Abstract
Heteroatom doping is an effective strategy to regulate electrocatalysts for the oxygen evolution reaction (OER). Nonmetal heteroatoms can effectively engineer geometric and electronic structures and activating surface sites of catalysts due to their unique radius and the electronegativity of nonmetal atoms. Hence, the surface geometric and electronic structure and activity of nonmetal atoms (X, X = B, C, N, O, P)-doped Ni3S2 (X-Ni3S2) were studied to screen high-performance Ni3S2-based OER electrocatalysts through density functional theory calculation. Theoretical results demonstrated that dopants in X-Ni3S2 can alter bond length and charge of surface, modify active sites for intermediates adsorption, and adjust the theoretical overpotential. Among all dopants, C can effectively modulate surface structure, activate surface sites, weaken the adsorption of key intermediates, decrease theoretical overpotential, and enable C-Ni3S2 with the best theoretical OER activity among all X-Ni3S2 with the lowest theoretical overpotential (0.46 eV). Further experimental results verified that the synthesized C-Ni3S2 performed an improved OER activity in the alkaline condition with a considerably enhanced overpotential of 261 mV at 10 mA cm−2 as well as a Tafel slope of 95 mV dec−1 compared to pristine Ni3S2.
1. Introduction
Electrochemical water splitting is identified as a promising technology for renewable energy conversion [1,2]. Due to the relatively high energy barrier and the inertia involved in the four electron processes, the oxygen evolution reaction (OER), the anode reaction of water splitting exhibits slow kinetics, large reaction overpotential, and high energy consumption, which impede the efficiency of the hydrogen evolution reaction (HER) on the cathode of water splitting. Efficient electrocatalysts are crucial for facilitating the sluggish and energy-intensive OER. Up to now, Ru- and Ir-based oxides are the most prevalent and efficient catalysts for OER [3]. However, their inadequacy and high cost stymie their large-scale commercial applications. Therefore, designing highly active, durable, and nonprecious materials as alternative OER electrocatalysts is the key to overcoming the technical obstacles of hydrogen production and an intensively challenge for extensive commercial application of water splitting technology [4,5,6].
Ni-based nonprecious materials, such as sulfides [7,8,9], nitrides [10,11], oxides [12,13], and phosphides [9,14], have been extensively explored for OER in recent years. Among them, Ni3S2 has been regarded as a latent catalyst to replace Ru- and Ir-based noble OER catalysts due to its facile synthesis and inherent metal Ni–Ni connecting network, giving it decent conductivity and praiseworthy catalytic activity. Nevertheless, the intrinsic activity of Ni3S2 is still inferior to the Ru and Ir oxides catalysts [15]. Strategies such as heteroatom doping, heterojunction, and morphology engineering have been devoted to promote the OER performance of catalysts [16]. Among these, heteroatom doping serves as an extremely effective method, and has been widely used to tune the structure and catalytic activity of Ni3S2. For example, introducing transition metal atoms (Fe, Co, Mn, V, etc.) into the catalysts’ lattice can change lattice structure [8,16], modulate electronic structure [17,18], as well as increase the number of active sites [19,20] of catalysts resulting in improved catalytic activities for Ni3S2 materials. Similar to metal heteroatom regulation, nonmetal heteroatoms can also engineer geometric and electronic structures and activate surface sites of catalysts. Moreover, as the radius and electronegativity of nonmetal atoms (especially the second and third period nonmetal atom) usually differ greatly from that of transition-metal atoms, nonmetal atoms are more likely bringing about changes in the local coordinate environment and cause changes in the local geometric and electronic structures of active sites of catalysts [21]. This makes nonmetal heteroatoms more attractive in adjusting the electrocatalytic activity of materials. For example, the N heteroatom can significantly modify the electronic structure and morphology of Ni3S2, which brings high exposure of active sites, enhances conductivity, and optimizes HER activity [22]. Previous studies also proved that some nonmetal atoms (X, such as O, C, N) can adjust the electronic structure and increase the number of active sites in terms of HER study [23,24]. In this regard, nonmetal doping would be a more promising way to regulate the electronic structure and OER activity of Ni3S2. Even though some studies focused on improving the HER activity of Ni3S2 by nonmetal atoms doping [22,25,26], up to now, there are fewer investigations on modulating the OER activity of Ni3S2 by doping nonmetal atoms. Therefore, systematically investigating and rationally screening the nonmetal-atoms-doped Ni3S2 catalysts are needed to develop high performance OER electrocatalysts. Generally, it is time-consuming and of high cost to systematically screen the superior OER catalysts in experiments. Fortunately, with the help of high-performance computers and advanced theories of computational chemistry, we can screen the promising electrocatalysts at atomic scale by calculating the adsorption of intermediates, as well as reaction overpotentials, and further prepare the excellent catalysts by advanced experimental methods [1,23,24,25]. Thus, exploring the potential of nonmetal-atoms-doped Ni3S2 catalysts by combining theoretical prediction and experimental preparation to improve OER’s catalytic activity can further develop the strategy for designing and constructing OER electrocatalysts.
Inspired by the above considerations, we systematically investigated the OER performances of five kinds of nonmetal-atoms (X, X = B, C, N, O, P)-doped Ni3S2 (X-Ni3S2) electrocatalysts and screened out the most promising X-Ni3S2 OER catalysts. The geometric and electronic structures of X-Ni3S2, intermediates adsorption, potential determining step (PDS), and theoretical overpotentials of OER were studied using the density functional theory (DFT) calculations. As a result, C can effectively disturb the local geometric and electronic structure of Ni3S2, thus adjusting the adsorption of key intermediates among all dopants, consequently enabling Ni3S2 to exhibit excellent OER performance with more active sites and much lower theoretical overpotentials. Additionally, C dopants can also improve the OER activity of nickel hydroxides when C-Ni3S2 incurs surface oxidation during the OER process. Guided by theoretical results, the C-Ni3S2 and Ni3S2 electrocatalysts with similar structure and morphology are synthesized in this work. C-Ni3S2 was verified to exhibit more optimized OER performance than Ni3S2 by means of electrochemical tests. Our theoretical prediction joins with experiment verification results provide a guide to designing promising catalysts for OER through nonmetal heteroatom doping engineering.
