) implemented in Orca (version 5.1) [30] is employed. The ground state optimization involves the use of Becke, three-parameter, Lee–Yang–Parr exchange–correlation (B3LYP) functional [31,32] with def2-SV(P) and def2/J basis sets, including frequency calculations, to ensure the obtained structures correspond to the true minima. The self-consistent field (SCF) convergence criteria and print options are carefully chosen to balance the accuracy and computational efficiency. Using the optimized equilibrium ground state structures (Figure 2), we compute the equilibrium energies, single-point energies, and EHOMO and ELUMO, which are used for calculating the photovoltaic and quantum chemical parameters, as described in our earlier work [28,29]. Finally, TD-DFT calculations implemented in Orca (version 5.1) [30], are performed to calculate the electronic transitions in each molecule. The B3LYP functional is again employed with the def2-SVP and def2/J basis sets, utilizing the RIJCOSX approximation and a tight SCF convergence. The TD-DFT calculations includes calculating 30 excited states, the maximum dimension of 200 for the Davidson diagonalization space, and the exclusion of the Tamm–Dancoff Approximation (tda false).
Using the calculated energies and of each of the optimized structures, the bandgap (), ionization potential (IP), electron affinity (EA), chemical potential (), electronegativity (χ), softness (S) and hardness (η) are obtained as follows [28,29,33]:
The reorganization energy of each molecule is calculated as follows [28,29]:
where and are the reorganization energies of electrons (e) and holes (h), respectively, and these are expressed as follows:
where is the single-point energy (SPE) of a molecule, and is the SPE of the corresponding anion state. is the SPE of anionic state of an anion molecule, is the SPE of an anion, is SPE of a cation, is the SPE of the corresponding cation state, and is the SPE of a cation.
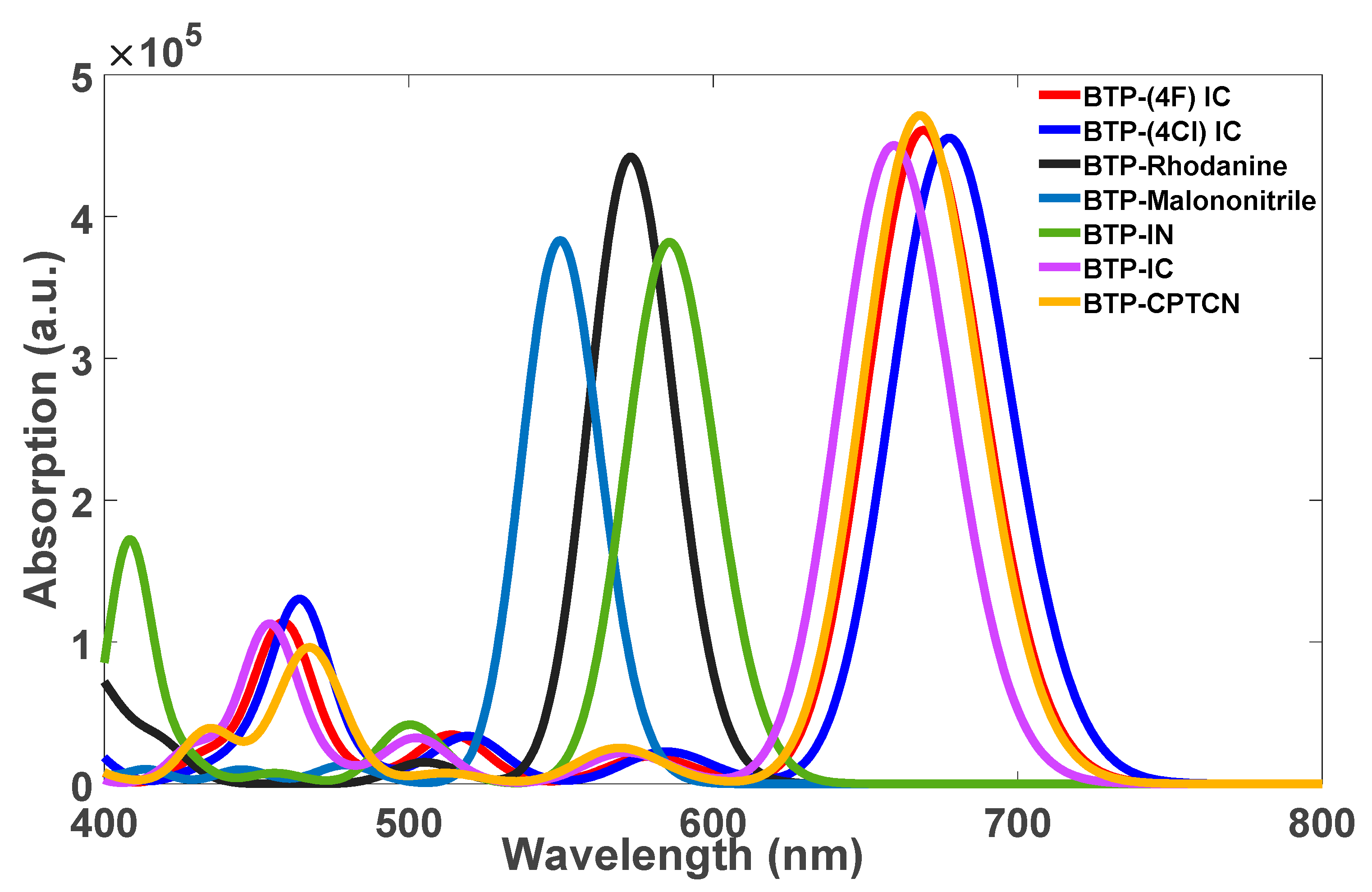
The calculated energies of HOMO and LUMO and the corresponding band gap energies obtained from Equation (1) for all the seven molecules are listed in Table 1. Using Equations (2)–(9), other parameters are also calculated and listed in Table 2 for all seven molecules. The absorption spectra of all seven molecules were obtained by calculating 30 excited state transitions, each corresponding to the absorption of light at distinct wavelengths using TD-DFT at the B3LYP level of theory; the resulting excitations along with their relative intensities were then plotted using MATLAB, as illustrated in Figure 3.

