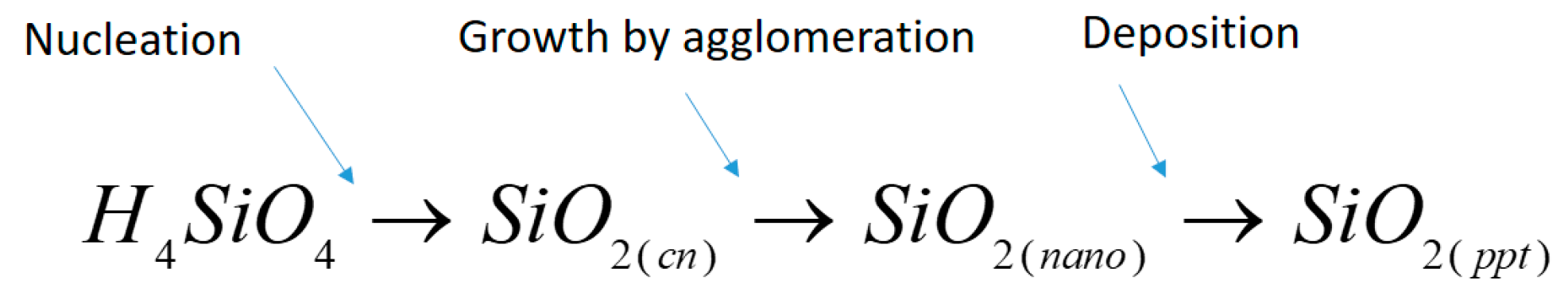1. Introduction
Geothermal energy is an important source of low-carbon emission, weather-independent, baseload energy in the near future. Geothermal energy from supercritical sources is superior in its low power cost if such resources can be utilized efficiently [
1,
2]. It is known that silica solubility in superheated steam increases with pressure [
3]. The kinetics of precipitation from pressurized steam differ from the solidification of silica in liquid water. Research on precipitation of silica from highly supersaturated superheated steam is scarce, as exploration of geothermal wells from supercritical waters, where silica content is expected to be high [
4], is relatively new. Knowledge of particle number density, size and time scales of growth in different depressurization scenarios is essential to handle deposits and avoid scaling in inconvenient parts of the steam processing.
The inspiration for the hot case well considered in this paper is the deep wells drilled into near magmatic areas as attempted by the Iceland Deep Drilling Project (IDDP) and several other projects in recent years [
5,
6,
7]. The IDDP1 well showed that production close to a magma plume was possible and hit the record as the hottest-producing well on the planet in 2010 [
8,
9]. If supercritical fluids similar to that modeled in the case study for this paper are used in a steam turbine where silica is extracted without energy loss, the electric power per well can be up to 49 MW for a well producing at 50 kg/s [
10]. The thermal efficiency,
, can be up to 50%, compared to a conventional well that may range between 8–20%. The concept, therefore, represents a significant decrease in energy cost per well. Conventional silica handling involves quenching of the fluid, which leads to a potential decrease of down to 30 MW of electric power per well [
10]. Knowledge that may lead to improved methods for silica handling is, thus, important to maximize power output. This, however, requires knowledge of the detailed process of precipitation from depressurized supercritical steam with relatively high mineral concentrations. References [
10,
11] compare different methods of utilizing the geothermal steam from a supercritical fluid reservoir considering silica precipitation and formation of hydrochloric acid, respectively.
The model presented in this paper describes the precipitation process of solid silica from steam at varying pressures and a constant enthalpy. The model quantifies the total deposited material in a specific system for varying initial supersaturation values. Supersaturation is herein defined as the ratio of the actual concentration of silicic acid in the solution to the equilibrium concentration for amorphous silica. The calculated deposition rate presented is based on the numerical integration of the classical nucleation theory and growth by agglomeration. To complete the model, the deposition of particles onto a surface and hydrodynamic effects are included. These three separate processes are co-dependent, as illustrated in
Figure 1.
The concentration of solid particles in the solution will affect the agglomeration rate. The size development of the particles determines the transport of solids to the wall of the pipeline. The deposition rate will, in turn, affect the concentration of solid particles in the solution. Critical nuclei and nanocolloids are both defined as solid forms of silica in the bulk. All size ranges of solid silica may deposit. However, the total number concentration in each size range affects the agglomeration rate. As the deposition velocity and agglomeration rates are different for each size range, the agglomeration rate will affect the further agglomeration and the deposition rate.
Although there is a lack of research data for silica nanocolloid formation in steam, a lot of information is gathered on experimental and theoretical research of silica solidification and growth in liquid water. Despite the vast amount of research on the kinetic process of silica polymerization and nanocolloid formation in both pure water [
12,
13,
14,
15,
16,
17,
18,
19,
20,
21] and in natural waters or simulated geothermal systems [
22,
23,
24,
25,
26,
27,
28,
29,
30,
31], there is still strong disagreement between the experimental results. As a result, the reaction rates and the kinetic models used to explain silica precipitation in aqueous solutions vary.
Observations made in the Icelandic Deep Drilling Project, especially the geothermal fluid produced from well IDDP1, give indications of the nature and morphology of the silica precipitate from superheated pressurized steam with this specific chemical composition. During the flow testing in 2010–2012, where geothermal steam of 145 bar and 450 °C was produced at the wellhead, precipitation was apparent within the first few days of testing [
9]. When the well reached 500 °C, white dust started depositing on the silencer rocks. The control orifices used to limit the well flow also showed signs of clogging due to the silica scale. The clogging of a 26.6 mm bed after only 24 days indicates high scaling rates [
32]. The scale was characterized as soft, porous and removable by compressed air. A scaling experiment performed by Hauksson Kemia and Markusson is described in [
9], where pressure drops were induced by a cascade of orifices with a pipe section in between. The work quantifies the precipitation on each orifice plate, but primary particle size development, concentration of solids versus dissolved material in each stage and deposition velocity in these conditions remain unknown. In the review presented by reference [
33], previous work relevant to silica precipitation behavior in supercritical fluids and superheated pressurized steam is analyzed further.
Figure 2 represents a pipe segment after a sudden depressurization where supersaturation is only affected by the change in concentration of dissolved silicic acid due to precipitation. The initial conditions, velocity
v, pressure
p, temperature
T, concentration of dissolved silicic acid
c, and the supersaturation S, are known. Supersaturation, S, is defined as the ratio of the actual concentration of silicic acid in the solution to the equilibrium concentration for amorphous silica. The cause of the increase in supersaturation can be a sudden pressure drop so that the time required to achieve the supersaturation is fast enough to be considered instantaneous.
2. Materials and Methods
Nucleation, by definition, applies to both a new thermodynamic phase and a self-organizing new structure. The standard theory that describes the behavior for the nucleation of a new thermodynamic phase from a stochastic process is called classical nucleation theory [
34]. The classical nucleation theory is a physical model with several assumptions that may or may not hold. It has been shown to give reasonable results when compared to computer simulations but may not be relied upon for accuracy [
35]. The nucleation theorem relates nucleation rate, the nucleus size and the supersaturation.
In classical nucleation theory, the total change in Gibbs free energy,
, during formation of a solid in a solution is the sum of the total change in Gibbs surface free energy and change in Gibbs volume free energy as given in Equations (1)–(4), respectively [
34].
