
Microscopic Mechanism of Fe2O3-Catalyzed NO Reduction during Sludge Combustion: A Density Functional Theory Study



Abstract
:1. Introduction
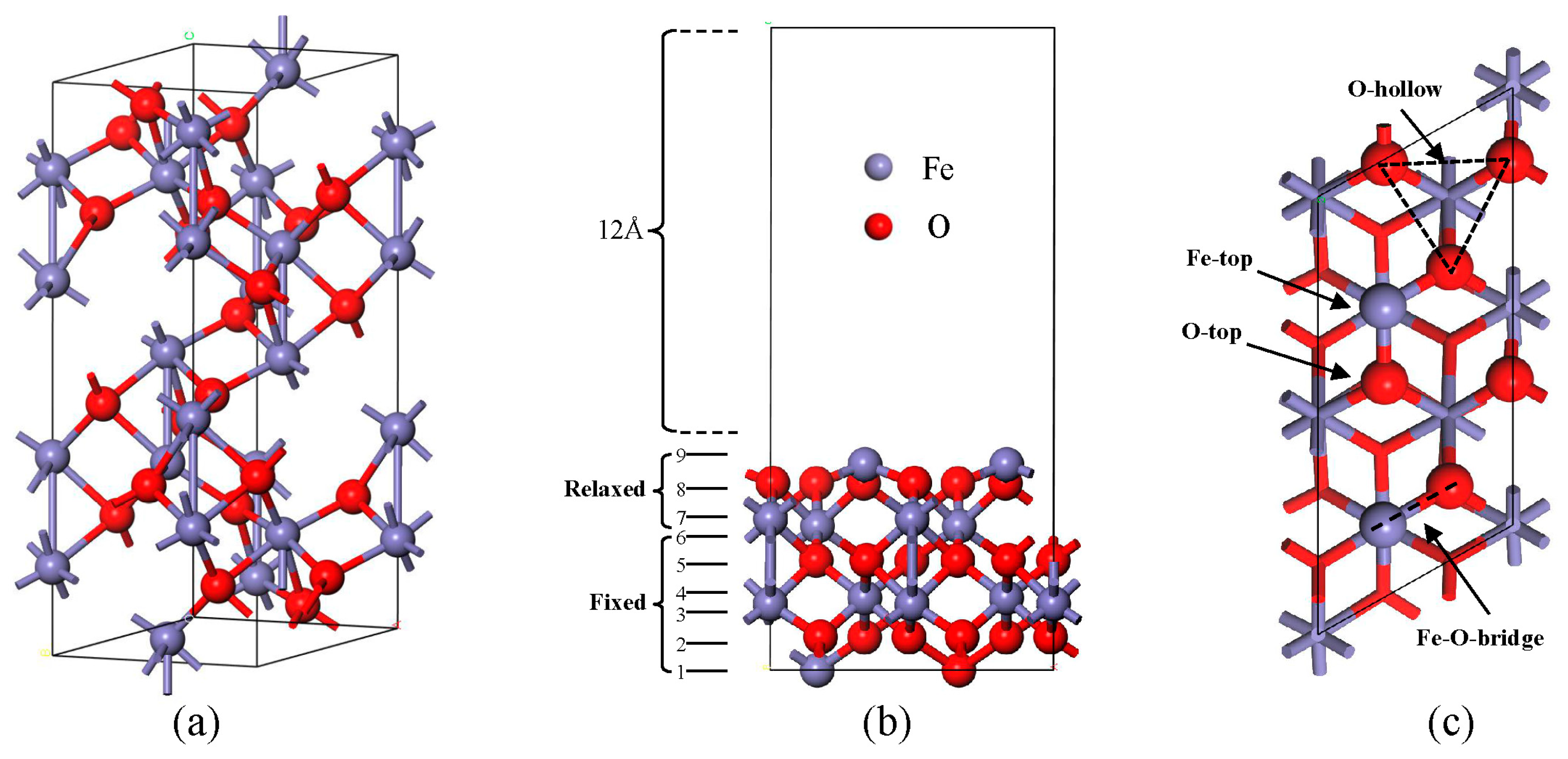
2. Model Construction and Computational Methods