Table 1.
DFT calculated energies of HOMO (EHOMO), LUMO (ELUMO) and bandgap (Eg) of the seven molecules. Also listed are a few available measured experimental values for comparison.
Table 2.
Calculated values of IP (Equation (2)), EA (Equation (3)), μ (Equation (4)), χ (Equation (5), η (Equation (4)), REe (Equation (8)), REh (Equation (9)).
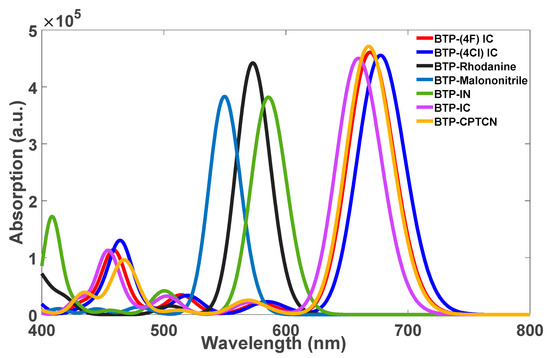
Figure 3.
TD-DFT-calculated absorption spectra of the optimized structures (shown in Figure 2) of the seven molecules: BTP-IC, BTP-(4F) IC, BTP-(4Cl) IC, BTP-IN, BTP-CPTCN, BTP-Malononitrile and BTP-Rhodanine.
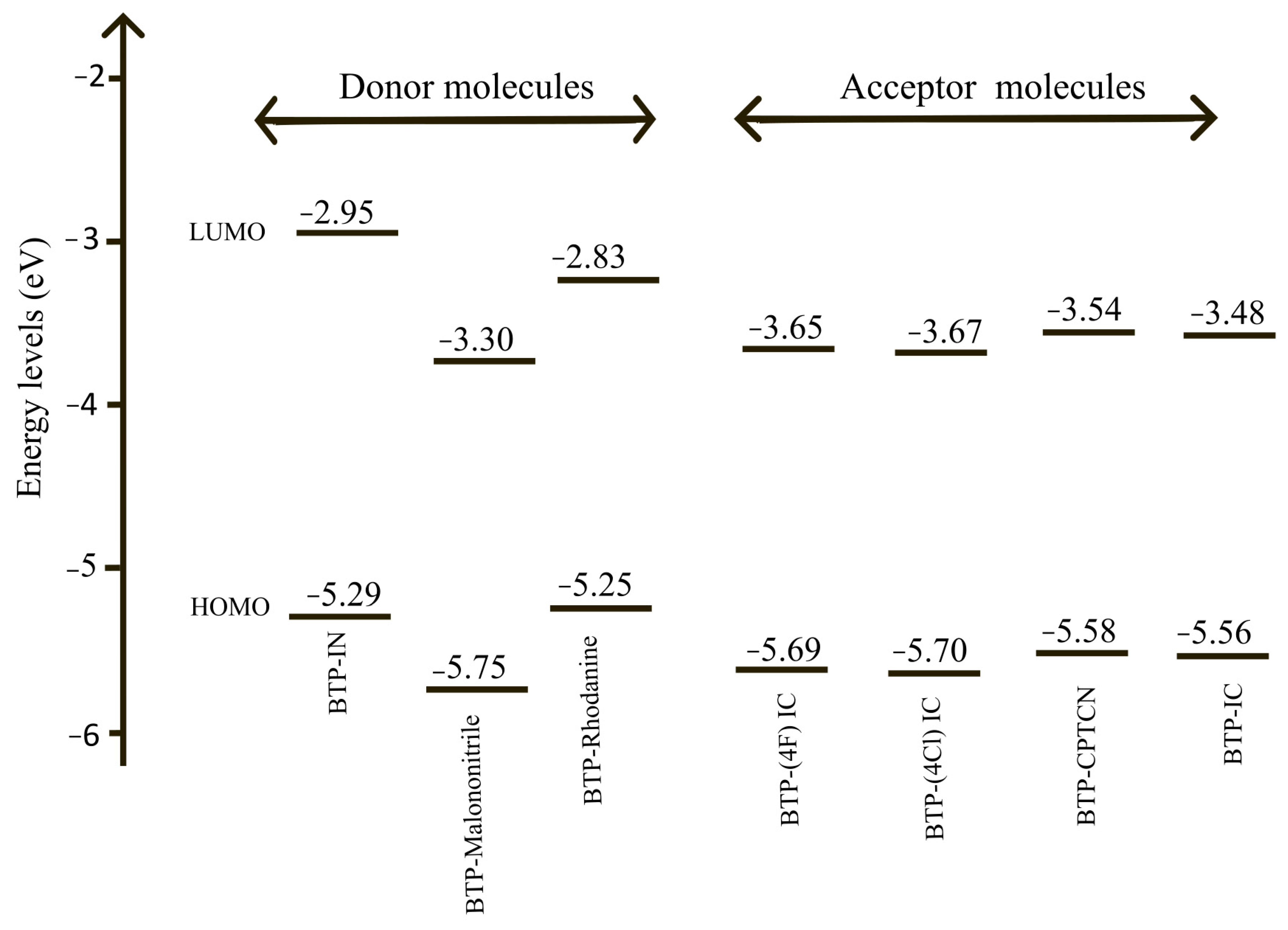
The energy level diagrams of the HOMO and LUMO energies for all the seven molecules listed in Table 1 are shown in Figure 4.
