1. Introduction
As a large country of coal resources, it is a great significance to make clean, efficient and rational use of the existing coal resources in China, which has abundant coal reserves, which are good for power, and low mining costs [
1,
2]. However, Zhundong coal has a special quality, with a high content of alkali and alkaline earth metals in the ash. The Na, Ca, and K salts are precipitated during the combustion process and easily react with other minerals in the coal ash, which further melts the compounds. The molten inorganic compounds condense on the lower temperature pipe wall to form a low-temperature eutectic, which makes melting and slagging in the high-temperature zone easy. Further, it results in slagging/fouling of the boiler and reduces its operation efficiency [
3,
4,
5].
As an important indicator to measure whether the boiler can operate normally, the melting characteristics are related to the composition of coal ash [
6]. Researchers have shown that Na reacts with SiO
2 during combustion to form a thin layer of silicate. Silicate further reacts with sodium to form portil, which condenses on the pipe wall at low temperatures to form a low-temperature eutectic [
7,
8,
9,
10,
11]. Some researchers have studied the deposits on the heating surface of the boiler and found that sulfates containing Na and Ca deposited on the heating surface are a prominent cause of slagging and fouling problems in Zhundong coal [
12,
13]. Previous studies have shown that wollastonite and sodium silicate generated by the reaction of Na
2O and CaO with SiO
2 were the main factors causing the slagging of Zhundong coal. However, the study on slagging characteristics is limited to the macroscopic viewing analysis of slag samples. Therefore, it is crucial to explore the influence of its micro-formation mechanism on the slagging and fouling of Zhundong coal. By calculating the electronic structure and adsorption energy of minerals, slagging, a trend caused by the reaction of Na
2O and CaO with SiO
2, is predicted, which provides data support for the discussion of slagging characteristics.
In recent years, with the development of quantum chemistry theory, the first-principle calculation has been widely used to determine the structure and ground state properties of materials [
14,
15]. It explains the reaction activity of minerals at the molecular level and provides a theoretical basis for analyzing the reaction mechanism of substances during the phase transition process [
16]. One way to study the relationship between a minerals’ structure and property based on quantum chemistry theory is to model the actual problem, and selection of model is key to solving the problem. The bilayer model can calculate the situation of individual molecules that are adsorbed on the surface and the interaction of molecules between layers, and it can explain the properties of minerals from the electronic structure and surface reaction. The researchers of [
17] applied quantum chemistry theory to reveal the macroscopic phenomenon of substances from the perspective of the microscopic characteristics of the molecular structure, which provided a theoretical basis for analyzing the physical and chemical properties in the reaction process. Researchers have studied the heterogeneous adsorption of HCN on the CaO(100) surfaces by using the first-principle calculation, indicating that HCN was more easily adsorbed on the Ca vacancy surface of the defective CaO(100) surface and the adsorption capacity was decreased with increasing temperature. Lu [
18] studied the action mechanism of Cd on the SiO
2(001) surface from the microscopic point of view and found that the O site is the active site on the SiO
2(001) surface. The adsorption mode of CdO on the SiO
2(001) surface was chemical adsorption, which made the reaction with the SiO
2(001) surface easier. The results have provided theoretical support for the removal of Cd pollutants. Lin [
19] used the density functional theory (DFT) to study the adsorption reaction of CO on different crystal surfaces of Fe
2O
3 in the chemical ring combustion process. The results showed that a CO adsorption reaction could occur on both (001) and (100) crystal surfaces of Fe
2O
3 and that the addition of Fe
2O
3 could promote the adsorption of CO.
The above research is the application of quantum chemistry theory to the adsorption and surface properties of a single molecule on the crystal surface. Compared with the single-molecule model, crystal-surface models with periodic boundary conditions are better to describe the solid surface. However, there are few studies on the molecular interactions between layers on crystal surfaces. Han [
20] et al. studied the interaction of surface modified colloidal silica with polyvinyl alcohol by molecular simulation, which showed that the interaction of modified silica with polyvinyl alcohol molecules was significantly enhanced. By using quantum chemistry theory, Yang [
21] studied the interaction between the NaCl surface and Fe
2O
3, Al
2O
3, and NiO surface in high-alkali coal and found that the adsorption energy was positively correlated with the slagging condition in the boiler. The greater the adsorption energy, the more serious the slagging.