In these equations,
AS is the surface area,
V is the volume of a particle of radius,
r,
is the interfacial free energy [J/m
2],
is the molecular volume
[m
3/molecule] and
is the chemical potential difference per solute mol. When looking at dilute solutions, the activity,
a, represents the free concentration of a chemical specie that, in an ideal solution, is equal to the concentration in molar units.
a*, is the equilibrium concentration and is a function of specific volume. The surface free energy,
, denotes the free energetic cost of creating a surface, and it increases with increasing interfacial free energy between the solid surface and solution and the particle radius. The average of the interfacial free energies reported is 0.092 J/m
2 for amorphous silica surfaces in water [
13,
36,
37]. The volume of excess free energy,
, depends on the change in chemical potential and, therefore, the degree of supersaturation in the solution.
The nucleus is a structural cluster of molecules above the critical radius
rcrit. Below this radius, the nucleus is unstable and will dissolve. Above this radius, the nucleus is stable and will continue to grow. The maximum of the Gibbs free energy will be at the critical radius, and the critical radius can, therefore, be defined by Equation (5).
Crystallization is viewed as a first-order phase transition along two order parameters, namely concentration and structure. In a situation where crystals nucleate from a melt, mainly structural fluctuations are needed. In the situation where crystallization occurs in a dilute solution, both structural and density fluctuations need to take place. In the classical approach to nucleation, these two processes are assumed to take place simultaneously [
34].
The nucleation rate,
J, was first proposed as proportional to the exponential of the energy required to form a nucleus over the Boltzmann constant and temperature by Volmer and Weber in 1929. Later, Becker, Döring and Zeldovic provided important insight into the kinetic nature of the prefactor, leading to the expressions in Equations (6) and (7) [
34].
J is then the product of a thermodynamic barrier and a kinetic barrier given by the rate of atomic attachment. In Equation (6),
A is the kinetic factor made up by
f*, the rate of transport and attachment of new molecules on the surface.
NS is the number of nucleation sites available.
Z is called the Zeldovich factor, and it is the probability that a nucleus at the top of the barrier will go on to form the new phase and not dissolve.
Z can be calculated [
38] but is often close to unity.
There are many expressions for the rate of attachment of new molecules on the surface found in the literature [
39]. Usually, in dilute solutions, either the transport of molecules to the surface of the particle, the condensation reaction on the surface of the particle, or the structural organization of the crystal will govern the kinetic factor. In the case of amorphous precipitation, both surface reactions and diffusion must be compared as the limiting factor. The Brownian motion diffusion coefficient for Silica,
, can be determined by the Stokes–Einstein equation (Equation (8)) [
40]. Equations (9) and (10) are provided for calculation of the volume diffusion-controlled kinetic factor,
, and the surface integration-controlled kinetic factor,
, respectively [
34]. The lesser of these will be the rate-limiting kinetic barrier. In Equations (8)–(10),
is the Boltzmann constant,
is the equilibrium concentration of silicic acid and
is the particle diameter.
Several experimental studies on homogenous nucleation in various systems have been compared to results from the classical nucleation theory in the last decade. Experimental studies have mostly confirmed classical nucleation theory to be fairly correct, at least for unary systems of non-polar molecules [
41,
42]. The theory has also been modified and extended with some success to address a wide variety of issues, including multicomponent nucleation [
43]. The calculated results remain sensitive to assumptions regarding the interfacial free energy, the value of which varies in the literature. Also, the assumption that the interfacial free energy of a nucleus is equivalent to that of a macroscopic flat surface is known to be true on a molecular level. Surface energy of nanocolloids depends on radius of curvature. Nevertheless, the assumptions allow for a reasonably accurate analysis [
35,
44]. An atomistic approach, like Molecular Dynamics simulation, takes care of the pathways of individual molecules in the nucleation process. However, this method is limited in the size of discrete cluster it can consider.
If a solid surface is available, heterogeneous primary nucleation will occur before homogenous nucleation. The activation energy barrier for nucleation is lower in this case due to the reduced energetic cost of creating the surface. Free energy required for heterogeneous nucleation is lower than homogenous nucleation by a factor related to the contact angle between the nuclei and the foreign substrate. When nucleation sites are available, the probability of nucleation increases as a result of the lower activation energy barrier. The nucleation rate is often enhanced due to the effects of the nucleation sites on kinetic factors of nucleation.
The following assumptions apply for calculation of the nucleation rate presented herein:
The thermodynamic barrier for formation of a nucleus is that of the total Gibbs free energy corresponding to the critical radius. This is calculated according to Equations (1)–(4).
Interfacial free energy is assumed to be constant with a value of 0.092 J/m2
The process is limited by transport of molecules to the surface of the nucleation site as opposed to dehydration and attachment. This assumption can be justified by the relatively low concentration of silicic acid molecules in the solution. Further, the transport of molecules to the surface is mainly by diffusion, and Stokes–Einstein equation (Equation (8)) can be used to determine the diffusion coefficient.
The kinetic barrier for formation of a nucleus can be determined by Equation (9) (volume-diffusion controlled kinetic factor), with dimension (number of nuclei). The kinetic factor for surface integration control was also calculated by Equation (10), but as this equation yields slightly higher values, the volume diffusion mechanism is, therefore, taken as limiting for all temperatures and supersaturations in the investigated range.
The number of available nucleation sites is equal to the initial total number of silicic acid molecules in the solution per cubic meter of water.
A realistic initial concentration of silicic acid is derived from quartz solubility data for this pressure and temperature condition, assuming that the reservoir is in equilibrium and neglecting the effect of other species in the water. The initial concentration of silicic acid is calculated according to the well-known correlation described in reference [
45] to be 244 mg/kg for quartz in pure water. Precipitation is evaluated for sudden depressurization to varying pressures. Amorphous silica solubility is used to determine precipitation potential.
The solubility of amorphous silica in the steam phase presented here and used further in the model relies mainly on the work of [
46] and the experimental values of [
47,
48]. The data are density-corrected and interpolated to obtain complete amorphous silica solubility data along the isenthalpic line [
33].
The further development of the population balance will be affected both by continuous chemically controlled polymerization, herein modeled by classical nucleation theory, and by growth of the nanocolloids formed. When comparing calculated agglomeration rates in a turbulent pipe flow with experimentally determined surface growth rates in liquid water [
18,
19,
21,
49], the latter is negligible. In a literature review for similar conditions, it is proposed that for precipitation from highly supersaturated steam, two-step dominate precipitation, nucleation of critical-sized particles and growth of these particles by agglomeration. To determine scaling on a surface or remaining precipitate in the fluid at any given point, a third step, namely deposition of the precipitate on a surface, is as important [
33].
Agglomeration is, by definition, the process of several particles coming together to form one larger particle. The process involves transport of particles, collision, interaction and loose attachment, either rupture and dissolution or formation of stronger bonds leading to partly or fully cementation [
34]. Fluid motion, particle interaction forces and turbulent diffusivity play an important role in the relative motion of particles and the resulting collision rate. Four types of collisions are often recognized: those due to Brownian motion (perikinetic collisions), those due to the velocity gradient in a laminar or turbulent flow field (orthokinetic collisions), those due to the gravitational settling of particles (inertial collisions) and those due to stirring. Only the two first are relevant for the particle sizes considered here. For the smaller particles, Brownian motion will be governing, then as particles grow, the effect of turbulent diffusivity increases exponentially.