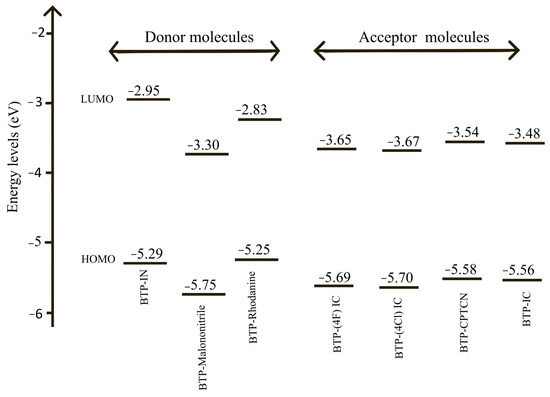
Figure 4.
Energy level diagrams of HOMO and LUMO energies of all seven molecules; three donor molecules: BTP-IN, BTP-Malononitrile and BTP-Rhodanine; and four acceptor molecules: BTP-(4F) IC, BTP-(4Cl) IC, BTP-CPTCN and BTP-IC.
2.2. Optical Properties
In addition to calculating the absorption spectra shown in Figure 3, it may be desirable to calculate the singlet (ES) and triplet (ET) excitation energies, corresponding binding energies, and , oscillator strength (f) and light-harvesting efficiency (LHE). The exciton binding energies and LHE are calculated using Equation (10) [29,36] and Equation (11) [37], respectively, as follows:
where is the excitation energy of singlet or triplet excitons.
The calculated energies ES, ET, and , oscillator strength f and LHE for all seven molecules are listed in Table 3.
Table 3.
Calculated (Cal) singlet and triplet excitation energies, binding energy of singlet (BES) and triplet excitons (BET), oscillator strength (f) and light-harvesting efficiency (LHE).
2.3. Design of BHJ OSCs
According to the absorption spectra shown in Figure 3, three molecules of BTP-IN, BTP-Malononitrile and BTP-Rhodanine show absorption in the high energy range and four molecules of, BTP-(4F) IC, BTP-(4Cl) IC, BTP-CPTCN and BTP-IC, show absorption in the lower energy range. Accordingly, the first three molecules can act as donors and the other four as acceptors if blended in the active layer of OSCs designed using these seven molecules, leading to the total possibility of 12 BHJ OSCs. However, according to Table 1 and Figure 4, only BTP-IN and BTP-Rhodanine have higher HOMO and LUMO energies compared to the four acceptor molecules of BTP-4F, BTP-4Cl, BTP-CPTCN and BTP-IC.