In order to more accurately describe the real situation, the adsorption models of oxides are established in this paper. Density functional theory plays a key role in the calculation of micro parameters such as adsorption energy, charge transfer, and the bond length of gases and solids on the coal surface. The adsorption capacity of different substances on the coal surface is obtained by calculation. The theory provides an important theoretical basis for the study of spontaneous coal combustion, gasification, and the combustion process. Through the analysis of adsorption characteristics, we can deeply understand the interaction mechanism between substances, that is, a higher adsorption capacity means a stronger intermolecular interaction.
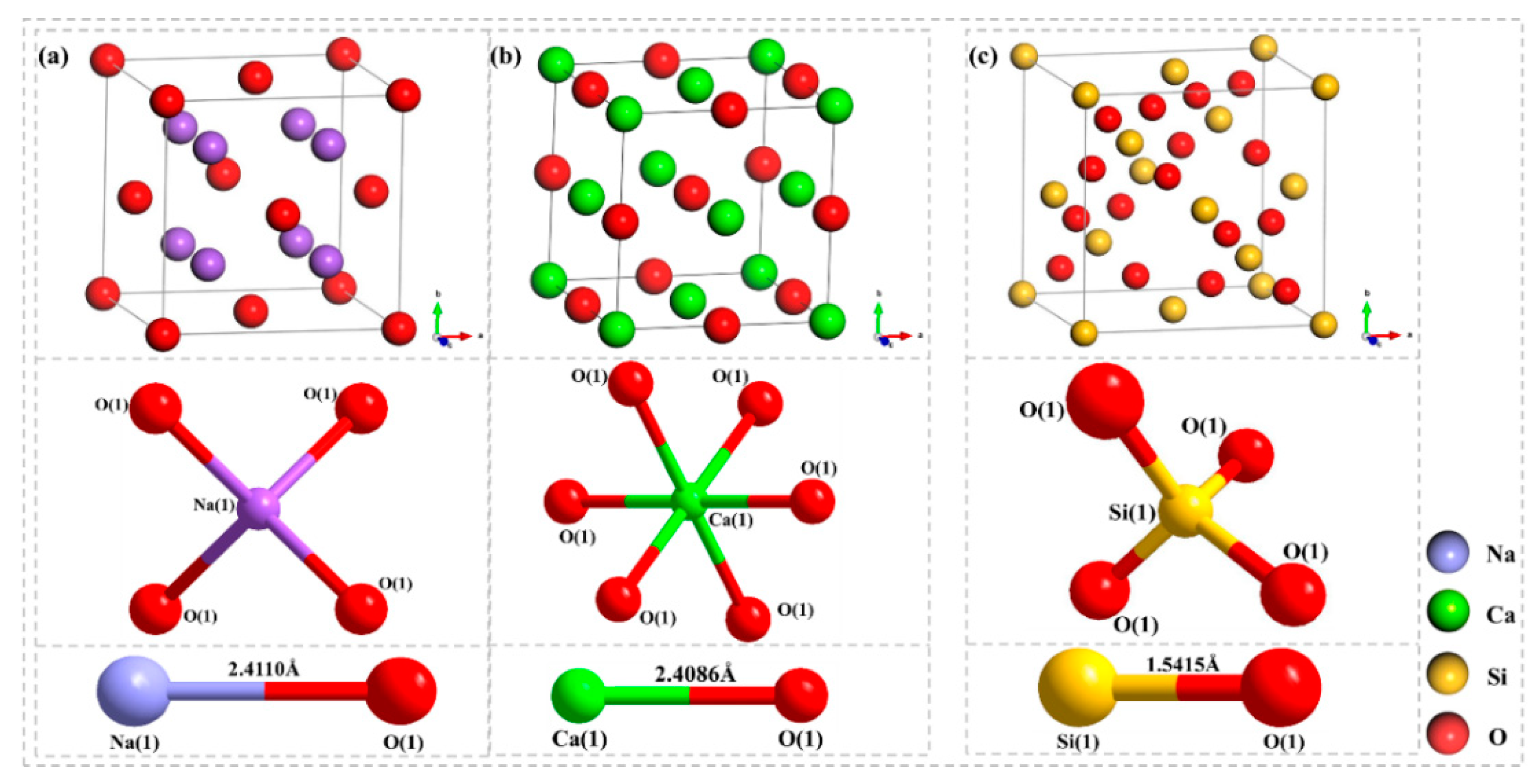
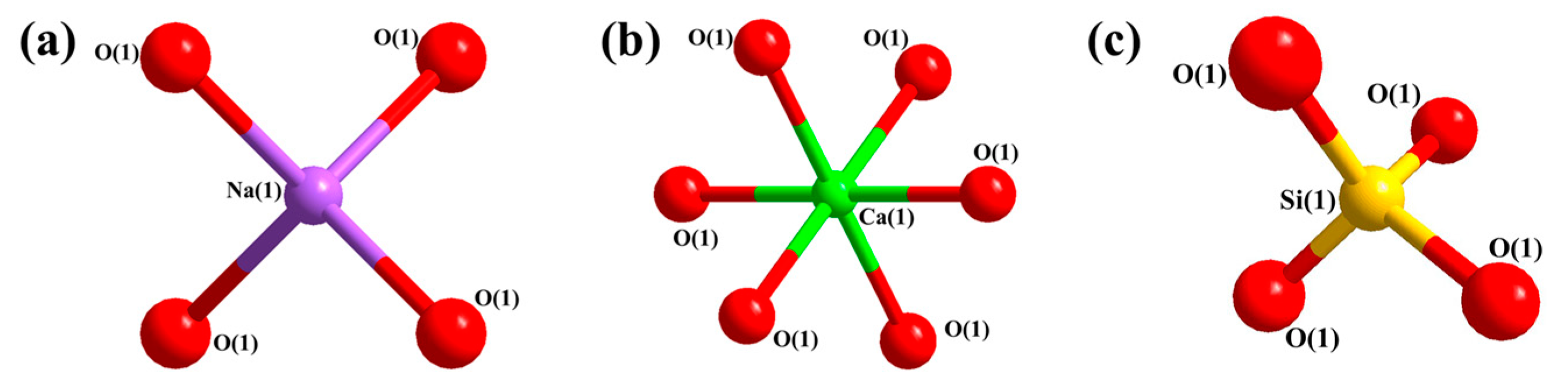
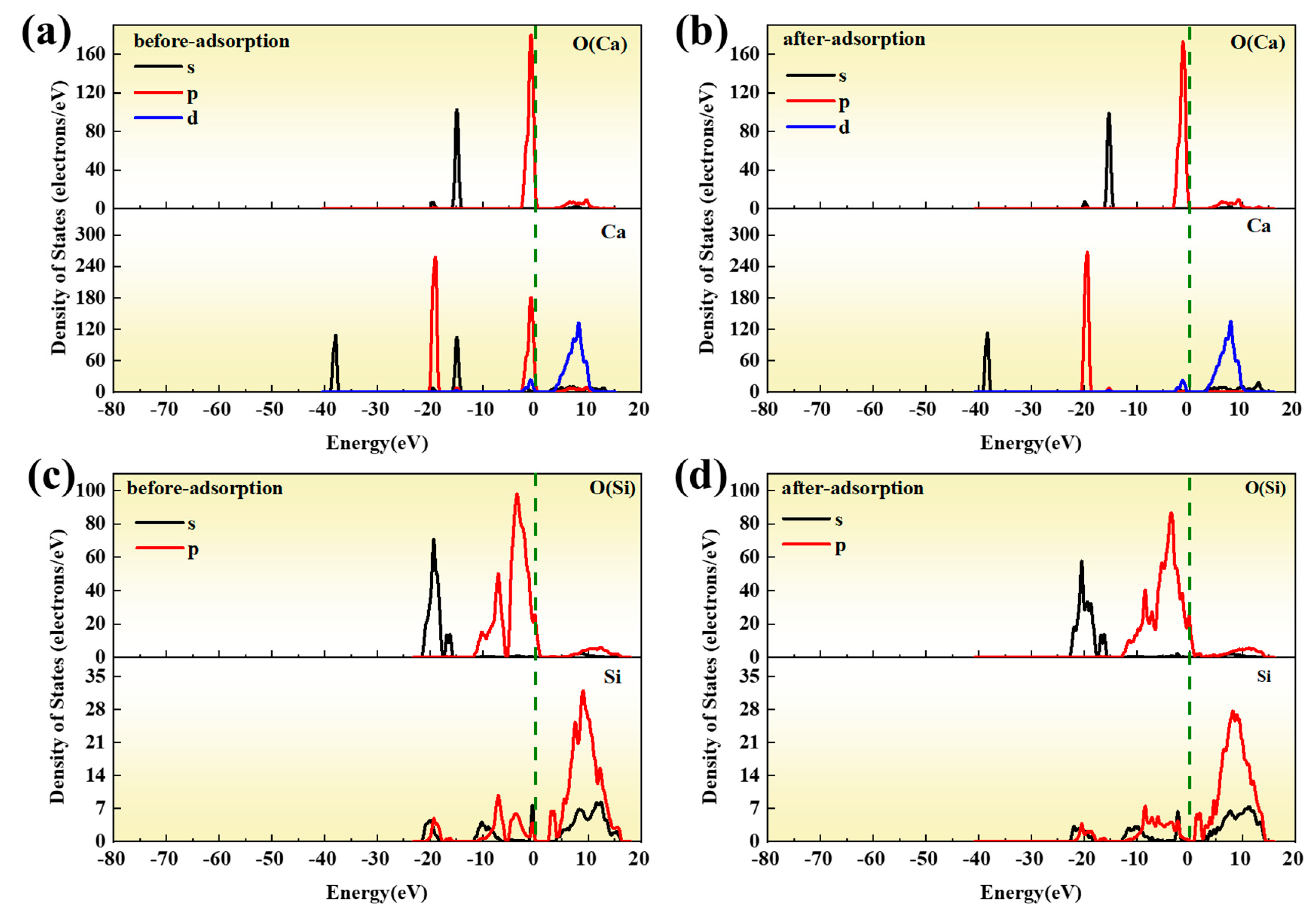
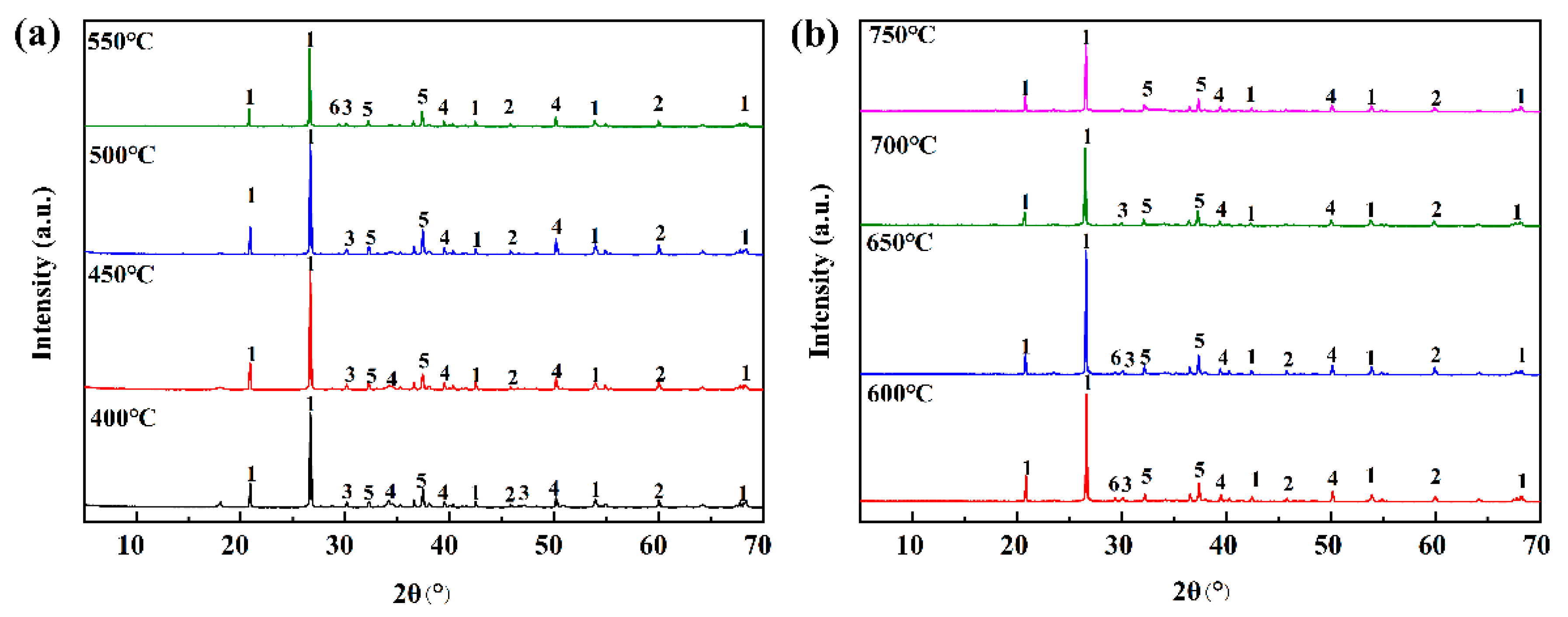
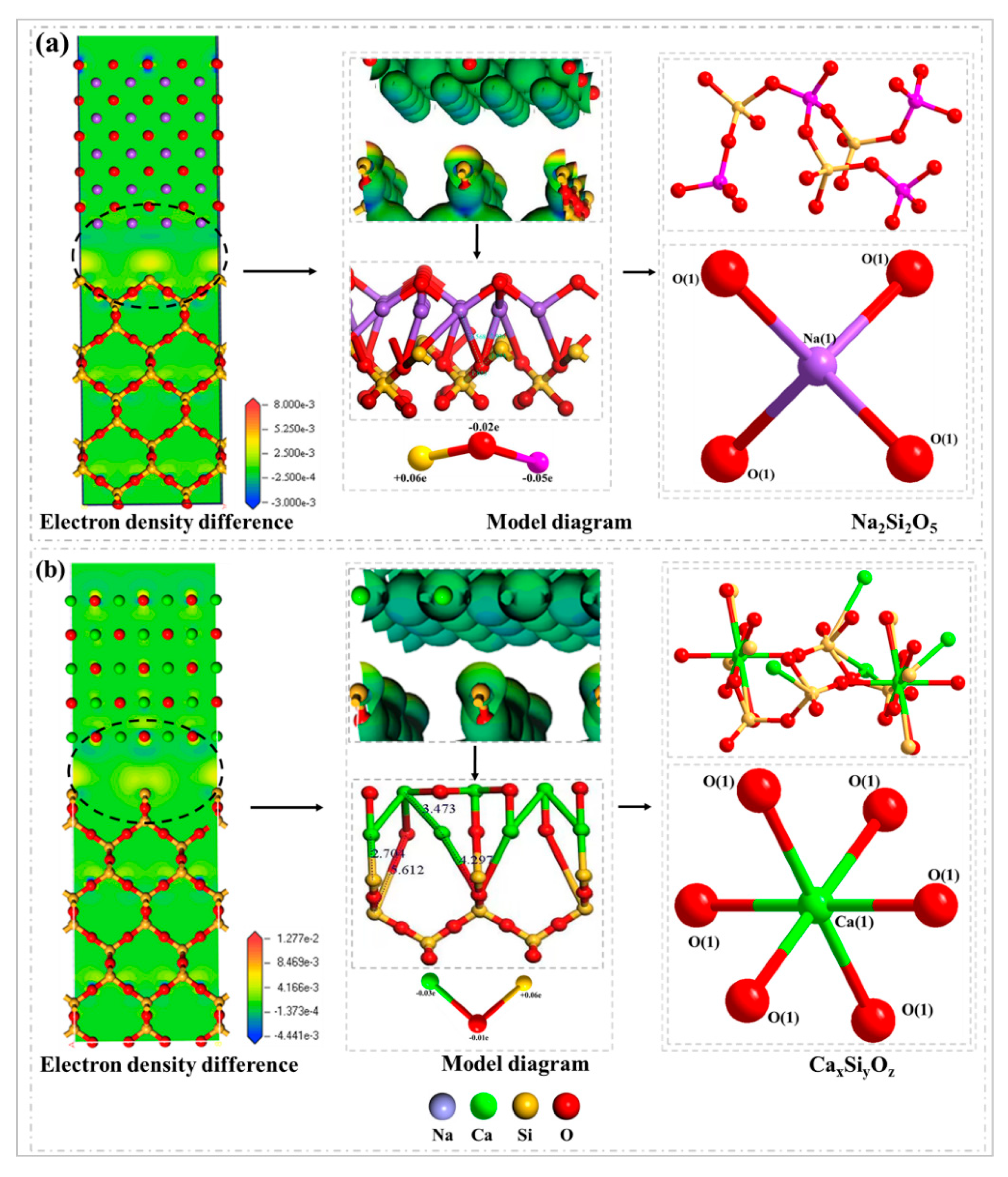
In this paper, the competition mechanism of alkaline metal oxides during the reaction between compounds in Zhundong coal is studied by means of simulations and experiments. Na2O(110)-SiO2(100) and CaO(100)-SiO2(100) models were established and the adsorptions were calculated using DFT. Orbital density and Mulliken population analysis were used to reveal the competitive relationship between Na2O and CaO, which reacted with SiO2 on the surface and predicted the trend of slagging caused by the reaction of Na2O and CaO with SiO2. In addition, the competition between Na2O and CaO in the mixture was studied by XRD results of the complex under different temperature conditions. Combined with the simulation results, it is shown that the formation of Na-containing minerals and other compounds in the high-temperature zone is more serious, which provides an effective way to solve the slagging and fouling problem of Zhundong coal.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}