3. Results and Discussion
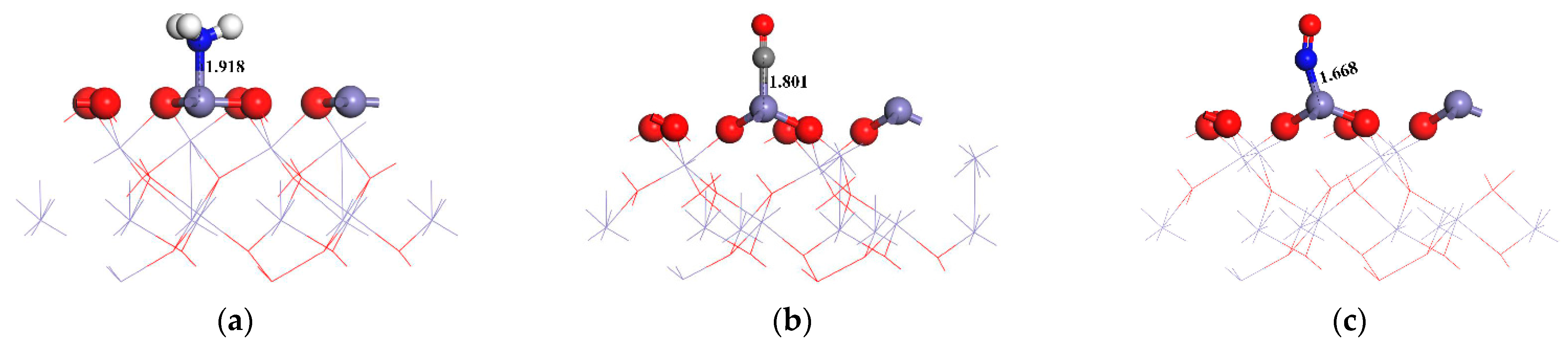
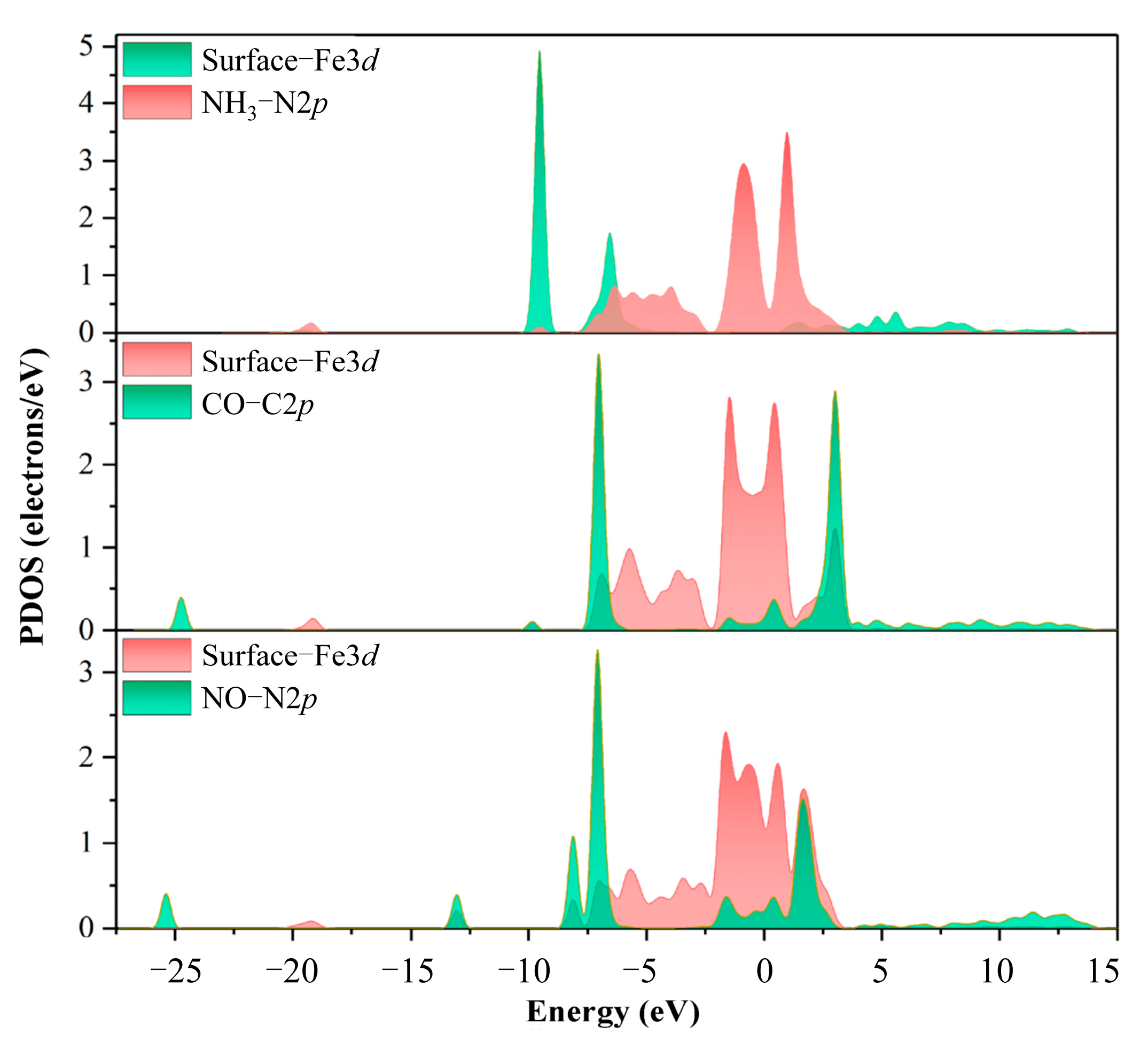
3.1. Adsorption of NH3, CO, and NO on α-Fe2O3(001) Surface
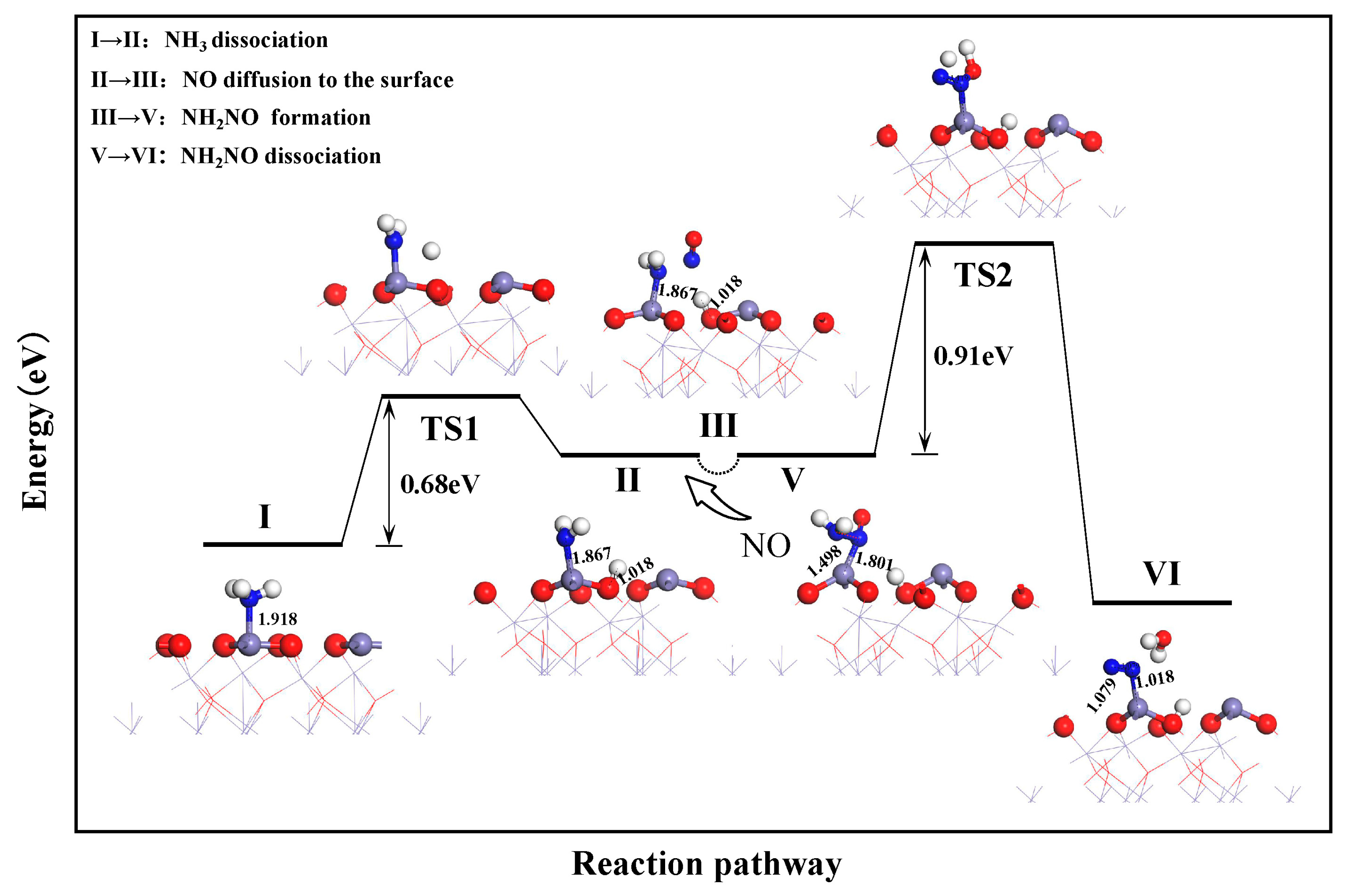
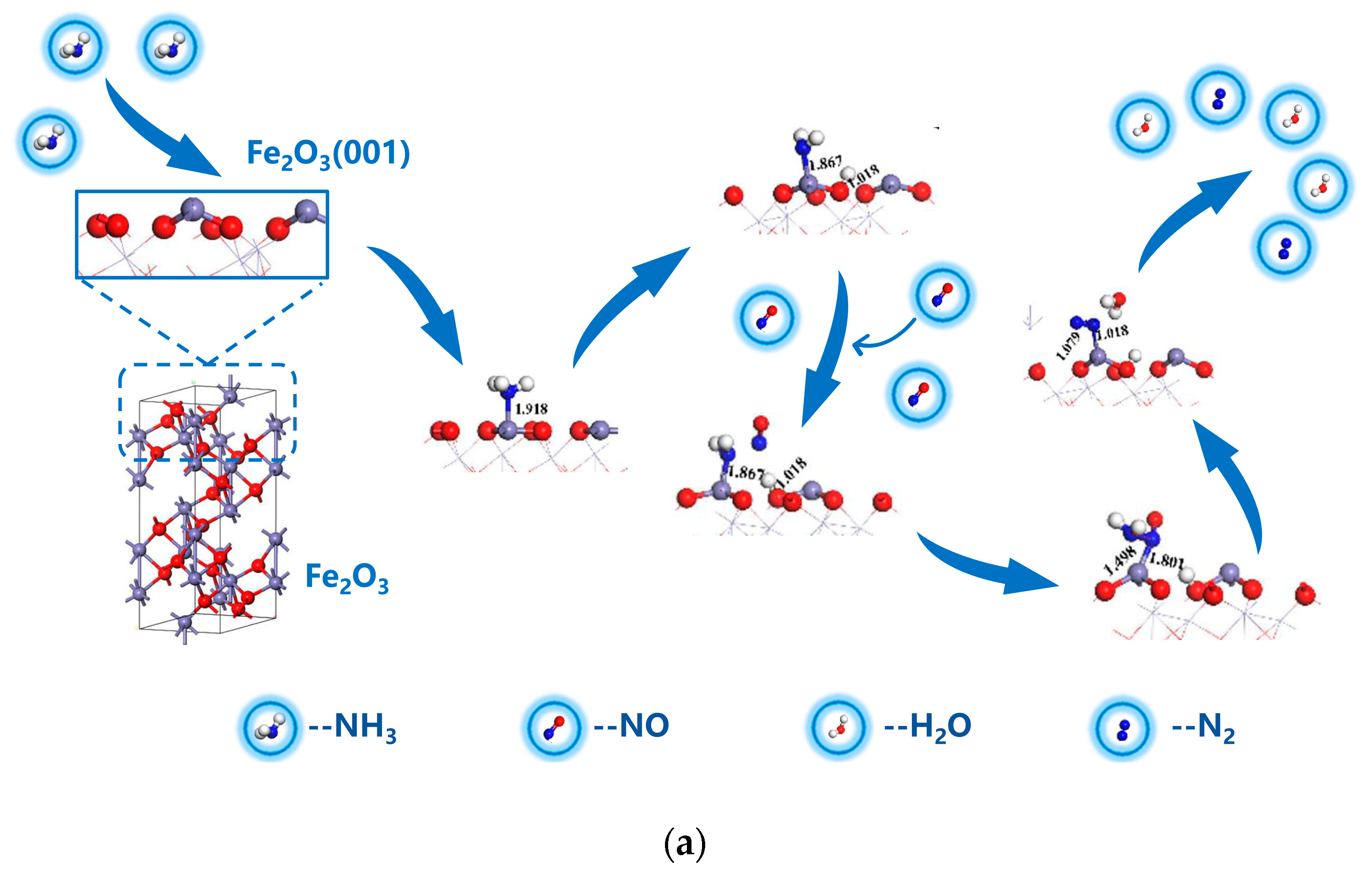