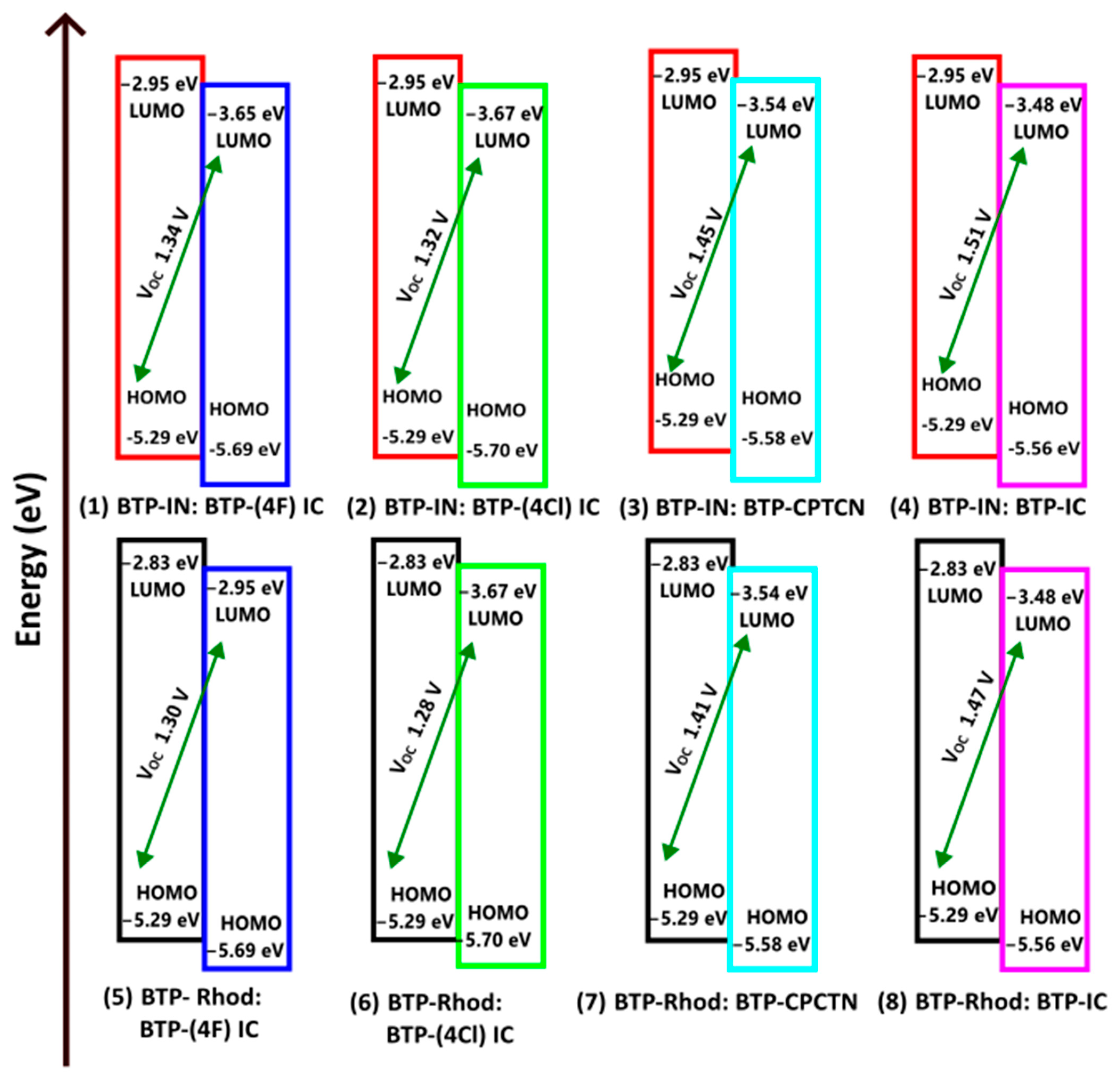
The possible third donor of BTP-Malononitrile has the lowest HOMO energy and, hence, it does not comply with the requirements to become a donor with the four selected acceptors shown in Figure 4. Accordingly, only BTP-IN and BTP-Rhodanine will act as donors and BTP-(4F) IC, BTP-(4Cl) IC, BTP-CPTCN and BTP-IC as acceptors if blended in the active layer of OSCs. It is therefore important not only to choose the donor and acceptor molecules based on the absorption spectra but also compare their HOMO and LUMO energies using the energy level diagram as shown in Figure 4. Thus, among the seven studied molecules, only two can act as donors and four as acceptors leading to the total possibility of designing eight BHJ OSCs. The energy level diagrams of the active layer of these eight BHJ OSCs are shown in Figure 5.
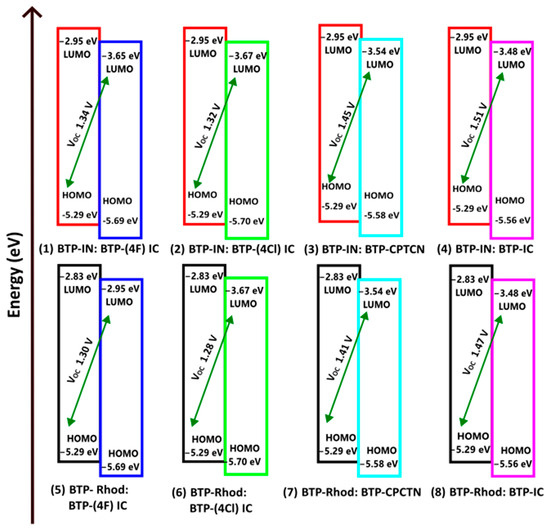
Figure 5.
Design of the active layer of eight BHJ OSCs using two donors of BTP-IN and BTP-Rhodanine and four acceptors of BTP-(4F) IC, BTP-(4Cl) IC and BTP-IC.
Thus, we have designed eight solar cells as shown in Figure 5. For an efficient dissociation of the charge transfer (CT) excitons formed at the interfaces, the energy differences in the HOMO and LUMO of the donor and acceptor at the interfaces, and are required to be greater than or equal to the binding energy of CT excitons (), as follows [38]:
Both energy offsets, and , for the eight selected donor and acceptor pairs have been calculated and listed in Table 4, which shows that the condition in Equation (12) is satisfied by all eight selected BHJ OSCs.
Table 4.
Calculated values of , , FF, PCE, and .