The process of loose attachment is largely governed by the collision rate and particle-particle interaction forces. While there are models for estimating the particle collision rate in turbulent flow, the accurate determination of the effect of repulsive forces may be difficult [
50]. The concept of collision efficiency takes into account that not all collisions are successful in forming an attachment. The cementation process is directly related to the growth rate in the corners and spaces between touching particles. The probability of attachment can be expressed as a power law as in Equation (11), where
P is the probability,
Eb is the energy barrier,
kb is the Boltzmann constant and
T is absolute temperature.
As it is difficult to model the steps of agglomeration separately, the overall agglomeration rate is mostly described with one size-dependent function combining the effects of collision, interaction, rupture and cementation. This is the agglomeration rate constant, also called agglomeration kernel
, calculated as per Equation (12) [
34]. This is often split into a size-independent part
, determined by process conditions and the size-dependent part
, which depends upon characteristic lengths.
In this equation,
and
, are characteristic lengths of the two approaching particles,
, is the turbulent energy dissipation and,
S, is supersaturation. Several models for the agglomeration kernel exist in the literature [
34], but most do not include supersaturation, which has been shown to also significantly affect the agglomeration rate. The size-dependent kernel function is mostly derived only from the collision mechanism. The kernels most commonly used for solid particle precipitation are given in Equations (13)–(16) [
34]. These are size-dependent Brownian motion agglomeration kernel, process-dependent Brownian motion agglomeration kernel, process-dependent turbulent shear agglomeration kernel and size-dependent turbulent shear kernel, respectively.
In these equations, as previously, kb is Boltzmann constant, T is absolute temperature, is the dynamic viscosity, is the turbulent dissipation, assumedly equal to 0.1, and is the kinematic viscosity.
Silica deposition in steam has been shown to vary with hydrodynamic conditions [
50,
51]. In some chemical and hydrodynamic conditions, protrusions of silica have been observed [
26,
50]. These types of scale formations will likely be sensitive to varying hydrodynamic conditions. Particle deposition from gas streams is a well-researched area.
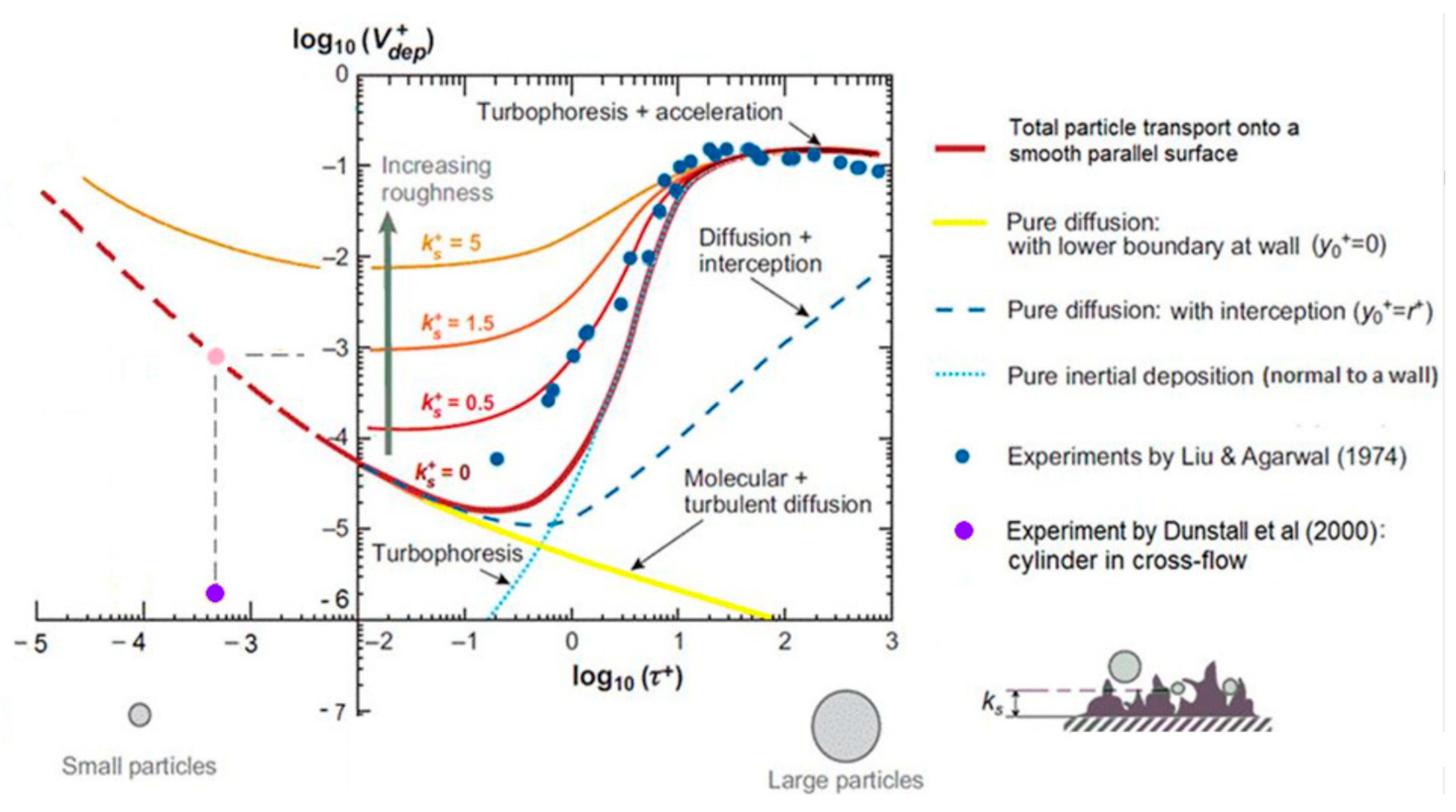
Figure 3 shows the dimensionless particle deposition velocity as a function of the dimensionless particle relaxation time, where also the effect of roughness is indicated [
50]. Both roughness and thermophoresis will significantly influence the deposition velocity in the relevant particle relaxation range [
52].
The dimensionless particle relaxation time is defined as , where is the friction velocity and Cc is the Cunningham slip correction factor. is wall shear stress, is the fluid density, is the particle density and is kinematic viscosity of the fluid. The dimensionless deposition velocity is defined as , where is the mass transfer rate in m/s.
Since the wall shear stress, , will depend on Reynolds number and velocity of the flow, it is necessary to introduce an example pipe to evaluate the particles generated at the supersaturations in steam provided in the previous sections. With a relatively small pipe (1.4 mm internal diameter) and a mass flow of 0.23 g/s at 150 bar pressure, Reynolds number will be approximately 8400, and the particle relaxation time for the particles generated will range from 10−7 to 10−3 in a 2 m pipe with the initial conditions used in the previous steps. For a constant mass flow rate, the Reynolds number will decrease with increasing pressure and increase with decreasing pressure. This is due to the change in viscosity of the fluid.
Silica nanoparticles are so small that they behave more like molecules than particles. By the dimensionless relaxation time range calculated, one can conclude that the particles will be in the diffusion-dominated regime. Deposition rates are expected to be low, with dimensionless deposition velocity between 1 and 10
−4 for the above example case. For particles in this size range, an analogy to heat transfer can be used to estimate the mass transport rate from the bulk of the fluid through the boundary layer and onto the wall. Corresponding to the yellow line in
Figure 3.
The mass transfer version of the correlation of Popov and Petukhov [
54], given in Equation (17), is valid for the considered Schmidt numbers,
, ranging from 0.03 to 1063, but the Reynolds number
Re, ranging from 7000 to 10,000 for the given mass flow, is slightly below the validity range for the high-pressure cases investigated. The Reynolds numbers are, however, still high enough that this correlation should give fairly accurate results. The Sherwood number,
Sh, is further used to determine the mass transfer coefficient,
, by Equation (18).