2. Computational Details
The periodic DFT calculations were carried out by employing the Vienna ab initio simulation package (VASP) [27]. The projector augmented wave (PAW) potentials [28] and the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional [29] were used to describe electronic-ion interactions and model the electron exchange-correlation, respectively. A cutoff energy of 450 eV for plane-wave basis set, Monkhorst–Pack grid k-points of 3 × 3 × 1, energy convergence criterion of 10−5 eV, and force convergence criterion of 0.02 eV/Å were employed for all slab models. When calculating the adsorption energy of intermediates, dipole corrections along the surface were considered, Van der Waals interactions were corrected by using Grimme’s DFT-D3 method [30], and the solvent effect was included by using VASPsol [31].
The optimized Ni3S2 bulk [23] with rhombohedral crystal structure, lattice constant of 4.07 Å, Ni-S, and Ni-Ni bond lengths of 2.27 Å and 2.52 Å, respectively, all of which are very close to the experimental values [32]. As the (100) surface has the same atomic arrangement as that of the (001) and (010) surfaces and is the most studied and regarded as the most stable low-index surface with the lowest surface energy (Figure S1), the (100) surface was selected as the catalyst surface of Ni3S2 in the following investigations. The catalyst surface was simulated using a (2 × 2) Ni3S2(100) slab cleaved from a Ni3S2 bulk cell. The dopant X includes B, C, N, O, and P, and all these X-Ni3S2 with 2.5 at% doping content (one of the S atoms in (2 × 2) Ni3S2(100) slab containing 48 atoms was replaced by X atom) materials were validated with the possibility to be prepared in practice [23]. Furthermore, two types of X-Ni3S2(100) surface models were constructed due to their similar surface stability, as our previous research mentioned [23]. They are Xout-Ni3S2(100) and Xin-Ni3S2(100), in which X substitutes one of the top-layer S atoms (Xout) and one of the sublayer S atoms (Xin) of Ni3S2(100), respectively. Considering the surface oxidation and phase transition of Ni3S2 during the OER process [17,33], the (2 × 2) Ni(OH)2(100) and (2 × 2) NiOOH(100) surfaces were also constructed using the same method to verify the most effective dopant among those mentioned above. For all slab models, a 15 Å vacuum layer on the vertical direction of slab was added.
The key OER steps include the following:
The free energies of each species were calculated according to the equation
where G, E, ZPE, and TS are the free energy, total energy, zero-point energy, and entropic contributions (T was set to be 300 K), respectively. ZPE and TS were calculated after obtaining the vibrational frequencies () [20]:
Here, the free energy of () can be regarded as , and the electrochemical potential of electron () is equal to −, where is electrode potential relative to the reversible hydrogen electrode (RHE). Due to the inaccuracy of DFT in estimating the energy of O2, the free energy of O2 was derived according to [34]. The adsorption Gibbs free energy of main intermediates were calculated as follows:
Correspondingly, the Gibbs free energy of Equations (1)–(4) were calculated as follow:
To evaluate the OER performance, the theoretical overpotential (η) is defined as
where is the most positive value from to .
Experimental details can be found in Supplementary Materials.
3. Results and Discussion
3.1. The Local Geometric Environment and Electronic Structure of X-Ni3S2
Our previous work has validated that X-Ni3S2 can be prepared in experimental conditions, and the topmost layer and sublayer doping on Ni3S2(100) (Xout-Ni3S2(100) and Xin-Ni3S2(100), respectively) show good surface stability and might coexist due to the small difference in surface energy between them [23]. In addition, X-Ni3S2 shows good electronic conductivity because the orbitals cross the Fermi level shown in the density of states (DOS) of Ni3S2 with 2.5 at% X doping content (Figure S2). X-Ni3S2 can be a candidate for OER electrocatalysts, which can benefit the charge transfer between the surface and the intermediates during electrocatalysis [35].
In Figure 1a, the stable top-most Ni-nonmetal atomic arrangement of each surface denotes that X doping can well maintain the original ordered crystal structure of Ni3S2(100). While due to the radius (Rx) and electronegativity(χx) of X are different from those of S, X doping induces disturbance on local structure of surface. Clearly, the distance between X and Ni1 atoms (dX-Ni1, Ni1 represents the nearest Ni atoms and labeled in Figure 1a) is different from the original dS-Ni1. As shown in Figure 1b, except for the P-Ni1 bond, the other X-Ni1 bonds are shorter than the S-Ni1 bond. Additionally, C- and N-Ni3S2(100) surfaces have the shortest dX-Ni1 than other X-Ni3S2(100) surfaces due to C and N having larger χx and smaller Rx. Interestingly, though the O atom has the smallest Rx and largest χx, the O-Ni1 bond is longer than the C-Ni1 and N-Ni1 bonds. This may be because O tends to form lone pair electrons when it bonds with the nearest metal atoms [24], which keeps it from getting any closer to Ni atoms. Moreover, the Ni1-Ni5 bonds (both Ni1 and Ni5 connect with X, Figure 1a) are also shorter than those on the pristine Ni3S2(100) surface (Table S1) due to the change of X-Ni1 bond. The change of X-Ni1 bond directly induces the change of adjacent Ni-Ni bond length, which further affects the bond length of adjacent Ni-S and distance between top-most Ni atoms. For example, the shorter C/N-Ni1 bond causes obviously shrinkage in the Ni1-Ni5 bond, which further elongates the Ni1-S3 and Ni5-S2 bonds, finally causing the slight displacement of topmost Ni atoms. Especially, among all X atoms, C and N cause more obvious changes in local Ni-Ni and Ni-S bond lengths, denoting that they can trigger prominent local structure disturbance on the Ni3S2(100) surface.
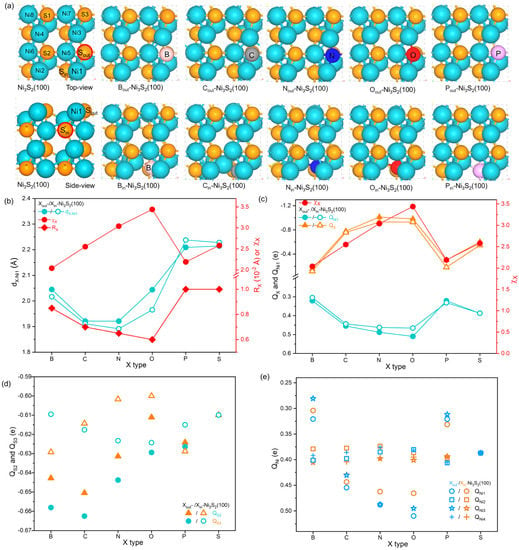
Figure 1.