2.4. Photovoltaic Properties
Here, we present the photovoltaic properties of the eight donor–acceptor molecular blends shown in Figure 5, which are (1) BTP-IN: BTP-(4F) IC, (2) BTP-IN: BTP-(4Cl) IC, (3) BTP-IN: BTP-CPTCN, (4) BTP-IN: BTP-IC, (5) BTP-Rhodanine: BTP-(4F) IC, (6) BTP-Rhodanine: BTP-(4Cl) IC, (7) BTP-Rhodanine: BTP-CPTCN and (8) BTP-Rhodanine: BTP-IC. The photovoltaic properties are then calculated for all eight OSCs first; , is calculated as follows [39]:
where and are the energies of the acceptors’ LUMO and donors’ HOMO, respectively. Following our previous work [28,29], we have calculated for all eight BHJ OSCs shown in Figure 5. The fill factor (FF) is calculated as follows [28,29,40]:
and finally, the PCE of each OSC is calculated as follows:
The , , FF and PCE thus calculated for all eight OSCs are given in Table 4. Accordingly, the OSC of BTP-Rhodanine: BTP-(4F) IC is found to have the lowest PCE (12.07% (and that of BTP-IN: BTP-CPTCN has the highest PCE (14.73%).
3. Discussions
3.1. Optical Properties
The PCE of BHJ OSCs depends on the energy of HOMO and LUMO of the donors and acceptors. Therefore, the positioning of these orbitals on the energy scale governs the optimal operation of BHJ OSCs. Additionally, the positioning also determines whether a molecule should serve as a donor or an acceptor in the BHJ blend, as described above. To further facilitate the material selection and design of donor and acceptor molecules, one may also analyze the following reactivity parameters: IP, EA, η and μ = −χ of all the seven molecules. Both BTP-IN and BTP-Rhodanine have the lowest ionization potential (5.56 eV) (see Table 2) and, hence, can act as donors, and molecules BTP-(4F) IC, BTP-(4Cl) IC, BTP-CPTCN and BTP-IC have higher electron affinities and can act as acceptors. Likewise, both BTP-Rhodanine and BTP-IN appear to have the lowest μ value (−4.04 eV; see Table 2), indicating these to be an efficient electron donor.
The reorganization energy (RE) calculated from Equation (7) for all seven molecules as presented in Table 2 shows a general trend that all acceptor molecules have lower electron reorganization energy (REe) and higher hole reorganization energy (REh), and all donor molecules have higher REe and lower REh [41]. This is expected because donor molecules transport holes and, hence, have lower REh, and acceptor molecules transport electrons, and thus have lower REe.
The absorption spectra presented in Figure 3 reveal that among the seven molecules considered, BTP-IN, BTP-Malononitrile, and BTP-Rhodanine exhibit maximum absorption in the UV region at wavelengths 586 nm, 550 nm, and 573 nm, respectively. In contrast, the acceptor molecules BTP-(4F) IC, BTP-(4Cl) IC, BTP-CPTCN, and BTP-IC have maximum absorption in the visible region at wavelengths 669 nm, 678 nm, 668 nm, and 659 nm, respectively. Although the core unit is identical in all four acceptor molecules (see Figure 2), the absorption peaks appear at different wavelengths, which are due to different substitutions. The larger redshift in BTP-4Cl may be related to the stronger intermolecular π–π packing caused by the larger atomic size of Cl and larger length of the chlorine–carbon bond [35]. As all four acceptor molecules have absorption peaks at a lower energy than that of all three donor molecules, all can be regarded as efficient acceptors. However, only two, BTP-IN and BTP-Rhodanine, of the three donor molecules can be classified as be good donors because, as required, their HOMO and LUMO are above the HOMO and LUMO of all acceptors (see Figure 4). However, the third donor of BTP-Malononitrile has higher LUMO but much lower HOMO than the HOMO of all four acceptor molecules, which will not allow the holes’ transfer from acceptors to donors to form charge transfer excitons, leading to their dissociation. Therefore, a BHJ OSC fabricated using BTP-Malononitrile as the donor (with these four acceptors) can operate only through the transfer of electrons from donor to acceptor without the transfer of holes from acceptor to donor. As a result, such BHJ OSCs may be expected to be, at best, only half as efficient as those using the other two donors. This is the reason for the selection of eight possible highly efficient BHJ OSCs formed from the combination of two donors and four acceptors and excluding the donor BTP-Malononitrile.
It is to be noted that the light-harvesting efficiency (LHE) of all molecules in our study approaches unity at their respective absorption maxima, as indicated in Table 3, which makes them excellent candidates for the construction of an active layer blend.
The binding energy of singlet and triplet excitons is calculated and given in Table 3. An interesting trend emerges from the halogenation of BTP-IC, BTP-(4F) IC and BTP-(4Cl) IC, which reduces both their singlet and triplet exciton binding energies. According to Equation (12), the energy offsets at the D-A interfaces, , must be greater than the exciton binding energy for efficient generation of free charge carriers in BHJ OSCs. According to Table 4, the condition given in Equation (12) is met for all eight BHJ OSCs designed in this paper.