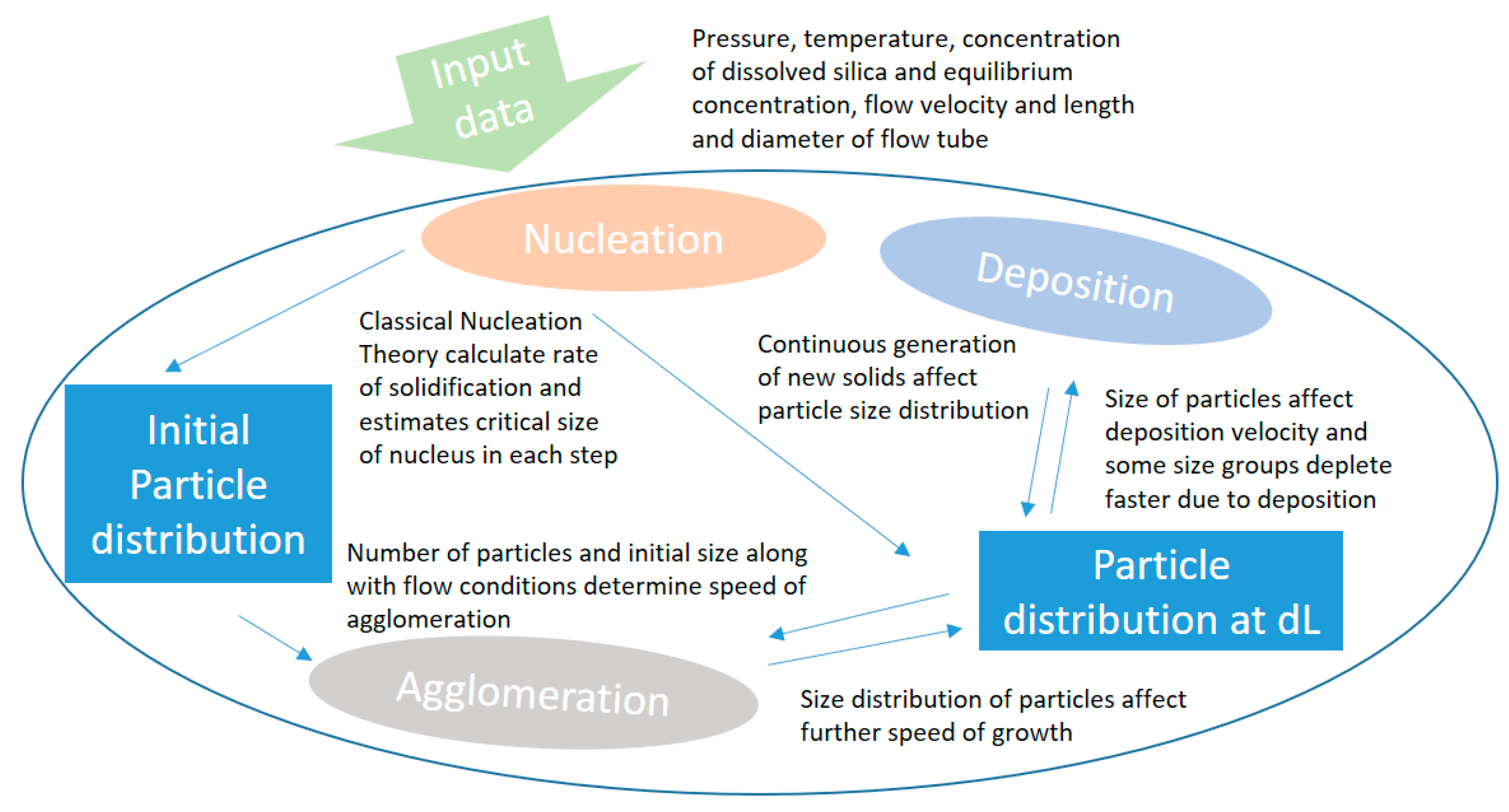
The three processes are combined numerically in a numerical model that calculates initial conditions and development in concentration, deposited material and remaining particle size development for a definite number of size groups in each step. A schematic illustration of the calculation process is given in
Figure 4.
4. Discussion
The presented combined model provides an opportunity to calculate the amount of scale in a specific region of a pipeline downstream, a pressure reduction from supercritical conditions and the remaining silica in the fluid. With correct input, vital information about where in the pipe region deposition will be most severe can be obtained for different initial values of supersaturation and system configurations. This information can be used to improve reservoir understanding processing techniques and, if silica scaling mitigation methods are improved as a result, increase plant energy utilization. The calculation will be sensitive to the chemical composition of the fluid in question as this affects solubility, kinetics and collision efficiency. The calculations are also sensitive to the geometries in question, as the hydrodynamic conditions in bends, valves and around obstacles vary from that of a straight pipe and this effect cannot be neglected.
An experiment was designed to measure deposition from depressurized supercritical pure water saturated with silica in a straight pipe. This experiment was designed with simple features such as straight pipe and purified water to enable validation of the mathematical model. A detailed description of the experimental work and its results can be found in [
56,
57] present preliminary results and comparison. The results reported correlate fairly well with the modeled results. A thorough comparison of this experiment and the modeled results for the same conditions with sensitivity analysis and specific improvement suggestions for the case scenarios evaluated is in progress [
58].
Other investigated deposition rates of silica particles from pressurized water vapor include the work reported in [
59]. The experimental setup produced larger particles than what can be expected based on the time frame evaluated in this paper. It was, however, concluded that the theory of solid particles in gas can be applied to calculate the deposition rates of silica in steam for the size range investigated.