(a) The structures of Ni3S2(100), Xout-Ni3S2(100), and Xin-Ni3S2(100) (surface Ni atoms are labeled as Ni1~Ni8, and Ni1~Ni4 represent top most Ni atoms, Ni5~Ni8 represent sublayer Ni atoms). (b) The bond length of X-Ni1 (dX-Ni1) corelates with the electronegativity (χx) and atom radius (Rx) of X (S on horizontal axis represent pristine Ni3S2(100)). (c) Bader charge of Ni1(QNi1) and X(QX) correlate with χx for both Xout-Ni3S2(100) and Xin-Ni3S2(100) systems. (d) The Bader charge of S2 and S3 atoms. (e) The Bader charge of Ni1~Ni4 atoms.
The local geometric structure was disturbed by the change of local coordination environment, which could be accompanied by changes in local electronic structure. Following this, the charges of surface atoms were calculated by Bader charge analysis [36] to examine the alteration of X on the electronic structure of the surface. As shown in Figure 1c, for all surfaces, the Ni1 atom which was directly affected by X possesses a positive charge (QNi1), while X shows a negative charge (QX), denoting the Ni atoms deliver the charge to X atoms. Notably, compared with QNi1 of pristine Ni3S2(100), QNi1 is altered by X, and X with larger χx results in a more positive QNi1. This charge transfer between X and Ni and the above-mentioned local structure change further affect the charge of adjacent S atoms (Figure 1d). For example, after Cout doping, a shorter C-Ni1 bond than the pristine S-Ni1 bond combines with more charge transfer between C and Ni1 than S and Ni1, resulting in the adjacent S (S3 atom in Figure 1a) atom bonding more strongly with sublayer Ni atoms (Figure S3); thus, it has slightly more charge than the corresponding S atom on Ni3S2(100). Furthermore, the charges of surface Ni sites (i.e., top-most four Ni atoms, Ni1, Ni2, Ni3, and Ni4 in Figure 1a) are also disturbed by the change of local coordination environment. As shown in Figure 1d, on one X-Ni3S2(100) surface, QNi1 and QNi3 sites are intensely altered, accompanied by QNi2 and QNi4 being slightly altered by X, resulting in the four Ni sites having different charges. For example, QNi1, QNi2, QNi3, and QNi4 of Cout-Ni3S2(100) are 0.46e, 0.48e, 0.43e, and 0.39e respectively, which are about 0.01~0.07e more positive than QNi1 of Ni3S2(100), indicating C dopants play a major role in alteration of electronic structure of Ni sites. Thus, according to above analysis, X dopants on Ni3S2(100) surface can disturb local geometric structure, change the charge distribution of surface, and modify the charge of Ni sites to varying degrees, which could further affect the OER intermediates’ adsorption performance and consequently modify the OER activity on Ni3S2(100).
3.2. The OER Activity of X-Ni3S2(100)
We further probe into the OER activity of X-Ni3S2(100) to screen the promising X-Ni3S2 catalysts. For OER, the *OH, *O, and *OOH are key intermediates for elementary reactions (Equations (1)–(4)) on nickel-based sulfide [37]. To evaluate the OER activity of the pristine Ni3S2 surface, the adsorption of the above intermediates and free energy change () of each elementary step on the pristine Ni3S2(100) surface were calculated, respectively, to recognize the adsorption site and the potential determining step (PDS) of OER. On pristine Ni3S2(100), *OH and *OOH tend to be absorbed on the Ni top site, while *O is likely to be absorbed on the Ni1-Ni2 bridge site as shown in Table S2; thus, the metal sites should be the reaction sites for OER. Accordingly, the OER pathway is illustrated in Figure 2a, and the free energy diagram is shown in Figure 2b. Apparently, all steps run uphill at U = 0 V vs. NHE and the third step, i.e., the *OOH formation (Equation (4)), is the PDS. This generates the highest of about 1.95 eV among four steps, corresponding to a theoretical overpotential (η) value of 0.72 V (or an onset potential value of 1.95 V), which is needed to make all steps of the free energy diagram go downhill. It means that the pristine Ni3S2(100) surface has a strong interaction with *OH and *O or too weak binding with *OOH. This is consistent with how the metal usually possesses a strong bonding interaction with *OH and *O [38]. Thus, it is deduced that properly weakening *O or OH* adsorption or strengthening *OOH binding would reduce the η and enhance the OER performance.
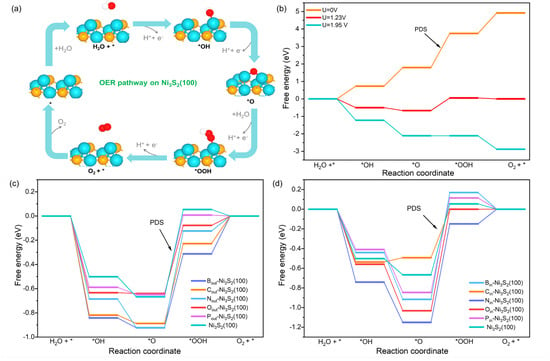
Figure 2.
(a) The OER process on Ni site of Ni3S2(100). (b–d) Free energy diagrams of OER on Ni1 site of Ni3S2(100), Xout-Ni3S2(100), and Xin-Ni3S2(100), respectively. (* means surface active site).