3.2. Photovoltaic Properties
According to Figure 5 and Table 4, all eight BHJ OSCs designed here are found to have high but the blend of BTP-IN: BTP-IC has the highest at 1.51 V. However, this OSC has a lower and, hence, its PCE is slightly lower than that of BTP-IN: BTP-CPTCN at a PCE of 14.73%. This agrees closely with the PCE of 14.5% achieved from the BHJ OSC fabricated with a blend of the acceptor BTP-(4Cl) IC, which is also called Y7, and two polymer donors, PTQ10 and PM6 [14].
There are other studies [12,22,42] which have simulated the photovoltaic properties of BHJ OSCs with such A-D-A-type NFAs. The acceptor designed using the fluorine end groups is found to have superior carrier mobility, molecular packing, other structural properties, electronic structure, exciton separation, and charge transport, which determine ultimate cell efficiency [12,22,42]. It is also found that NFAs with fluorination end groups, like in Y6-based A-DA’D-A cores, exhibit a PCE of up to 17.10%. Although our OSCs (see Table 4) designed from the halogenated acceptors BTP-IN: BTP-(4F) IC(Y6) and BTP-Rhodanine: BTP-(4F) IC (Y6), do not give the highest PCE, they are not far behind the highest obtained from BTP-CPTCN as acceptor. The development of the A-D-A-type NFA structures has also led to the discovery that symmetric A-D-A or A-DA’D-A NFAs exhibit better PCE [12], but the reason for this universality is not yet fully understood. The symmetry may, however, affect the acceptor properties, crystallinity, charge dynamics, and exciton transport. The results of our study may be considered to be consistent with this symmetry effect as all seven acceptors are symmetric and show similar efficiency in the range of 13 to 15%.
4. Conclusions
We have investigated the effect of five organic and two halogenated end groups, namely, 1,3-Indandione (IN), 2-thioxothiazolidin-4-one (Rhodanine), propanedinitrile (Malononitrile), (2-(6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene)malononitrile) (CPTCN) and 2-(3-oxo-2,3-dihydroinden-1-ylidene (IC), (4F) IC, and (4Cl) IC, on the photovoltaic properties of the BTP DA’D core structure. Our investigation has revealed that the substitution of IN, Rhodanine and Malononitrile end groups onto the BTP core makes these molecules capable of absorbing electromagnetic radiation in the higher energy region. Additionally, these three molecules exhibit higher LUMO energies, thus favoring their use as donors. In contrast, the absorption spectra of molecules with substituted end groups (4F) IC, (4Cl) IC, CPTCN, and IC occur in the lower energy spectral region, favoring their use as acceptors. Thus, among the seven studied molecules, three appear to act as donors and four as acceptors. However, the third possible donor of BTP-Malononitrile has the lowest energy of HOMO and, hence, it does not comply with the requirements to become a donor with the four selected acceptors. A BHJ OSC fabricated using BTP-Malononitrile as the donor can only operate through the electron transfer from donor to acceptor but no hole transfer from acceptor to donor. As a result, such an OSC can be expected to be half efficient at best and, hence, it has been excluded from the BHJ OSC designs, and only eight BHJ OSCs with the two donors and four acceptors combinations are designed.
The designed BHJ OSC with the active layer of BTP-IN: BTP-CPTCN has revealed the highest PCE (14.73%), while BTP-Rhodanine: BTP-(4F) IC exhibited the lowest PCE (12.07%). These findings indicate promising prospects for fabricating high-efficiency BHJ OSCs by blending both donor and acceptor components based on the same core structure. Finally, it is expected that this strategy of employing an identical core structure for both donor and acceptor components offers the potential of synthesizing these molecules for the fabrication of highly efficient BHJ OSCs.
Author Contributions
Conceptualization, D.D.Y.S.; Methodology, D.D.Y.S.; Software, D.D.Y.S., K.S.R., H.M.-R. and D.O.; Investigation, D.D.Y.S.; Writing–original draft, D.D.Y.S.; Writing–review & editing, K.S.R., H.M.-R., D.O. and J.S.; Supervision, J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Obobisa, E.S. Achieving 1.5 °C and net-zero emissions target: The role of renewable energy and financial development. Renew. Energy 2022, 188, 967–985. [Google Scholar] [CrossRef]