3.2. Reaction Pathways of NH3 and NO on α-Fe2O3(001) Surface
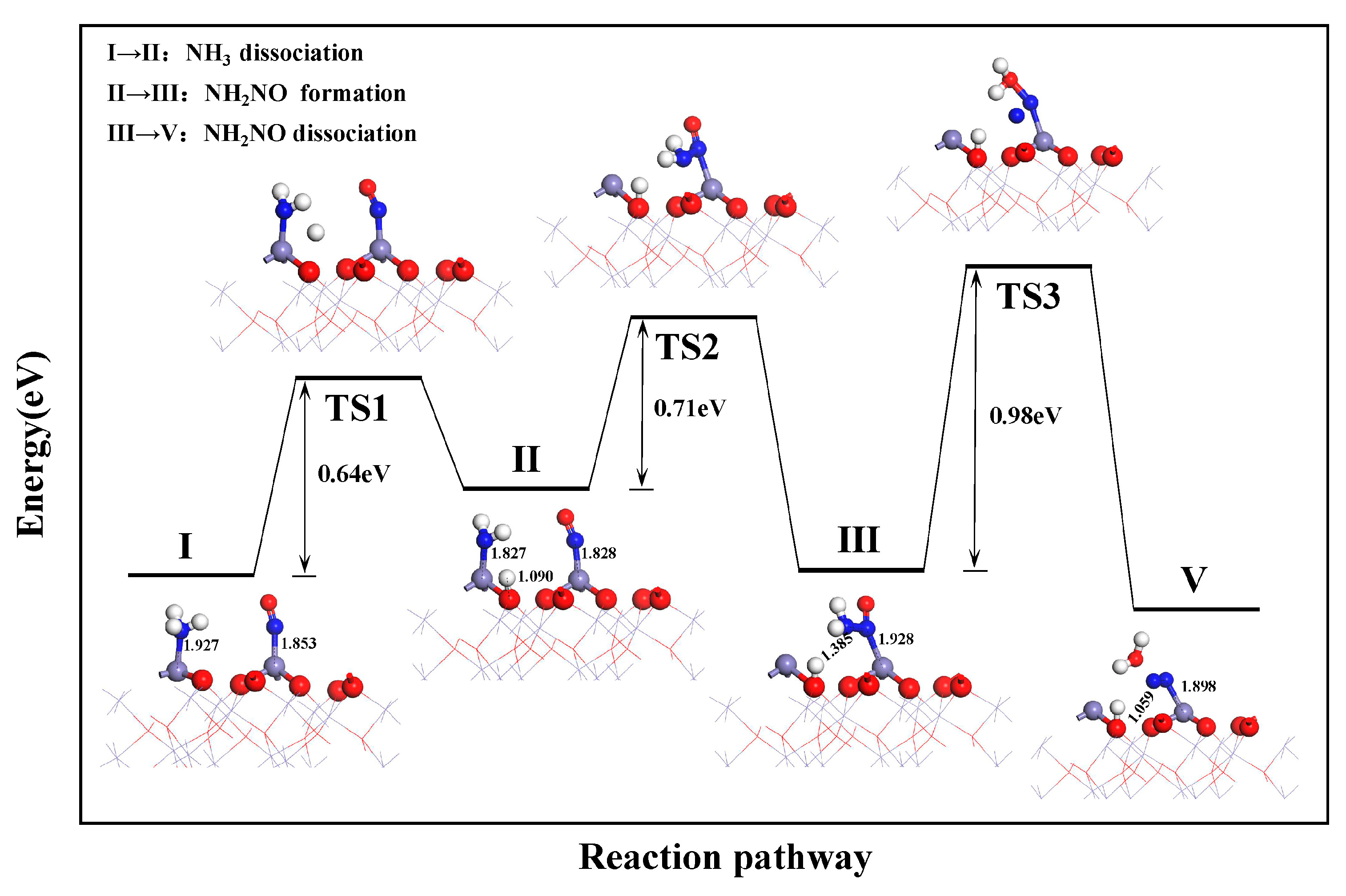
3.2.1. E-R Mechanism Pathway
3.2.2. L-H Mechanism Pathway
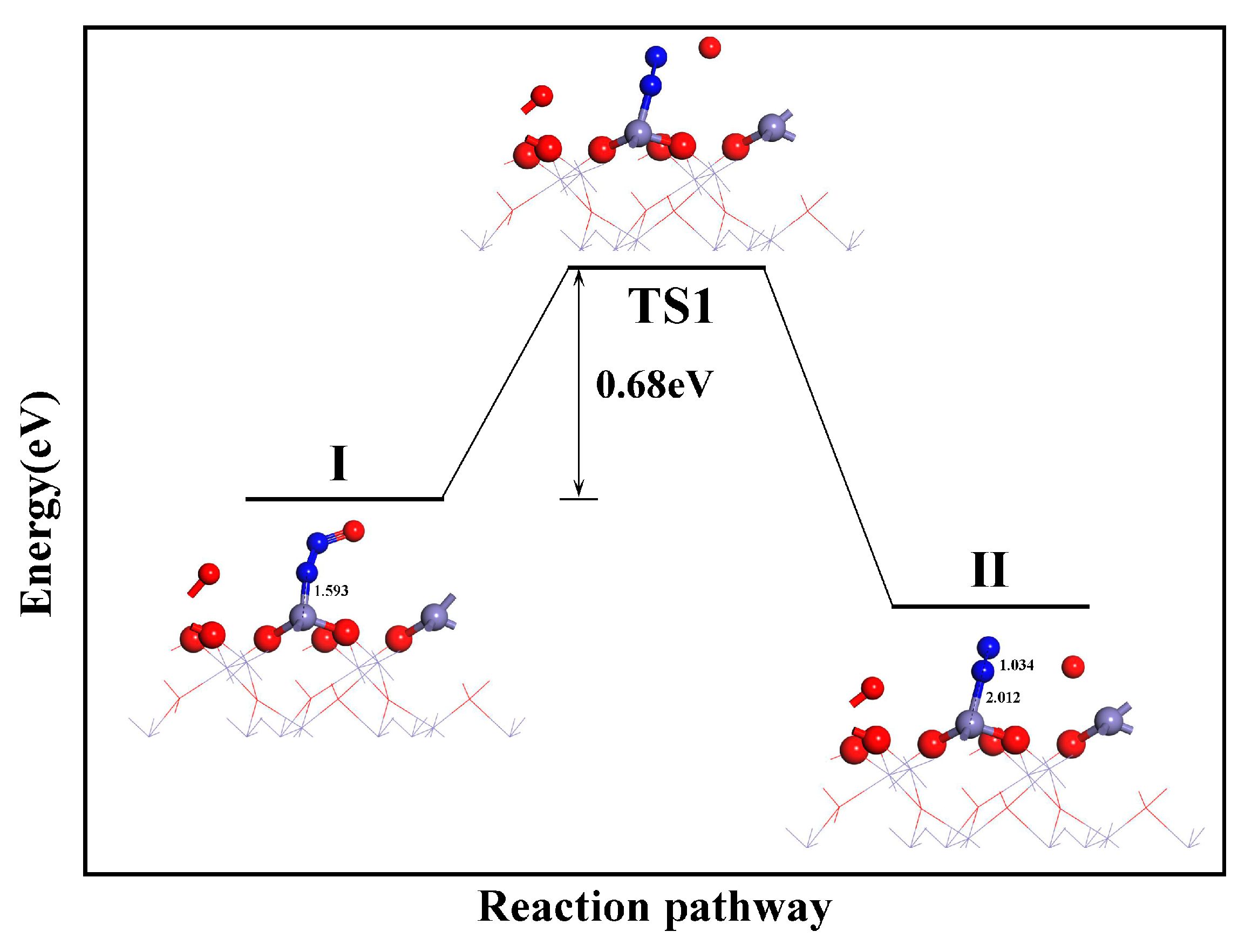
3.3. α-Fe2O3(001) Surface: Reaction Pathway of CO with NO