Further, we calculated the OER activity on X-Ni3S2. We first focused on the OER activity of Ni1 site, as it is directly bonded with X. Similar to the intermediate adsorption on the pristine Ni3S2(100) surface, the *OH, *O, and *OOH adsorbed on the Ni1 top, Ni1-Ni2 bridge, and Ni1 top sites, respectively. The free energy diagrams of the OER steps on the Ni1 site of Xout and Xin-Ni3S2(100) are shown in Figure 2c,d, respectively. For Xout-Ni3S2(100), all X dopants enhanced the *OH and *OOH adsorption, and B, C, and N enhanced the O adsorption, while O and P doping slightly weakened the O adsorption. This maintains the third step as the PDS, and the of PDS follows the trend of O > B > C > P > S > N (S represents pristine Ni3S2(100)), with the η trend of O (0.56 V) > B (0.61 V) > C (0.66 V) > P (0.67 V) > S (0.72 V) > N (0.80 V). Thus, except for N, all other Xout doping can enhance the OER activity on the Ni1 site of Ni3S2(100), especially the Oout doping, which almost enhances the OER activity on the Ni1 site by reducing η by 0.16 V compared with Ni3S2(100).
For Xin-Ni3S2(100), except B and P which weakened OH and OOH adsorption, all other X dopants enhanced the OH and OOH adsorption, and except C which weakened O adsorption, all other X dopants strengthened O adsorption. The PDS on the Ni1 site of Xin-Ni3S2(100) is still the *OOH formation step. The of PDS follows the trend of C > S > P > N > O > B, with the overpotential trend of C (0.49 V) > S (0.72 V) > P (0.96 V) > N (1.00 V) > O (1.03 V) > B (1.09 V). Thus, only Cin doping can enhance the OER activity on Ni1 site by reducing η by 0.23 V compared with Ni3S2(100), while other Xin doping may hinder the OER activity on the Ni1 site. According to the OER activity on the Ni1 site of both Xout- and Xin-Ni3S2(100), C dopant is beneficial for improving the OER activity of its neighbor Ni site, while other X atoms enhance the OER activity of their neighbor Ni site only when doped at the topmost S site. As Xin and Xout may coexist in physical catalysts, the promising X-Ni3S2 catalysts should satisfy that both Xin-doped Ni3S2 and Xout-doped Ni3S2 have better OER activity than pristine Ni3S2. In this condition, the C dopant should be a good candidate for doping on the Ni3S2(100) surface due to how both Cin and Cout can enhance the OER activity of their adjacent Ni sites.
On both Xout- and Xin-Ni3S2(100) surfaces, Ni1 sites are directly affected by X, exhibiting different OER activity from that on pristine Ni3S2(100). Considering that the surface geometric and electronic structure are disturbed by X dopants as discussed in Section 3.1, the intermediates’ adsorption, and hence OER activity, on other Ni sites (Ni2, Ni3, and Ni4) may also be affected by X. Correspondingly, other Ni sites have the potential be more efficient OER active sites. Thus, OER activity on other Ni sites of each surface should be calculated and the number of efficient active sites needs to be counted to better screen out the most effective X dopants. Nevertheless, the adsorption of *O tends to coordinate with more metal atoms and has more complicated adsorption sites than *OH and *OOH (Figure S4 and Table S3), which make the calculation and screening of the effective adsorption site for OER intermediates too complex and high-cost. Usually, the adsorption free energy of *OH () can be scaled, correlated with that of *OOH () on the same site [39,40]. Thus, it is possible to conveniently estimate the OER activity of different sites on X-Ni3S2(100) by calculating the difference between the adsorption energy of *OOH or *OH and *O (). Based on this, the relationship between and on Ni1 sites of all X-Ni3S2(100) were tested. As shown in Figure 3a, the larger corresponds to larger , validating the linear scaling relation between and , which follows the equation of
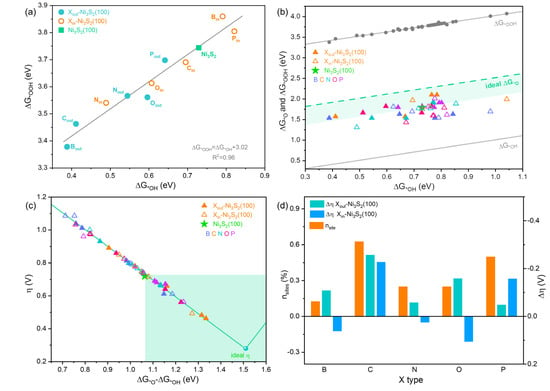
Figure 3.
(a) The scale relation between and . (b) The correlation between (or ) and , and (c) the η vs. value of on different X-Ni3S2(100) surfaces (the highlighted area in (b) and (c) denote the relative optimum and small η values of X-Ni3S2(100), respectively). (d) The improved active site proportion (nsite) on X-Ni3S2(100) and the difference between minimum η of each surface and η of Ni3S2(100).
Thus, the sum free energy change of the second and third steps is fixed, that is
Hence, we can deduce that when is in the middle of these two adsorption energies, that is, or equals to the critical value of 1.51 eV, the lowest η of about 0.28 V for OER is obtained. Therefore, it suggests that the value of or could be used as a descriptor to evaluate the OER activity. If the difference value of is smaller than the critical value, the formation of *OOH is the PDS, such as that on pristine Ni3S2(100), and the η at standard conditions is
Conversely, if the difference value of is larger than the critical value, the formation of *O (the second step) becomes the PDS, and the theoretical overpotential at standard conditions is
Accordingly, all possible and corresponding on X-Ni3S2(100) were calculated thoroughly (*OH on top sites, and *O on bridge sites shown in Figure S4) to evaluate the OER activity more accurately. The corresponding was calculated using the linear scaling equation of . Figure 3b presents the location of , and the difference value between and . All on X-Ni3S2(100) are close to the , denoting that the *OOH formation is still the PDS on pristine Ni3S2(100). Though the *O adsorption strength on X-Ni3S2(100) is a little stronger compared to the ideal *O adsorption strength, there are still many active sites showing weaker on X-Ni3S2(100) than on pristine Ni3S2(100), indicating a possible higher OER activity of the former. The relationship between and η is further plotted to confirm how many active sites of X-Ni3S2(100) have higher OER activity than pristine Ni3S2(100). In Figure 3c, there are 1, 5, 2, 2, and 4 of 8 sites on B-, C-, N, O-, and P-Ni3S2(100), respectively that have lower η than that of pristine Ni3S2(100), meaning that all X dopants can activate at least one surface Ni site, and C dopant contributes to the most active sites among five dopants. Moreover, the proportion of improved active site (nsite) and change value of η (η, more negative value means better activity than pristine Ni3S2(100)) on the best active site of each X-Ni3S2(100) are listed in Figure 3d. The nsite follow the sequence of C (62.5%) > P (50.0%) > O = N (25%) > B (12.5%), and η follows Pout (−0.05 V) > Nout (−0.06 V) > Bout (−0.11 V) > Oout (−0.16 V) > Cout (−0.26 V), and Oin (0.11 V) > Bin (0.06 V) > Nin (0.02 V) > Pin (−0.16 V) > Cin (−0.23 V) for Xout-Ni3S2(100) and Xout-Ni3S2(100), respectively. These values indicate that C is prominent among all dopants, can efficiently alter the OER active site and activate 62.5% of the surface Ni sites, and enable best active sites of both Cout- and Cin-Ni3S2(100) which have a much lower η relative to η of Ni3S2(100). Especially, Cout doping endows the surface with the lowest η of 0.46 V (Figure 3c), which is even comparable to 0.37 V of RuO2 [41] and 0.51 V of IrO2 [42]. Thus, considering both the number of active sites and activity of the Ni site, the C dopant can effectively activate the surface OER active site and can be selected as most effective dopant for X-Ni3S2 OER catalysts among all X dopants.