- Chen, L.X. Organic Solar Cells: Recent Progress and Challenges. ACS Energy Lett. 2019, 4, 2537–2539. [Google Scholar] [CrossRef]
- Facchetti, A. Polymer donor–polymer acceptor (all-polymer) solar cells. Mater. Today 2013, 16, 123–132. [Google Scholar] [CrossRef]
- Ram, K.S.; Ompong, D.; Rad, H.M.; Setsoafia, D.D.Y.; Singh, J. An Alternative Approach to Simulate the Power Conversion Efficiency of Bulk Heterojunction Organic Solar Cells. Phys. Status Solidi A 2021, 218, 2000597. [Google Scholar] [CrossRef]
- Weng, K.; Ye, L.; Zhu, L.; Xu, J.; Zhou, J.; Feng, X.; Lu, G.; Tan, S.; Liu, F.; Sun, Y. Optimized active layer morphology toward efficient and polymer batch insensitive organic solar cells. Nat. Commun. 2020, 11, 2855. [Google Scholar] [CrossRef] [PubMed]
- Coropceanu, V.; Chen, X.-K.; Wang, T.; Zheng, Z.; Brédas, J.-L. Charge-transfer electronic states in organic solar cells. Nat. Rev. Mater. 2019, 4, 689–707. [Google Scholar] [CrossRef]
- Benanti, T.L.; Venkataraman, D. Organic Solar Cells: An Overview Focusing on Active Layer Morphology. Photosynth. Res. 2006, 87, 73–81. [Google Scholar] [CrossRef]
- Alahmadi, A.N.M. Design of an Efficient PTB7:PC70BM-Based Polymer Solar Cell for 8% Efficiency. Polymers 2022, 14, 889. [Google Scholar] [CrossRef]
- Yu, H.; Wang, Y.; Zou, X.; Yin, J.; Shi, X.; Li, Y.; Zhao, H.; Wang, L.; Ng, H.M.; Zou, B.; et al. Improved photovoltaic performance and robustness of all-polymer solar cells enabled by a polyfullerene guest acceptor. Nat. Commun. 2023, 14, 2323. [Google Scholar] [CrossRef]
- Khalid, M.; Khan, M.U.; Razia, E.-t.; Shafiq, Z.; Alam, M.M.; Imran, M.; Akram, M.S. Exploration of efficient electron acceptors for organic solar cells: Rational design of indacenodithiophene based non-fullerene compounds. Sci. Rep. 2021, 11, 19931. [Google Scholar] [CrossRef]
- Yan, C.; Barlow, S.; Wang, Z.; Yan, H.; Jen, A.K.Y.; Marder, S.R.; Zhan, X. Non-fullerene acceptors for organic solar cells. Nat. Rev. Mater. 2018, 3, 18003. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, X.; Sun, R.; Xu, L.-Y.; Chen, Z.; Zhang, M.; Zhu, H.; Min, J. All-small-molecule organic solar cells with 18.1% efficiency and enhanced stability enabled by improving light harvesting and nanoscale microstructure. Joule 2023, 7, 2845–2858. [Google Scholar] [CrossRef]
- Wang, T.; Brédas, J.-L. Organic Solar Cells Based on Non-fullerene Small-Molecule Acceptors: Impact of Substituent Position. Matter 2020, 2, 119–135. [Google Scholar] [CrossRef]
- Wang, H.-C.; Chen, C.-H.; Li, R.-H.; Lin, Y.-C.; Tsao, C.-S.; Chang, B.; Tan, S.; Yang, Y.; Wei, K.-H. Engineering the Core Units of Small-Molecule Acceptors to Enhance the Performance of Organic Photovoltaics. Sol. RRL 2020, 4, 2000253. [Google Scholar] [CrossRef]
- Park, J.S.; Sun, C.; Han, Y.; Kim, G.-U.; Phan, T.N.-L.; Kim, Y.-H.; Kim, B.J. 2D Outer Side Chain-Incorporated Y Acceptors for Highly Efficient Organic Solar Cells with Nonhalogenated Solvent and Annealing-Free Process. Adv. Energy Sustain. Res. 2022, 3, 2200070. [Google Scholar] [CrossRef]
- Suman; Singh, S.P. Suman; Singh, S.P. Impact of end groups on the performance of non-fullerene acceptors for organic solar cell applications. J. Mater. Chem. A 2019, 7, 22701–22729. [Google Scholar] [CrossRef]
- Xu, W.; Chang, Y.; Zhu, X.; Wei, Z.; Zhang, X.; Sun, X.; Lu, K.; Wei, Z. Organic solar cells based on small molecule donor and polymer acceptor. Chin. Chem. Lett. 2022, 33, 123–132. [Google Scholar] [CrossRef]
- Yuan, J.; Zou, Y. The history and development of Y6. Org. Electron. 2022, 102, 106436. [Google Scholar] [CrossRef]
- Wen, Z.-C.; Yin, H.; Hao, X.-T. Recent progress of PM6:Y6-based high efficiency organic solar cells. Surf. Interfaces 2021, 23, 100921. [Google Scholar] [CrossRef]
- Liu, F.; Zhou, L.; Liu, W.; Zhou, Z.; Yue, Q.; Zheng, W.; Sun, R.; Liu, W.; Xu, S.; Fan, H.; et al. Organic Solar Cells with 18% Efficiency Enabled by an Alloy Acceptor: A Two-in-One Strategy. Adv. Mater. 2021, 33, 2100830. [Google Scholar] [CrossRef]