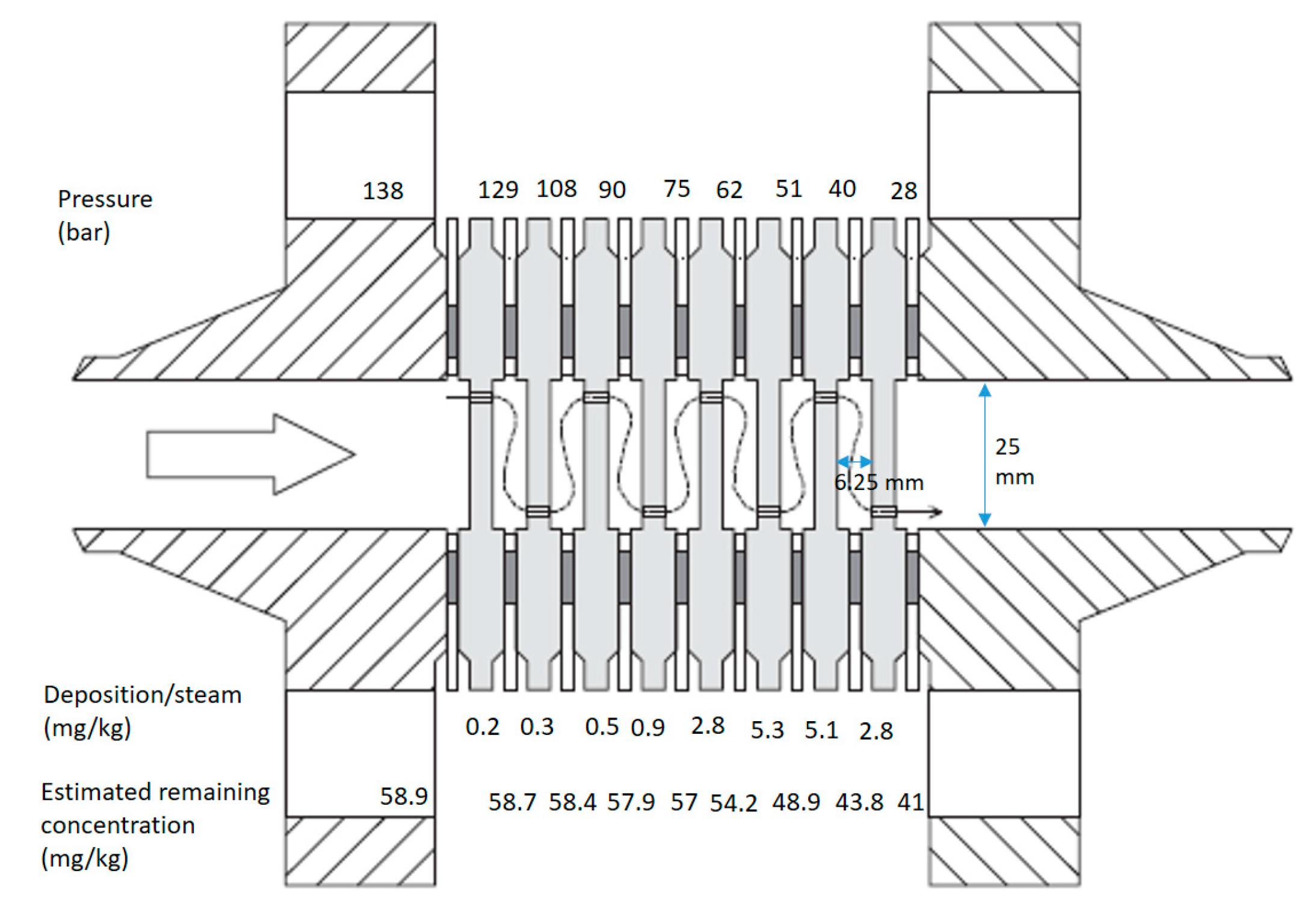
In the experiment reported by [
9], pressure drops were induced by a cascade of orifices with a pipe section in between, as illustrated in
Figure 11. Here, the pressures in each chamber are given on top, and the measured deposited material is given below the drawing of each chamber. From the measured deposits on each disk, a sharp increase in scaling was observed on the 3.5 mm orifice disk, where pressure was reduced below 75 bar. There are uncontrolled parameters in the experiment, such as previously precipitated material in the fluid, the effect of other minerals, initial population distribution and exact turbulence and flow conditions along the walls inside the chambers. This limits the possibility of pinpointing exactly what mechanisms are dominating.
The characteristics of the scale were analyzed and varied from “flaky iron oxide” at 138 bar, “granular silica and iron oxide” at 95 bar and “spherical and threadlike scale” at 34 bar [
9]. Microstructural analysis in electron microscopes shows great variation in scale structure and degree of cementation. Morphology was analyzed down to 100 nm, where 10–100 nm size particle shapes can barely be differentiated. The number of newly generated nuclei of size 1–10 nm and whether the larger structures are made up of smaller distinct units is difficult to determine in this resolution. The deposits contained traces of iron and iron chloride. From orifice 6, the composition of the deposit is 100% SiO
2. On orifice 7, the precipitate starts forming threadlike structures of 100 nm diameter. Further, on orifice 8, thicker threads of up to one-micron diameter are observed in addition to the thinner threads.
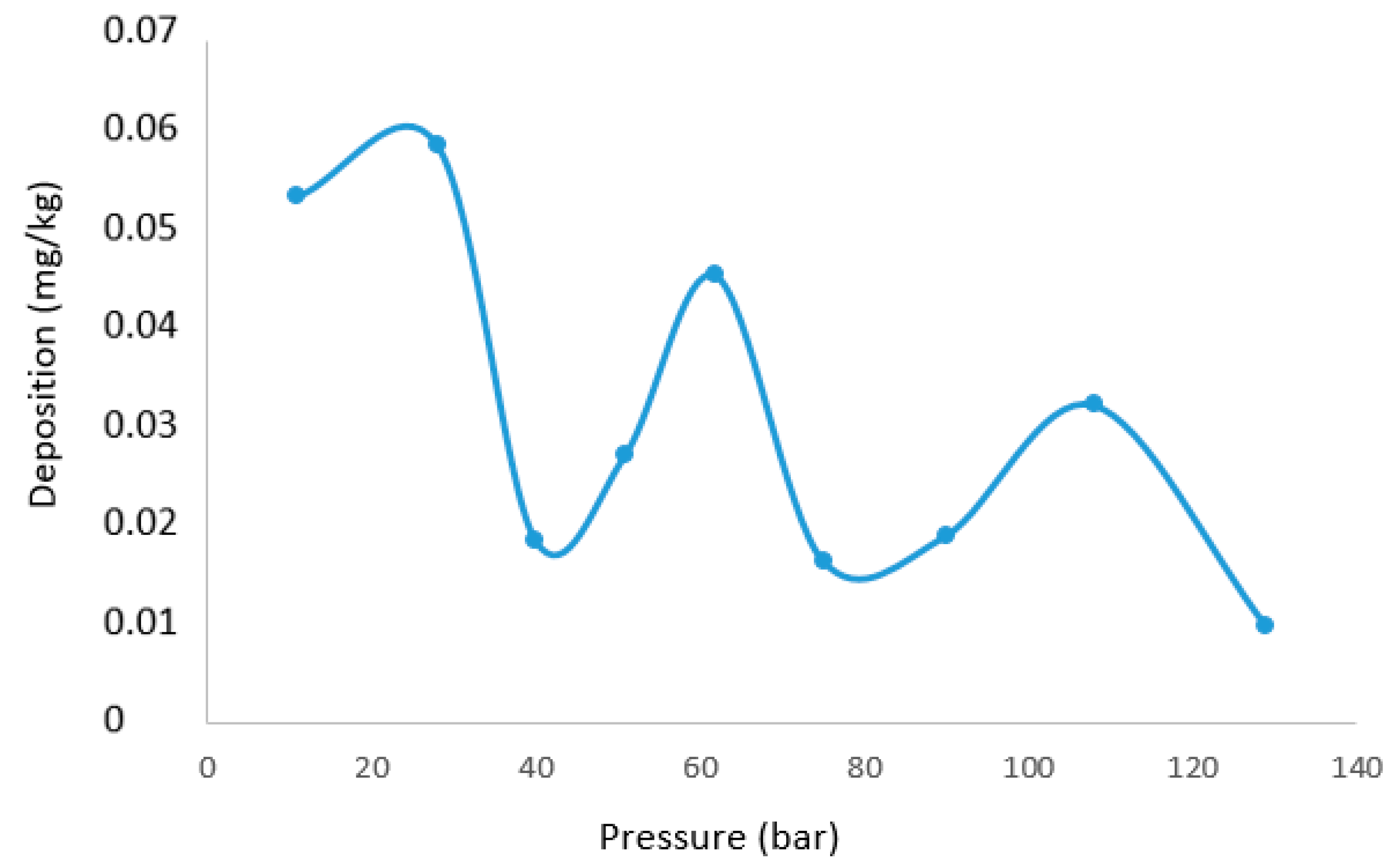
Solid conversion in a straight pipe under otherwise similar process conditions has been calculated using the proposed model for the various pressure reductions in the experiment. The calculation is not quantitatively comparable to the experiment, as the hydrodynamics in a straight pipe, and therefore, turbulence and deposition velocity, differ from a flow through an orifice arrangement. The results are intended to give an indication as to how the three discussed processes interplay in a system with several pressure reductions. The measured silica concentration in this experiment was 62 mg/kg before the filter, and this is used as model input. 75 bar at an enthalpy of 3180 kJ/kg corresponds to an equilibrium concentration of 6.5 mg/kg by approximating from experimental solubility data as discussed in [
33], where a linear correction for the lower density compared to the density of the experimental solubility measurement is used. The effects of other species in the mixture on solubility and kinetics are not accounted for.