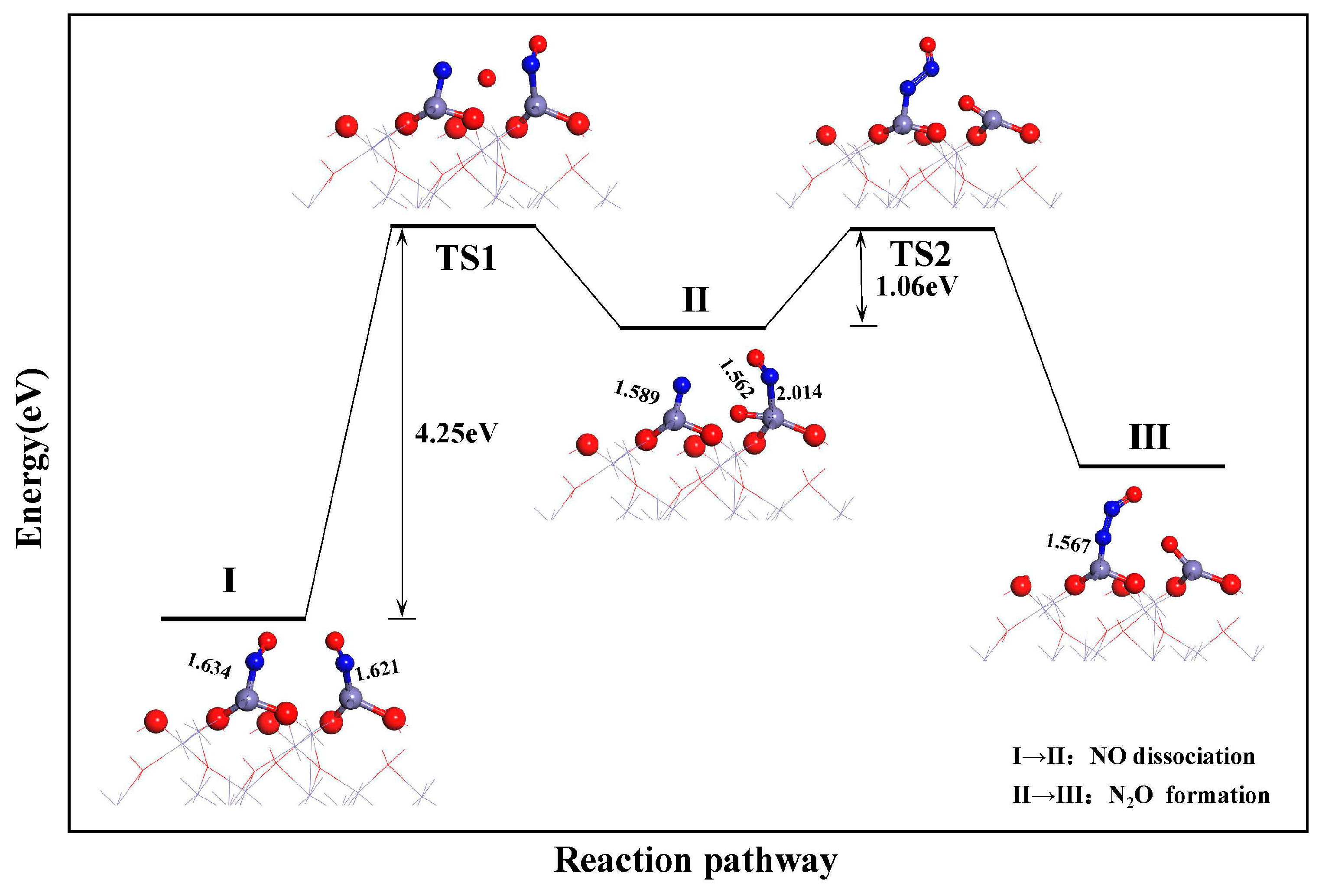
3.3.1. Dissociation of NO
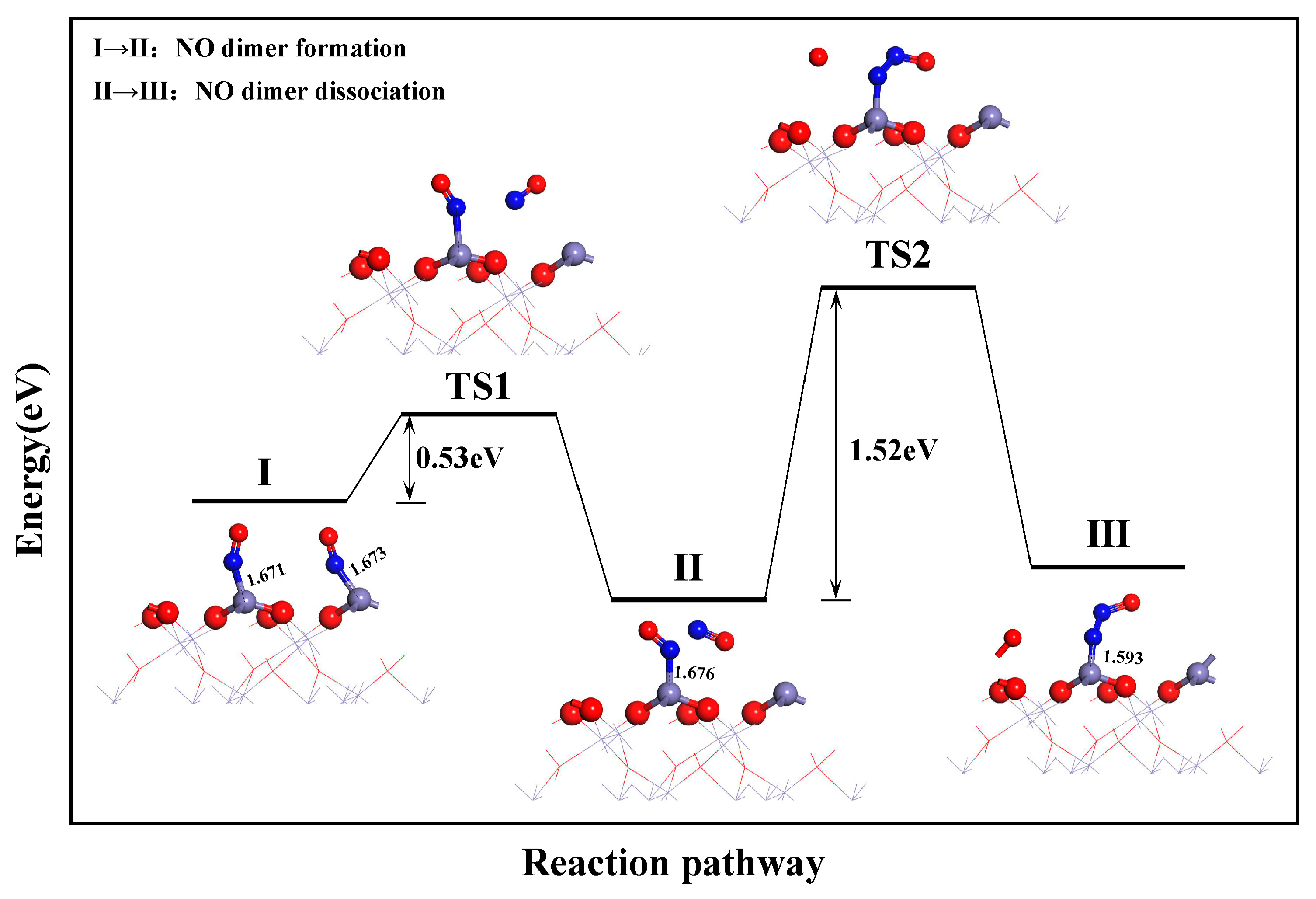
3.3.2. Catalytic Effect of Fe2O3 on NO Dissociation
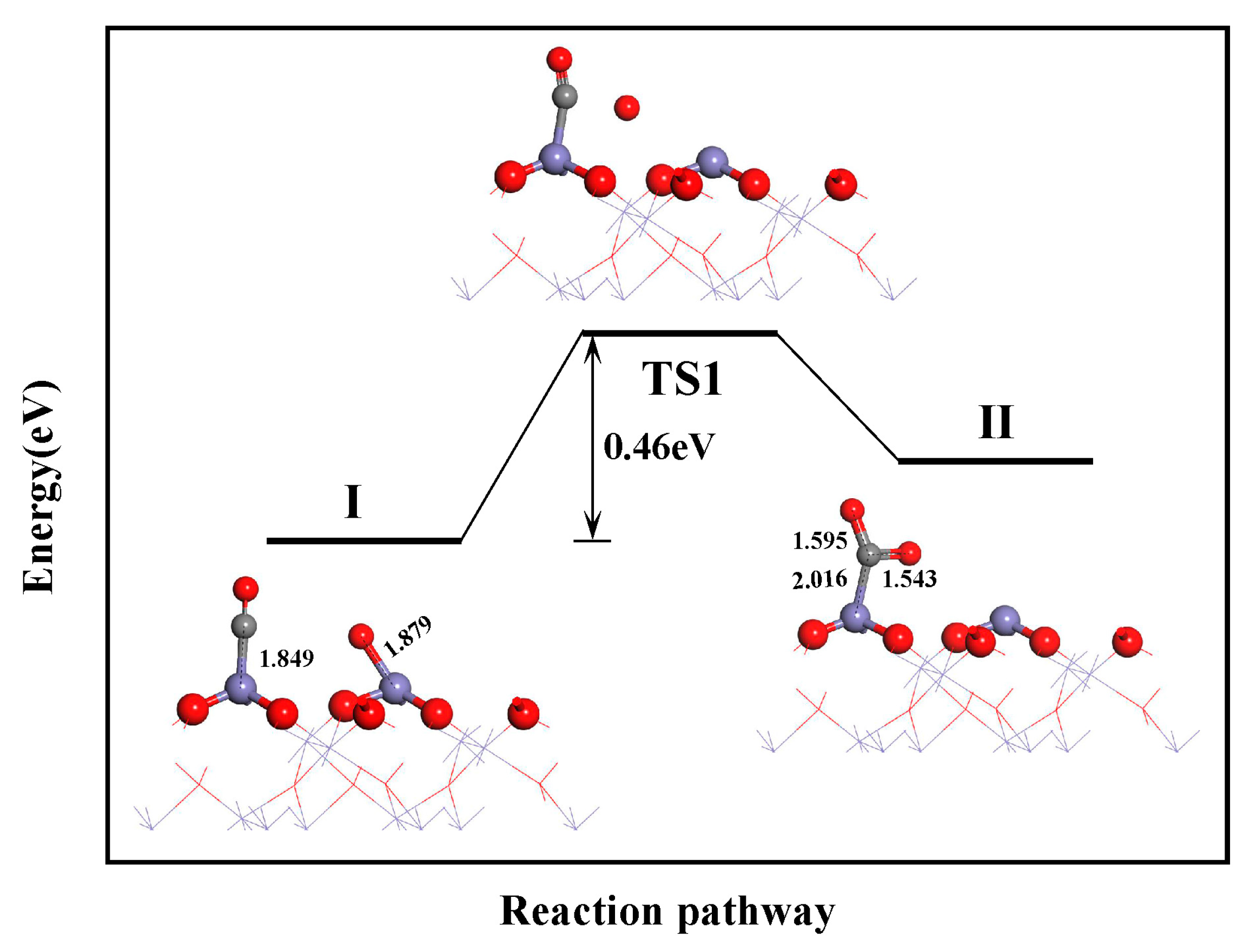
3.3.3. CO2 and N2 Formation
3.4. Mechanism of NH3/CO Reduction of NO on α-Fe2O3(001) Surface
4. Conclusions
- (1)
- The α-Fe2O3(001) surface can undergo chemical adsorption of NH3, CO, and NO preferentially on the surface Fe top sites. Among these, NO exhibits the strongest adsorption with an energy of −2.34 eV and a charge transfer of −0.58 e. The partial DOS analysis indicated significant hybridization between the surface Fe 3d orbitals and the N 2p orbitals of NO around −8, −7, the Fermi level, and 2 eV.