- Dang, D.; Yu, D.; Wang, E. Conjugated Donor–Acceptor Terpolymers Toward High-Efficiency Polymer Solar Cells. Adv. Mater. 2019, 31, 1807019. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, X.; Jones, L.O.; Alzola, J.M.; Mukherjee, S.; Feng, L.-w.; Zhu, W.; Stern, C.L.; Huang, W.; Yu, J.; et al. Systematic Merging of Nonfullerene Acceptor π-Extension and Tetrafluorination Strategies Affords Polymer Solar Cells with >16% Efficiency. J. Am. Chem. Soc. 2021, 143, 6123–6139. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Ufimkin, P.; Aniés, F.; Hu, X.; Kafourou, P.; Rimmele, M.; Rapley, C.L.; Ding, B. Molecular engineering of Y-series acceptors for nonfullerene organic solar cells. SusMat 2022, 2, 591–606. [Google Scholar] [CrossRef]
- Ryu, H.S.; Park, S.Y.; Lee, T.H.; Kim, J.Y.; Woo, H.Y. Recent progress in indoor organic photovoltaics. Nanoscale 2020, 12, 5792–5804. [Google Scholar] [CrossRef]
- Cai, G.; Chen, Z.; Xia, X.; Li, Y.; Wang, J.; Liu, H.; Sun, P.; Li, C.; Ma, R.; Zhou, Y.; et al. Pushing the Efficiency of High Open-Circuit Voltage Binary Organic Solar Cells by Vertical Morphology Tuning. Adv. Sci. 2022, 9, 2200578. [Google Scholar] [CrossRef] [PubMed]
- Azzouzi, M.; Kirchartz, T.; Nelson, J. Factors Controlling Open-Circuit Voltage Losses in Organic Solar Cells. Trends Chem. 2019, 1, 49–62. [Google Scholar] [CrossRef]
- Jiang, M.; Zhi, H.-F.; Zhang, B.; Yang, C.; Mahmood, A.; Zhang, M.; Woo, H.Y.; Zhang, F.; Wang, J.-L.; An, Q. Controlling Morphology and Voltage Loss with Ternary Strategy Triggers Efficient All-Small-Molecule Organic Solar Cells. ACS Energy Lett. 2023, 8, 1058–1067. [Google Scholar] [CrossRef]
- Setsoafia, D.D.Y.; Sreedhar Ram, K.; Mehdizadeh-Rad, H.; Ompong, D.; Murthy, V.; Singh, J. DFT and TD-DFT Calculations of Orbital Energies and Photovoltaic Properties of Small Molecule Donor and Acceptor Materials Used in Organic Solar Cells. J. Renew. Mater. 2022, 10, 2553–2567. [Google Scholar] [CrossRef]
- Setsoafia, D.D.Y.; Ram, K.S.; Mehdizadeh-Rad, H.; Ompong, D.; Singh, J. Density Functional Theory Simulation of Optical and Photovoltaic Properties of DRTB-T Donor-Based Organic Solar Cells. Int. J. Energy Res. 2023, 2023, 6696446. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Ma, H.; Liu, N.; Huang, J.-D. A DFT Study on the Electronic Structures and Conducting Properties of Rubrene and its Derivatives in Organic Field-Effect Transistors. Sci. Rep. 2017, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, Y.; Zhou, L.; Zhang, G.; Yip, H.-L.; Lau, T.-K.; Lu, X.; Zhu, C.; Peng, H.; Johnson, P.A.; et al. Single-Junction Organic Solar Cell with over 15% Efficiency Using Fused-Ring Acceptor with Electron-Deficient Core. Joule 2019, 3, 1140–1151. [Google Scholar] [CrossRef]
- Cui, Y.; Yao, H.; Zhang, J.; Zhang, T.; Wang, Y.; Hong, L.; Xian, K.; Xu, B.; Zhang, S.; Peng, J.; et al. Over 16% efficiency organic photovoltaic cells enabled by a chlorinated acceptor with increased open-circuit voltages. Nat. Commun. 2019, 10, 2515. [Google Scholar] [CrossRef] [PubMed]
- Bredas, J.-L. Mind the gap! Mater. Horiz. 2014, 1, 17–19. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, S.; Zhai, Y.; Xu, M.; Li, M.; Zhang, X. Optical-electronic performance and mechanism investigation of dihydroindolocarbazole-based organic dyes for DSSCs. Results Phys. 2021, 23, 103939. [Google Scholar] [CrossRef]
- Singh, J.; Narayan, M.; Ompong, D.; Zhu, F. Dissociation of charge transfer excitons at the donor–acceptor interface in bulk heterojunction organic solar cells. J. Mater. Sci. Mater. Electron. 2017, 28, 7095–7099. [Google Scholar] [CrossRef]
- Elumalai, N.K.; Uddin, A. Open circuit voltage of organic solar cells: An in-depth review. Energy Environ. Sci. 2016, 9, 391–410. [Google Scholar] [CrossRef]
- Qi, B.; Wang, J. Fill factor in organic solar cells. Phys. Chem. Chem. Phys. 2013, 15, 8972–8982. [Google Scholar] [CrossRef]
- Wen, G.; Hu, R.; Su, X.; Chen, Z.; Zhang, C.; Peng, J.; Zou, X.; He, X.; Dong, G.; Zhang, W. Excited-state properties of Y-series small molecule semiconductors. Dye. Pigment. 2021, 192, 109431. [Google Scholar] [CrossRef]
- Li, G.; Feng, L.-W.; Mukherjee, S.; Jones, L.O.; Jacobberger, R.M.; Huang, W.; Young, R.M.; Pankow, R.M.; Zhu, W.; Lu, N.; et al. Non-fullerene acceptors with direct and indirect hexa-fluorination afford >17% efficiency in polymer solar cells. Energy Environ. Sci. 2022, 15, 645–659. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).