Furthermore, considering the surface oxidation and phase transition of Ni3S2 to exist as Ni-based hydroxides (such as Ni(OH)2 and NiOOH) during OER processes [17,33], and to further verify the efficiency of the promising C dopant, we evaluated the OER activity of C-doped Ni-based hydroxides (the adsorption structures are shown in Figure S5). As shown in Figure S6a,b, when C is doped on both Ni(OH)2 and NiOOH, it can enhance the adsorption of OER intermediates, which is especially beneficial to the O adsorption. Consequently, the decrease the free energy of the PDS generates the η of 0.07 and 0.13 V lower than those of pristine Ni(OH)2 and NiOOH, respectively. Thus, C dopants not only help to improve the OER activity of pristine Ni3S2 but also contribute to the OER activity of Ni-based hydroxides, denoting that C-Ni3S2 can serve as a promising OER electrocatalyst.
Based on these theoretical prediction results, the C-doped Ni3S2 nanosheets (C-Ni3S2 NSs), as well as Ni3S2 nanosheets (Ni3S2 NSs), were synthesized in the same way used in previous study [23] (also see Supplementary Matrials for more detail) to verify its application in OER. Our previous work has demonstrated that C-Ni3S2 NSs and pristine Ni3S2 NSs have similar nanosheet morphology and the same crystal structure and uniform elemental distribution. Thus, we mainly focused on studying the OER performance of these two samples in this study. The OER activities of samples were evaluated in 1 M KOH. Figure 4a,b shows the LSV of the above samples and a comparison of their catalytic activities for OER, respectively. The C-Ni3S2 NSs exhibit higher OER activity than pristine Ni3S2 NSs, needing a lower overpotential of 261 mV and 480 mV to reach current density of 10 mA cm−2 and 900 mA cm−2, respectively, compared with Ni3S2 NSs (310 mV, 590 mV, respectively). The corresponding Tafel slope (Figure S7a) of C-Ni3S2 NSs (96.18 mV dec−1) is lower than that of pristine Ni3S2 NSs (109.35 mV dec−1), indicating the favorable reaction kinetics of C-Ni3S2 NSs. Nyquist plots in Figure S7b illustrate that C-Ni3S2 NSs have faster charge transfers between electrolyte and electrode. Furthermore, C-Ni3S2 NSs also show higher exchange current density (J0) (Figure 4b and Figure S7c) and average turnover frequency (TOF) of 0.02 mA cm−2 and 0.31, respectively, than those of Ni3S2 NSs (0.02 mA cm−2 and 0.06, respectively). Additionally, C-Ni3S2 NSs exhibits similar electrochemical double-layer capacitance with that of Ni3S2 NSs (Figure S7d). Moreover, the OER activity of the as-prepared catalyst is comparable to that of most reported nickel-based OER catalysts (Table S4).These experimental results validate that the C-Ni3S2 NSs have improved OER performance compared with pristine Ni3S2 NSs, which is in line with the above DFT results, highlighting the effectiveness of C dopant modification in the OER activity of Ni3S2.
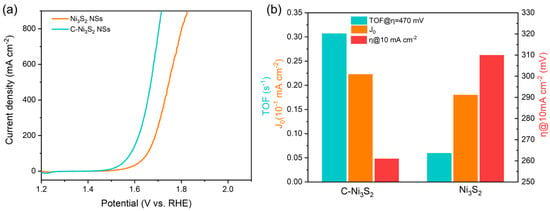
Figure 4.
(a) The OER polarization curves and (b) overpotential (η) at current density of 10 mA cm−2, average turnover frequency (TOF), and exchange current density (J0) of Ni3S2 NSs and X-Ni3S2 NSs electrocatalysts.
3.3. The Origin of Improved OER Activity of C-Doped Ni3S2(100)
For further insight into the improved OER activity of C-doped Ni3S2(100), the geometric and electronic structure of key OER intermediates on the most effective site, i.e., the Ni3 site of Cout-Ni3S2(100), were discussed as an example. As shown in Figure S8, for OH and OOH, they both adsorbed on the Ni3 top site to form a single O-Ni bond, and the bond length of all O-Ni bonds on Cout-Ni3S2(100) are longer than that on Ni3S2(100). The charge density difference displayed in Figure 5a shows O of *OH or *OOH can acquire charge through O-Ni bods, due to how charge can transfer from Ni to O and some charge can also transfer from C and S to O through Ni. The Bader charge in Figure 5a indicates that O of *OH or *OOH acquires less charge from the Ni3 site of Cout-Ni3S2(100) than from the Ni site of Ni3S2(100). Furthermore, the DOS of the O-Ni atoms pair in *OH shown in Figure 4b display that the main states below fermi level (−3.0~−4.5 eV) of the O atom on Cout-Ni3S2(100) are slightly less than that of the O atom on Ni3S2(100), verifying less charge transfer from the Ni3 site of Cout-Ni3S2(100) to *OH/*OOH. Thus, the C dopant can slightly elongate the O-Ni3 bond and decrease the charge transfer from Ni3 to O, consequently helping to impair the O-Ni3 interaction and weaken the OH and OOH adsorption (Figure 4c). For *O adsorption on Ni3 of Cout-Ni3S2(100), it tends to bond with Ni3 and the sublayer Ni atom (Ni7 in Figure 1a), forming two Ni-O bonds after optimization, which are different from those of *O bonds with the topmost two Ni atoms (Ni3 and Ni4) on Ni3S2(100). This makes the average Ni-O bond of *O on Cout-Ni3S2(100) longer than that of *O on Ni3S2(100), and *O acquires less charge from Ni (Ni3 and Ni7) of Cout-Ni3S2(100) than from Ni (Ni3 and Ni4) of Ni3S2(100) (Figure 5a). Furthermore, the states below Fermi level of O on Cout-Ni3S2(100) are less than that of *O on Ni3S2(100), manifesting the lower charge transfer between O-Ni. More importantly, the electronic states interaction between O and Ni of Cout-Ni3S2(100) occur at a higher energy level than that of Ni3S2(100), confirming the weaker O-Ni interaction, and consequently, the weaker *O adsorption on the Ni3 site of Cout-Ni3S2(100) than that on the Ni site of Ni3S2(100).