- (2)
- On the α-Fe2O3(001) surface, the reduction of NO by NH3 follows the E–R mechanism: NH3 adsorption → NH3 dissociation → NO diffusion to the surface → NH2NO formation → NH2NO dissociation. The dissociation of NH2NO is the rate-determining step in the reduction reaction, with an energy barrier of 0.91 eV.
- (3)
- During the reduction of NO by CO on the α-Fe2O3(001) surface, the formation of N2O follows the L-H mechanism: NO + NO → (NO)2 → (NO)2 → N2O + O. The dissociation of the NO dimer is the rate-determining step for the CO reduction of NO, with an energy barrier of 1.53 eV. The formation of CO2 and N2 requires energy barriers of 0.46 and 0.68 eV, respectively.
- (4)
- During sludge combustion, intrinsic α-Fe2O3 can catalyze the reduction of NO in a reducing atmosphere, and the surface Fe atoms function as catalytically active sites. Specifically, the catalytic effect includes a reduction in the energy barrier from 0.89 to 0.68 eV for NH3 dissociation, from 1.53 to 0.91 eV for NH2NO dissociation, and from 2.04 to 1.53 eV for NO dimer dissociation.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Fe2O3 | Iron oxide |
| NH3 | Ammonia |
| CO | Carbon monoxide |
| NO | Nitric oxide |
| DFT | Density functional theory |
| DOS | Density of state |
| PDOS | Partial density of states |
References
- Wang, Y.L.; Jia, L.; Guo, B.H.; Wang, B.R.; Zhang, L.; Zheng, X.; Xiang, J.; Jin, Y. Effects of CaO-Fe2O3-Fe3(PO4)2 in sewage sludge on combustion characteristics and kinetics of coal slime. Fuel 2022, 322, 124267. [Google Scholar] [CrossRef]
- Liu, H.M.; Wang, Y.C.; Zhao, S.L.; Hu, H.Y.; Cao, C.Y.; Li, A.J.; Yu, Y.; Yao, H. Review on the current status of the co-combustion technology of organic solid waste (OSW) and coal in china. Energy Fuels 2020, 34, 15448–15487. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Q.; Hu, H.Y.; Liu, P.; Hu, X.W.; Li, A.J.; Yao, H. Catalytic role of conditioner CaO in nitrogen transformation during sewage sludge pyrolysis. P. Combust. Inst. 2015, 35, 2759–2766. [Google Scholar] [CrossRef]
- Guo, S.C.; Zhang, K.X.; Liu, S.J.; Yang, S.; Du, G.W.; Zhang, H.X.; Shang, G.J.; Zhi, G.R. Progresses of research on the formation and transformation of NOX as atmospheric pollutant during coal burning. Appl. Chem. Ind. 2020, 49, 3205–3212+3245. [Google Scholar]
- Wang, Y.L.; Jia, L.; Guo, B.H.; Shen, X.; Zheng, X.; Xiang, J.; Jin, Y. Investigation of interaction mechanisms during co-combustion of sewage sludge and coal slime: Combustion characteristics and NO/SO2 emission behavior. Sci. Total Environ. 2022, 851, 158166. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.Y.; Hu, L.L.; Song, T.X.; Zhang, Y.; Zhang, H.; Ma, S.X.; Lyu, J.F. Occurrence transformation of iron oxides and their catalytic reduction of NO under fluidized bed temperature and CO. Chem. Ind. Eng. Prog. 2020, 39, 4474–4479. [Google Scholar]
- Zhao, Z.B.; Li, B.Q. Influence of Mineral Matter in Coal on NO-Char Reaction. J. Fuel Chem. Technol. 2001, 29, 129–134. [Google Scholar]
- Sun, J.; Zhao, B.T.; Su, Y.X. Advanced control of NO emission from algal biomass combustion using loaded iron-based additives. Energy 2019, 185, 229–238. [Google Scholar] [CrossRef]
- Hu, J.W.; Yan, Q.P.; Song, Y.Y.; Liu, J.Y.; Evrendilek, F.; Buyukada, M. Catalytic combustions of two bamboo residues with sludge ash, CaO, and Fe2O3: Bioenergy, emission and ash deposition improvements. J. Clean. Prod. 2020, 270, 122418. [Google Scholar] [CrossRef]
- Zhao, Z.L.; Li, J.W.; Chen, B.H. Effect of Fe2O3 crystal structure on the performance of Fe2O3/Cr2O3 high temperature water gas shift catalyst. Mod. Chem. Ind. 2006, S2, 143–146. [Google Scholar]
- Larrubia, M.; Ramis, G.; Busca, G. An FT-IR study of the adsorption and oxidation of N-containing compounds over Fe2O3-TiO2 SCR catalysts. Appl. Catal. B-Environ. 2001, 30, 101–110. [Google Scholar] [CrossRef]
- Yang, S.J.; Wang, C.Z.; Li, J.H.; Yan, N.Q.; Ma, L.; Chang, H.Z. Low temperature selective catalytic reduction of NO with NH3 over Mn-Fe spinel: Performance, mechanism and kinetic study. Appl. Catal. B-Environ. 2011, 110, 71–80. [Google Scholar] [CrossRef]
- Liu, F.D.; He, H.; Zhang, C.B.; Shan, W.B.; Shi, X.Y. Mechanism of the selective catalytic reduction of NOx with NH3 over environmental-friendly iron titanate catalyst. Catal. Today 2011, 175, 18–25. [Google Scholar] [CrossRef]
- Zhang, X.X.; Lyu, X.X.; Ww, H.X.; Xie, M.; Lin, R.Y.; Zhou, Z.J. Microscopic mechanism for effect of sodium on NO heterogeneous reduction by char. J. Fuel Chem. Technol. 2020, 48, 663–673. [Google Scholar] [CrossRef]