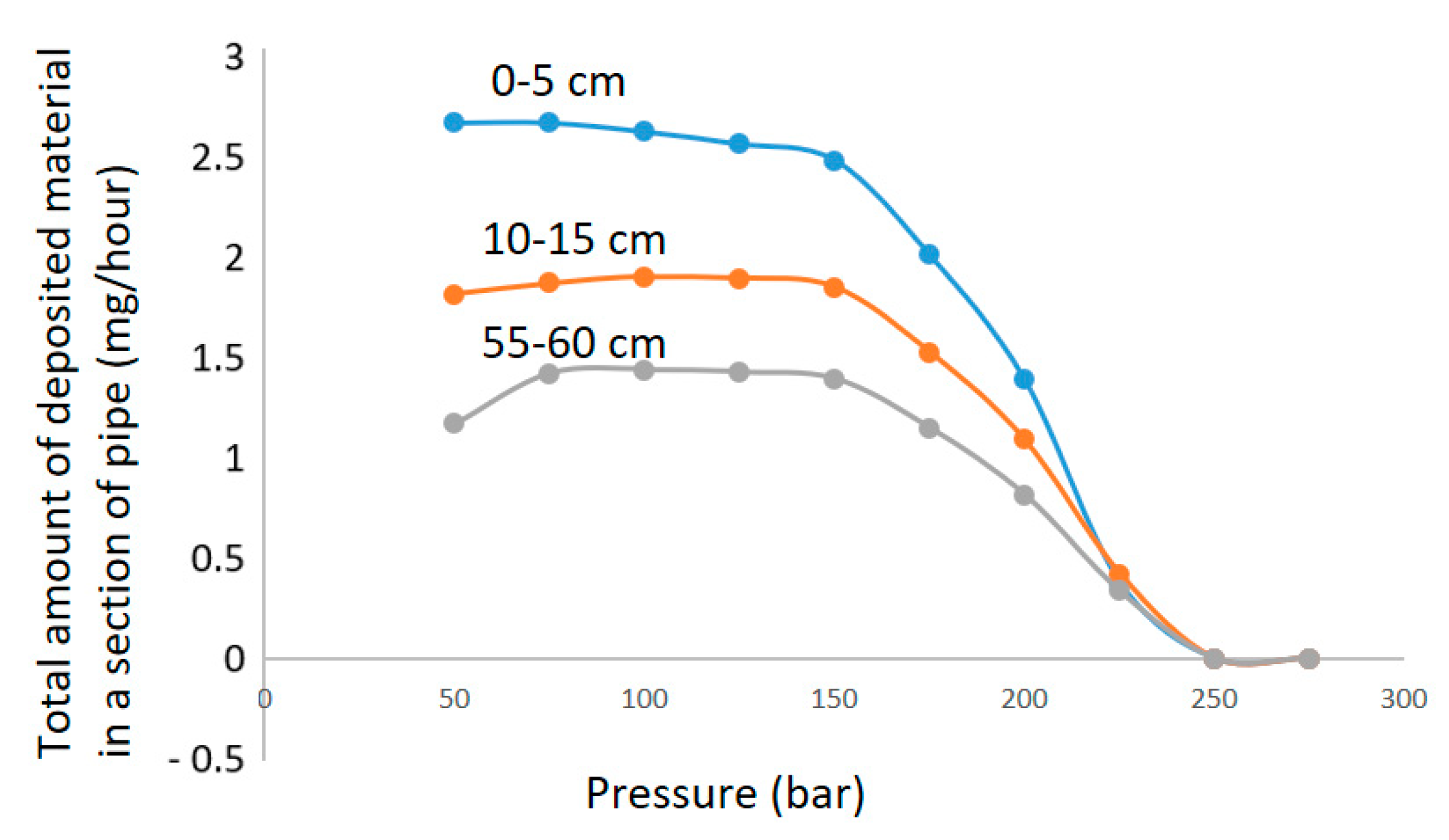
Each chamber between depressurization will be the space between two cylindrical plates. The velocity will be high right after the orifice, and there will be regions of lower velocity, while in a straight pipe, the velocity is fairly uniform. With an approximation of the velocity and volume inside the chamber, one can evaluate what happens in each chamber and what mechanisms are dominant. To quantitatively compare these results, however, an appropriate CFD model for the geometry, taking into account local turbulence, has to be verified. A sensitivity analysis would also have to be performed to evaluate the impact of other components in the fluid on all parts of the analysis. A sensitivity analysis looking at the effect of surface growth and heterogeneous nucleation directly onto the larger particles could also contribute to a better fit for the measured deposition. The expected deposition in each pipe section after each pressure drop, with surface area and velocity comparable to the chamber in the experiment, is given in
Figure 12.