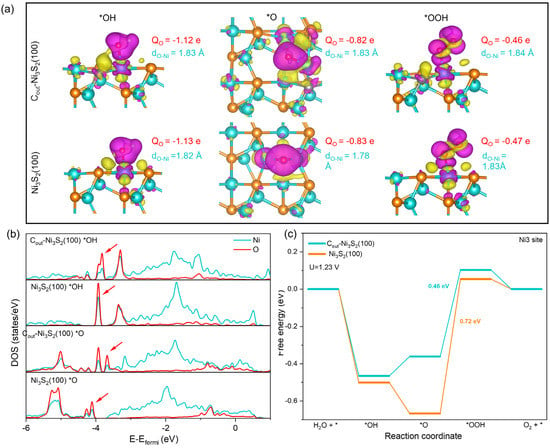
Figure 5.
(a) The Bader charge of O (QNi), bond legth of O-Ni (dO-Ni), and the charge density difference of OER intermediates adsorption on Ni3 site of Cout-Ni3S2(100) and Ni site of Ni3S2(100) (The purple part represents charge accumulation, while yellow part represents charge depletion. The isosurface level is set as 0.002 eV Å−3). (b) The density of states (DOS) for OH and O adsorption on the Ni3 site of Cout-Ni3S2(100) and Ni site of Ni3S2(100). (c) The OER free energy diagram of Ni3 site of Cout-Ni3S2(100) and Ni site of Ni3S2(100).
The weakened OER intermediate adsorption on the Ni3 site of Cout-Ni3S2(100) mainly results from an electronic structure alerted by C dopants. C dopant makes the charge of Ni3 site more positive and the d band center of Ni3 site negatively shift when compared with those of the Ni site on Ni3S2(100) (Table S5). Consequently, this decreases the charge transferred from Ni to O and weakens the O-Ni bonds. Moreover, the surface structure also contributes to weakening the O adsorption. Specifically, the disturbance on local structure caused by Cout makes the Ni3-Ni4 distance (4.68 Å) longer than the pristine Ni3-Ni4 distance (4.07 Å), which leads O to prefer bonding with Ni3 and the Ni7 atoms rather than Ni4 atom. Due to the more saturated coordination and lower position in the Z direction of the Ni4 atom, O forms a longer and weaker Ni-O bond with the Ni atom. Thus, the special surface geometric and electronic structure induced by C doping can weaken OER intermediates’ (especially O) adsorption on the Ni3 site of Cout-Ni3S2(100) and reduce the of the PDS (), consequently yielding a better OER activity with η as low as 0.46 V (Figure 5c).
4. Conclusions
In summary, the geometric structure, charge transfer, and OER activity of nonmetal atoms X (X = B, C, N, O, P)-doped Ni3S2 were studied systematically using DFT calculations. This demonstrated that the geometric structure and electronic structure, hence OER activity, are disturbed by the introduction of X dopants. Among all X dopants, C can be a promising dopant for Ni3S2 materials due to the way it can efficiently alter the local geometric structure and electronic structure of the Ni site, hence adjusting the key intermediates adsorption. This consequently enables the modified Ni3S2(100) surface to have the most effective active sites and exhibits the best OER activity, with the lowest theoretical overpotential of 0.46 V, which is 0.26 V lower than that of pristine Ni3S2(100). Further electrochemical experiments verified that C-doped Ni3S2 indeed presents higher intrinsic OER activity than pristine Ni3S2, which needs the 49 mV lower overpotential than that of pristine Ni3S2 to deliver a geometrical current density of 10 mA cm−2 in 1 M KOH. Above all, the DFT calculation and experimental results jointly highlight the promising C dopant in promoting the OER performance of Ni3S2. Our study offers guidance for screening and fabricating promising OER catalysts through nonmetal doping engineering, which can inspire more exploration of nonmetal doping of other electrocatalysts.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/en16020881/s1. Figure S1. The surface energy () of (100), (111) and (110) surfaces. (As the (001) and (010) have same structure as (100) and (101) has same structure as (110), we do not consider (001), (010) and (101) anymore. The is calculated according to , where is the energy of slab, , and represent the number of Ni, S and X atoms respectively, and A is the surface area of a slab). Figure S2. (a) The partial density of states for pure Ni3S2. (b) The total density of states for X doped Ni3S2(only half of DOS is displayed). Figure S3. the Bond length of S3-Ni and X-Ni bonds on Cout-Ni3S2(100) and Ni3S2(100). Figure S4. The adsorption site on (a) Xout-Ni3S2(100) surface or pristine Ni3S2(100) surface and (b) Xin-Ni3S2(100) surface. (Top sites are for OH or OOH adsorption, while bridge sites are for O adsorption). Figure S5. The structure of OER intermediats adsorption on prinstin and C doped Ni(OH)2 and NiOOH. Figure S6. (a,b) Free energy diagram of OER on pristine and C doped Ni(OH)2 and NiOOH respectively. Figure S7. (a) The Tafel slop, (b) Nyquist plots obtained by electrochemical impedance spectroscopy. (c) turnover frequency (TOF) and (d) electrochemical double-layer capacitance of C- Ni3S2 NSs and Ni3S2 NSs. Figure S8. The structures of OER intermediates on Ni3 site of Cout-Ni3S2(100) and pristine Ni3S2(100). Table S1. The bond length of X-Ni1 (dX-Ni1) and Ni1-Ni5 (dNi1-Ni5) bonds, corresponding bonds length deviation after X doping (ΔdX-Ni1 and ΔdNi1-Ni5, more nagetive value means shorter bond), electronegativity (χx) and atom radius (Rx) of X. Table S2. The adsorption free energy of OH, O and OOH on Ni3S2(100) surface. Table S3. The and of Cout-Ni3S2(100) and Cin-Ni3S2(100) (as examples). Table S4. The comparison of OER activity between catalyst in this work and in recent works. Table S5. Bader charge and b band center of Ni3 site and Ni site of Cout-Ni3S2(100) and Ni3S2(100).