- Li, Y.; Niu, S.L.; Lu, C.M.; Wang, J.X.; Peng, J.S. Molecular simulation study of NO heterogeneous reduction by biomass reburning. J. Fuel Chem. Technol. 2020, 48, 689–697. [Google Scholar]
- Cao, F.; Su, S.; Xiang, J.; Wang, P.Y.; Hu, S.; Sun, L.S.; Zhang, A.C. NO/NH3 adsorption properties on γ-Al2O3(110) surface during SCR process. J. Chem. Ind. Eng. 2014, 65, 4056–4062. [Google Scholar]
- Tian, K.; Tu, X.Y.; Dai, S.S. Density Functional Theory Study of NO+CO on Rh(111). Chem. J. Chinese U 2008, 29, 2360–2364. [Google Scholar]
- Finger, L.W.; Hazen, R.M. Crystal structure and isothermal compression of Fe2O3, Cr2O3, and V2O3 to 50 kbars. J. Appl. Phys. 1980, 51, 5362–5367. [Google Scholar] [CrossRef]
- Fang, Q.L.; Zhu, B.Z.; Sun, Y.L.; Zhu, Z.C.; Xu, M.G.; Ge, T.T. Mechanistic insight into the selective catalytic reduction of NO by NH3 over α-Fe2O3(001): A density functional theory study. Catal. Sci. Technol. 2019, 9, 116–124. [Google Scholar] [CrossRef]
- Li, Y.; Wan, Y.; Li, Y.P.; Guan, Q.X.; Tian, Y. Low-Temperature Selective Catalytic Reduction of NO with NH3 over Mn2O3-Doped Fe2O3 Hexagonal Microsheets. ACS Appl. Mater. Inter. 2016, 8, 5224–5233. [Google Scholar] [CrossRef]
- Song, W.Y.; Liu, J.; Zheng, H.L.; Ma, S.C.; Wei, Y.C.; Duan, A.J.; Jiang, G.Y.; Zhao, Z.; Hensen, J.M. A mechanistic DFT study of low temperature SCR of NO with NH3 on MnCe1-xO2(111). Catal. Sci. Technol. 2016, 6, 2120–2128. [Google Scholar] [CrossRef]
- Zhou, W.B.; Niu, S.L.; Wang, D.; Lu, C.M.; Han, K.H.; Li, Y.J.; Zhu, Y. Promoting e ffect of Ti in the Ti-modified γ-Fe2O3 catalyst on its performance in the selective catalytic reduction of NO with ammonia, a DFT calculation study. J. Fuel Chem. Technol. 2020, 48, 1224–1235. [Google Scholar]
- Zhou, Y.S.; Gao, F.Y.; Tang, X.L.; Yi, H.H.; Meng, J.X. Research progress on NO reduction by CO over metal oxide catalysts. Chem. Ind. Eng. Prog. 2019, 38, 4941–4948. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbates | Configuration Number | Configuration Position | Ead (eV) | d (Å) | q (e) |
|---|---|---|---|---|---|
| NH3 | 1 | Adsorbed at the O top site with N-end | −0.92 | 3.097 | −0.12 |
| 2 | Adsorbed at the Fe top site with N-end | −1.67 | 1.918 | −0.34 | |
| 3 | Adsorbed at the Fe-O bridge site with N-end | −1.52 | 2.159 | −0.29 | |
| 4 | Adsorbed at the O vacancy site with N-end | −0.81 | 3.124 | −0.05 | |
| CO | 1 | Adsorbed at the O top site with C-end | −0.58 | 3.241 | −0.01 |
| 2 | Adsorbed at the Fe top site with C-end | −1.84 | 1.801 | −0.42 | |
| 3 | Adsorbed at the Fe top site with O-end | −0.59 | 3.221 | −0.01 | |
| 4 | Adsorbed at the Fe-O bridge site with C-end | −1.51 | 2.124 | −0.35 | |
| 5 | Adsorbed at the Fe-O bridge site with O-end | −1.09 | 2.001 | −0.27 | |
| 6 | Adsorbed at the O vacancy site with C-end | −0.41 | 3.215 | −0.01 | |
| NO | 1 | Adsorbed at the O top site with N-end | −0.56 | 3.241 | −0.02 |
| 2 | Adsorbed at the Fe top site with N-end | −2.34 | 1.668 | −0.58 | |
| 3 | Adsorbed at the Fe top site with O-end | −1.82 | 1.984 | −0.43 | |
| 4 | Adsorbed at the Fe-O bridge site with N-end | −2.01 | 1.859 | −0.50 | |
| 5 | Adsorbed at the Fe-O bridge site with O-end | −1.57 | 2.109 | −0.29 | |
| 6 | Adsorbed at the O vacancy site with N-end | −0.69 | 2.967 | −0.07 |
| Reaction Step | Homogeneous | Heterogeneous |
|---|---|---|
| I → II (NH3 Dissociation) | 0.89 eV | 0.68 eV |
| V → VI (NH2NO Dissociation) | 1.53 eV | 0.91 eV |
| Pathway | Step 1 | Step 2 |
|---|---|---|
| E-R Mechanism Pathway | NO = N + O | N + NO = N2O |
| L-H Mechanism Pathway | NO + NO= N2O2 | N2O2 = N2O + O |
| Reaction Step | Homogeneous | Heterogeneous |
|---|---|---|
| Step 1 (Dimer Formation) | 0.56 | 0.53 |
| Step 2 (Dimer Dissociation) | 2.04 | 1.53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wang, Y.; Hu, X. Microscopic Mechanism of Fe2O3-Catalyzed NO Reduction during Sludge Combustion: A Density Functional Theory Study. Energies 2024, 17, 165. https://doi.org/10.3390/en17010165
Li J, Wang Y, Hu X. Microscopic Mechanism of Fe2O3-Catalyzed NO Reduction during Sludge Combustion: A Density Functional Theory Study. Energies. 2024; 17(1):165. https://doi.org/10.3390/en17010165
Chicago/Turabian StyleLi, Jingkuan, Yanlin Wang, and Xinhua Hu. 2024. "Microscopic Mechanism of Fe2O3-Catalyzed NO Reduction during Sludge Combustion: A Density Functional Theory Study" Energies 17, no. 1: 165. https://doi.org/10.3390/en17010165
APA StyleLi, J., Wang, Y., & Hu, X. (2024). Microscopic Mechanism of Fe2O3-Catalyzed NO Reduction during Sludge Combustion: A Density Functional Theory Study. Energies, 17(1), 165. https://doi.org/10.3390/en17010165