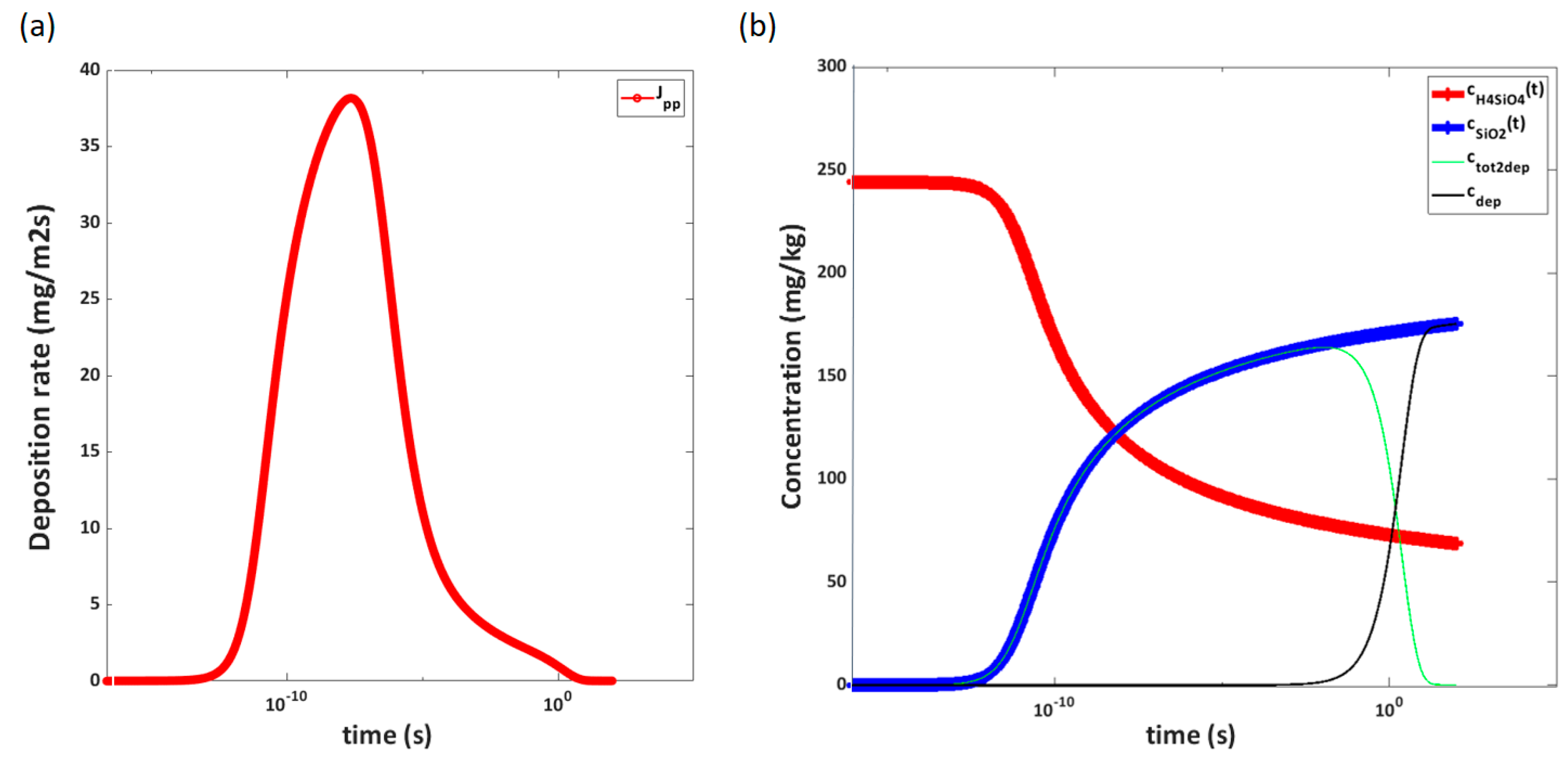
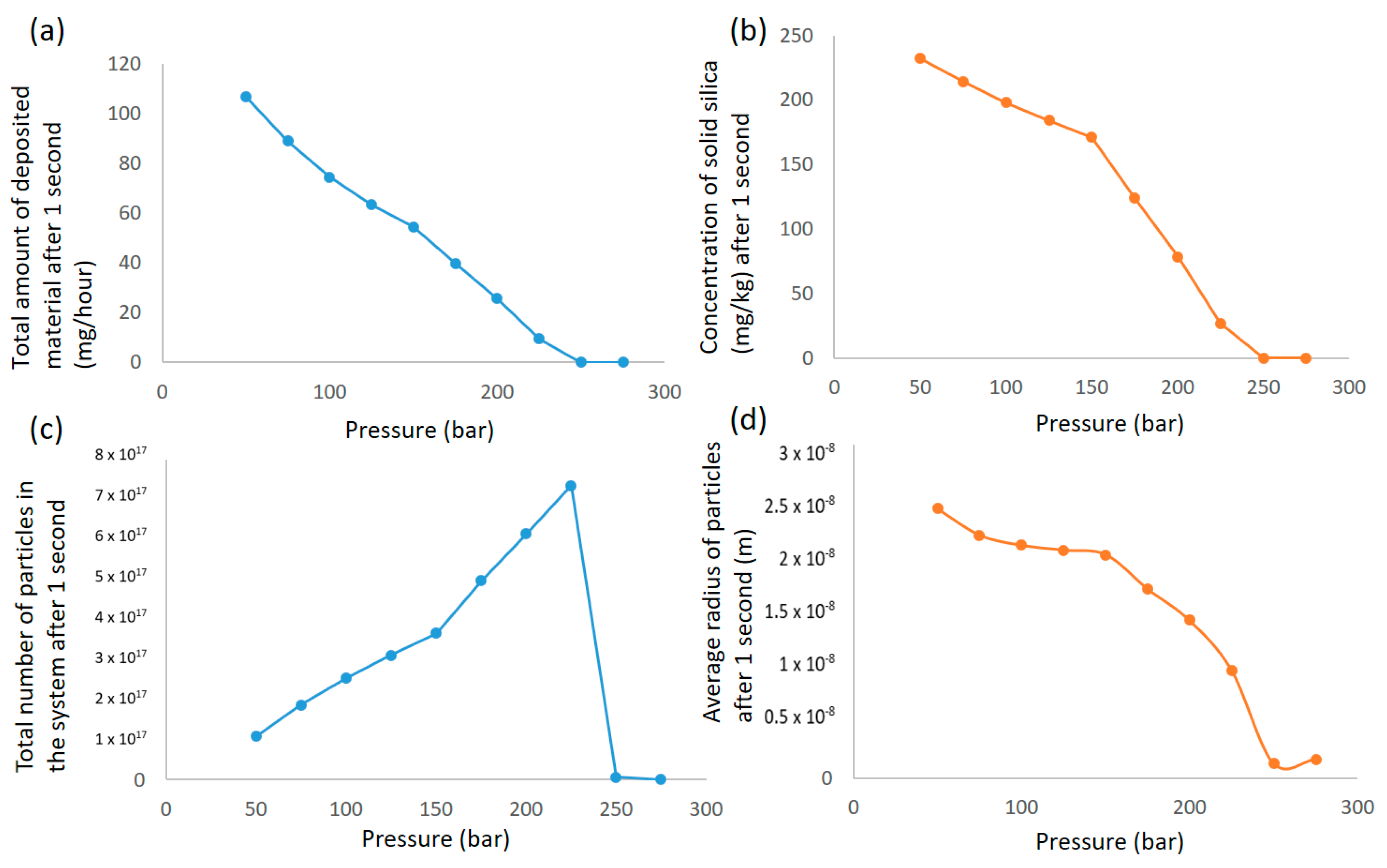
It is calculated that 74% of the silicic acid has transformed into solid particles already after the second orifice, where pressure is reduced from 129 to 108 bar. Deposition is thus the governing mechanism for the measured deposition in the following chambers. While the experiment indicates one peak deposition at the 62 to 51 bar pressure reduction plate and a lowering of deposition as pressure is further reduced below 40 bar, these are not general rules according to the calculations. The calculations show several peaks of deposition. Each peak results in the depletion of solids until pressure again reduces enough to establish significant supersaturation. The maximum deposition peak calculated is two orders of magnitudes lower than the experimental maximum measured. The overall deposition calculated is also lower than the overall deposit measured. It is likely that the overall deposition velocity will be higher in the chamber cascade than in a straight pipe.
The sticking probability in the calculation is assumed to be 100%. Slower agglomeration/increase in stability of particles due to particles’ negative surface charge will slow down growth, but it would also reduce deposition when a thin layer of silica has formed on the surface of the walls. The presence of foreign substances that affect the surface charge of the particles, solubility and interfacial free energy in the experiment could partly explain the discrepancy. Increased growth rate, presence of particles with significant inertia, and increased deposition velocity would not, however, explain the lack of deposition on the first four orifices seen in the experiment. Particles depositing in areas of high velocity will be exposed to high shear forces, and re-entrainment may occur.