Author Contributions
Conceptualization, X.Z.; methodology, X.Z.; validation, L.Z.; data curation, W.H.; writing—original draft preparation, X.Z.; writing—review and editing, L.L. and S.L.; visualization, X.Z.; supervision, L.L.; funding acquisition, X.Z., L.L. and S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. 52200076), the National Key R&D Program (2021YFB4000301), the Research Foundation of Chongqing University of Science and Technology (Grant No. ckrc2022026), Natural Science Foundation of Chongqing (Grant No. CSTB2022NSCQ-BHX0035), Special Research Assistant Program of Chinese Academy of Science.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Babar, P.; Mahmood, J.; Maligal-Ganesh, R.V.; Kim, S.-J.; Xue, Z.; Yavuz, C.T. Electronic structure engineering for electrochemical water oxidation. J. Mater. Chem. A 2022, 10, 20218–20241. [Google Scholar] [CrossRef]
- Lu, S.; Hummel, M.; Gu, Z.; Wang, Y.; Wang, K.; Pathak, R.; Zhou, Y.; Jia, H.; Qi, X.; Zhao, X.; et al. Highly efficient urea oxidation via nesting nano-nickel oxide in eggshell membrane-derived carbon. ACS Sustain. Chem. Eng. 2021, 9, 1703–1713. [Google Scholar] [CrossRef]
- Reier, T.; Oezaslan, M.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction (OER) on Ru, Ir, and Pt Catalysts: A Comparative Study of Nanoparticles and Bulk Materials. ACS Catal. 2012, 2, 1765–1772. [Google Scholar] [CrossRef]
- Kwon, T.; Hwang, H.; Sa, Y.J.; Park, J.; Baik, H.; Joo, S.H.; Lee, K. Cobalt Assisted Synthesis of IrCu Hollow Octahedral Nanocages as Highly Active Electrocatalysts toward Oxygen Evolution Reaction. Adv. Funct. Mater. 2017, 27, 1604688. [Google Scholar] [CrossRef]
- Lee, Y.; Suntivich, J.; May, K.J.; Perry, E.E.; Shao-Horn, Y. Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, H.; Yan, Y.; Cao, J.; Li, X.; Zhou, S.; Peng, Z.; Zeng, J. Engineering the Electrical Conductivity of Lamellar Silver-Doped Cobalt(II) Selenide Nanobelts for Enhanced Oxygen Evolution. Angew. Chem. Int. Ed. Engl. 2017, 56, 328–332. [Google Scholar] [CrossRef]
- Gu, C.; Hu, S.; Zheng, X.; Gao, M.R.; Zheng, Y.R.; Shi, L.; Gao, Q.; Zheng, X.; Chu, W.; Yao, H.B.; et al. Synthesis of Sub-2 nm Iron-Doped NiSe2 Nanowires and Their Surface-Confined Oxidation for Oxygen Evolution Catalysis. Angew. Chem. Int. Ed. Engl. 2018, 57, 4020–4024. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Hummel, M.; Gu, Z.; Gu, Y.; Cen, Z.; Wei, L.; Zhou, Y.; Zhang, C.; Yang, C. Trash to treasure: A novel chemical route to synthesis of NiO/C for hydrogen production. Int. J. Hydrogen Energy 2019, 44, 16144–16153. [Google Scholar] [CrossRef]
- Yan, C.; Yang, X.; Lu, S.; Han, E.; Chen, G.; Zhang, Z.; Zhang, H.; He, Y. Hydrothermal synthesis of vanadium doped nickel sulfide nanoflower for high-performance supercapacitor. J. Alloys Compd. 2022, 928, 167189. [Google Scholar] [CrossRef]
- Park, S.H.; Kang, S.H.; Youn, D.H. Direct One-Step Growth of Bimetallic Ni2Mo3N on Ni Foam as an Efficient Oxygen Evolution Electrocatalyst. Materials 2021, 14, 4768. [Google Scholar] [CrossRef]
- Luo, X.; Ma, H.; Gao, J.; Yu, L.; Gu, X.; Liu, J. Nickel-Rich Ni3N Particles Stimulated by Defective Graphitic Carbon Nitrides for the Effective Oxygen Evolution Reaction. Ind. Eng. Chem. Res. 2022, 61, 2081–2090. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Ji, W.-B.; Hu, B.-C.; Liang, H.-W.; Xu, X.-X.; Yu, Z.-L.; Li, B.-Y.; Yu, S.-H. Partially oxidized Ni nanoparticles supported on Ni-N co-doped carbon nanofibers as bifunctional electrocatalysts for overall water splitting. Nano Energy 2018, 51, 286–293. [Google Scholar] [CrossRef]
- Xia, B.; Wang, T.; Jiang, X.; Li, J.; Zhang, T.; Xi, P.; Gao, D.; Xue, D. N+-ion irradiation engineering towards the efficient oxygen evolution reaction on NiO nanosheet arrays. J. Mater. Chem. A 2019, 7, 4729–4733. [Google Scholar] [CrossRef]
- Kim, B.K.; Kim, S.-K.; Cho, S.K.; Kim, J.J. Enhanced catalytic activity of electrodeposited Ni-Cu-P toward oxygen evolution reaction. Appl. Catal. B 2018, 237, 409–415. [Google Scholar] [CrossRef]
- Wu, Y.; Li, G.-D.; Liu, Y.; Yang, L.; Lian, X.; Asefa, T.; Zou, X. Overall Water Splitting Catalyzed Efficiently by an Ultrathin Nanosheet-Built, Hollow Ni3S2-Based Electrocatalyst. Adv. Funct. Mater. 2016, 26, 4839–4847. [Google Scholar] [CrossRef]
- Zhao, Y.; You, J.; Wang, L.; Bao, W.; Yao, R. Recent advances in Ni3S2-based electrocatalysts for oxygen evolution reaction. Int. J. Hydrogen Energy 2021, 46, 39146–39182. [Google Scholar] [CrossRef]
- Ai, T.; Wang, H.; Bao, W.; Feng, L.; Zou, X.; Wei, X.; Ding, L.; Deng, Z.; Rao, B. Fe-V synergistic doping effect of hierarchical Ni3S2 oblate-nanorod arrays for efficient electrocatalytic oxygen evolution reaction. Chem. Eng. J. 2022, 450, 138358. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y.; Xiang, H.; Lei, H.; Xu, B.B.; Xing, L.; Yu, E.H.; Liu, T.X. Mass transfer effect to electrochemical reduction of CO2: Electrode, electrocatalyst and electrolyte. J. Energy Storage 2022, 52, 104764. [Google Scholar] [CrossRef]
- Zhong, B.; Cheng, B.; Zhu, Y.; Ding, R.; Kuang, P.; Yu, J. Hierarchically porous nickel foam supported Fe-Ni3S2 electrode for high-current-density alkaline water splitting. J. Colloid Interface Sci. 2022, 629, 846–853. [Google Scholar] [CrossRef]
- Liu, Q.; Xie, L.; Liu, Z.; Du, G.; Asiri, A.M.; Sun, X. A Zn-doped Ni3S2 nanosheet array as a high-performance electrochemical water oxidation catalyst in alkaline solution. Chem. Commun. 2017, 53, 12446–12449. [Google Scholar] [CrossRef]
- Wang, H.-F.; Tang, C.; Li, B.-Q.; Zhang, Q. A review of anion-regulated multi-anion transition metal compounds for oxygen evolution electrocatalysis. Inorg. Chem. Front. 2018, 5, 521–534. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, T.; Zhang, M.; Tong, Y.; Zhong, C.; Zhang, N.; Zhang, L.; Wu, C.; Xie, Y. 3D Nitrogen-Anion-Decorated Nickel Sulfides for Highly Efficient Overall Water Splitting. Adv. Mater. 2017, 29, 1701584. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, L.; Huang, J.; Peng, L.; Deng, M.; Li, L.; Li, J.; Chen, H.; Wei, Z. Boosting Hydrogen Evolution Reaction of Nickel Sulfides by Introducing Nonmetallic Dopants. J. Phy. Chem. C 2020, 124, 24223–24231. [Google Scholar] [CrossRef]
- Zheng, X.Q.; Peng, L.S.; Li, L.; Yang, N.; Yang, Y.J.; Li, J.; Wang, J.C.; Wei, Z.D. Role of non-metallic atoms in enhancing the catalytic activity of nickel-based compounds for hydrogen evolution reaction. Chem. Sci. 2018, 9, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Tang, J.; Wang, J.; Shao, M.; Chai, J.; Wang, S.; Yang, M.; Yang, Y.; Wang, N.; Wang, S.; et al. 3D heterostructured pure and N-Doped Ni 3 S 2 /VS 2 nanosheets for high efficient overall water splitting. Electrochim. Acta 2018, 269, 55–61. [Google Scholar] [CrossRef]
- Ding, Y.; Li, H.; Hou, Y. Phosphorus-doped nickel sulfides/nickel foam as electrode materials for electrocatalytic water splitting. Int. J. Hydrogen Energy 2018, 43, 19002–19009. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phy. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector Augmented-Wave Method. Phy. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Fleet, M.E. The crystal structure of heazlewoodite, and metallic bonds in sulfide mineral. Am. Miner. 1977, 62, 341–345. [Google Scholar]
- Lu, S.; Hummel, M.; Kang, S.; Pathak, R.; He, W.; Qi, X.; Gu, Z. Density Functional Theory Investigation of the NiO@Graphene Composite as a Urea Oxidation Catalyst in the Alkaline Electrolyte. ACS Omega 2022, 6, 14648–14654. [Google Scholar] [CrossRef]
- Peng, L.S.; Wang, J.; Nie, Y.; Xiong, K.; Wang, Y.; Zhang, L.; Chen, K.; Ding, W.; Li, L.; Wei, Z.D. Dual-Ligand Synergistic Modulation: A Satisfactory Strategy for Simultaneously Improving the Activity and Stability of Oxygen Evolution Electrocatalysts. ACS Catal. 2017, 7, 8184–8191. [Google Scholar] [CrossRef]
- Ghadge, S.D.; Velikokhatnyi, O.I.; Datta, M.K.; Damodaran, K.; Shanthi, P.M.; Kumta, P.N. Highly Efficient Fluorine Doped Ni2P Electrocatalysts for Alkaline Mediated Oxygen Evolution Reaction. J. Electrochem. Soc. 2021, 168, 064512. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Feng, L.L.; Yu, G.; Wu, Y.; Li, G.D.; Li, H.; Sun, Y.; Asefa, T.; Chen, W.; Zou, X. High-index faceted Ni3S2 nanosheet arrays as highly active and ultrastable electrocatalysts for water splitting. J. Am. Chem. Soc. 2015, 137, 14023–14026. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Back, S.; Jung, Y. Importance of Ligand Effects Breaking the Scaling Relation for Core-Shell Oxygen Reduction Catalysts. Chemcatchem 2017, 9, 3173–3179. [Google Scholar] [CrossRef]
- Liang, W.; Chen, J.; Liu, Y.; Chen, S. Density-Functional-Theory Calculation Analysis of Active Sites for Four-Electron Reduction of O2on Fe/N-Doped Graphene. ACS Catal. 2014, 4, 4170–4177. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Sun, W.; Song, Y.; Gong, X.Q.; Cao, L.M.; Yang, J. An efficiently tuned d-orbital occupation of IrO2 by doping with Cu for enhancing the oxygen evolution reaction activity. Chem. Sci. 2015, 6, 4993–4999. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).