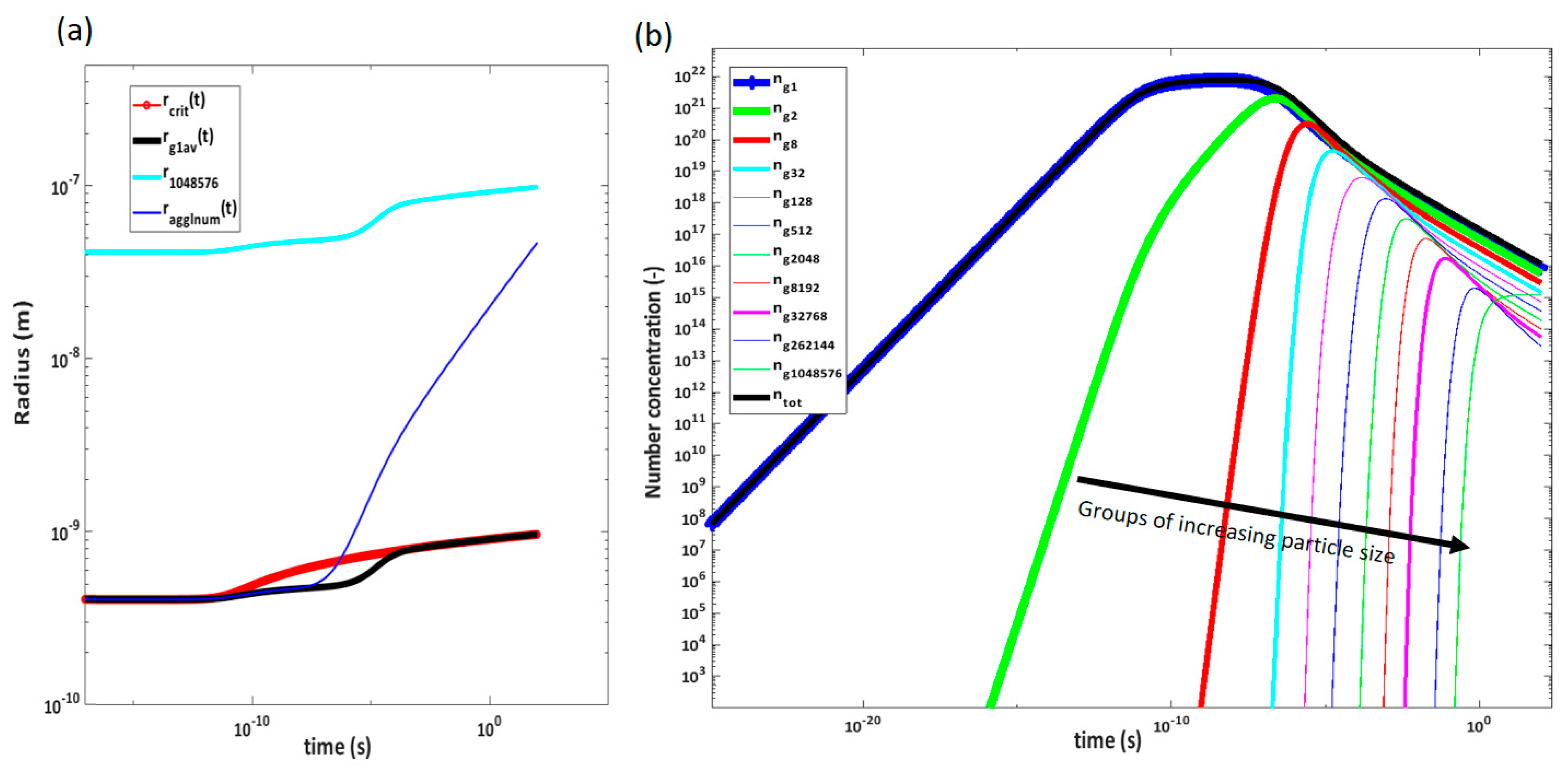
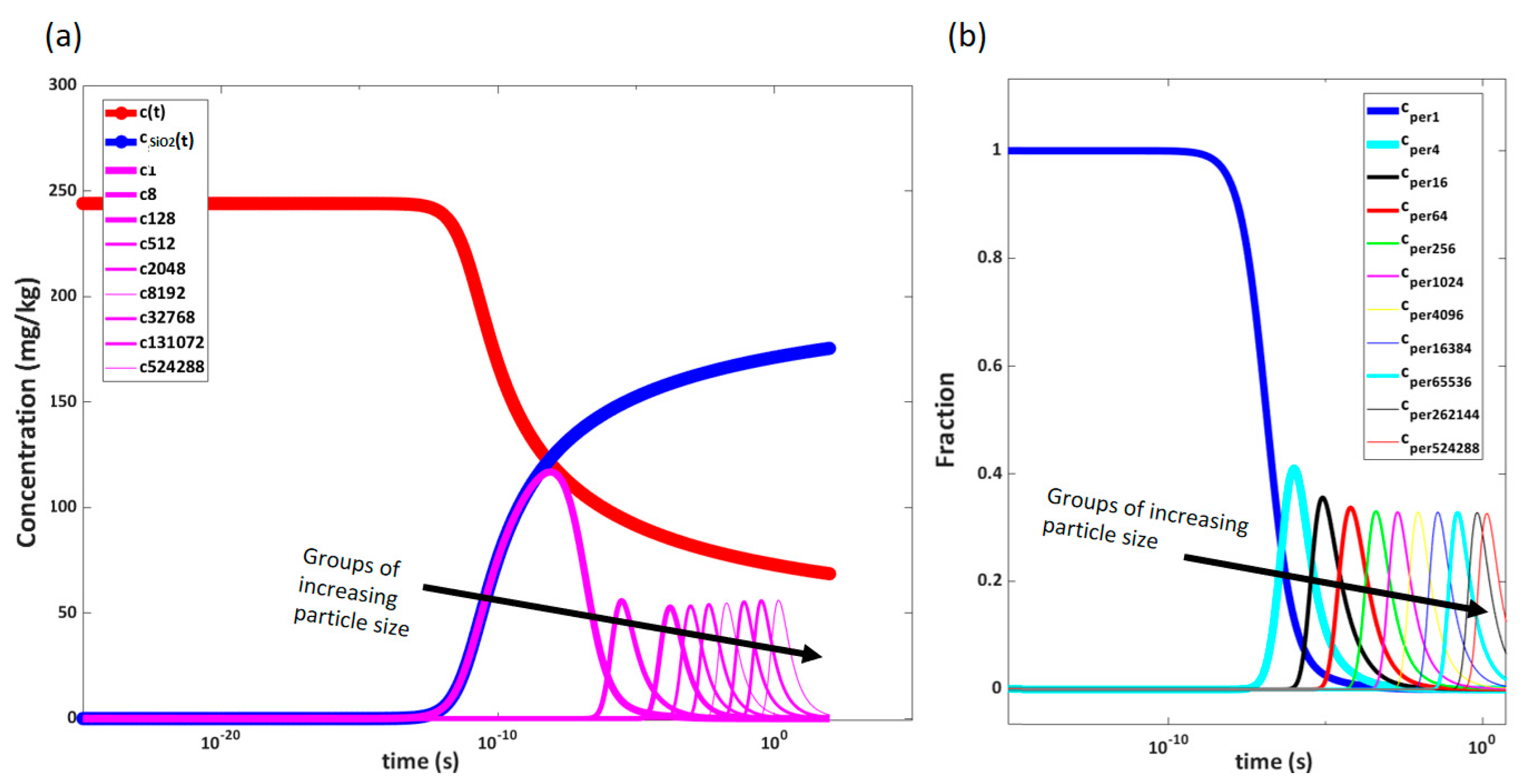
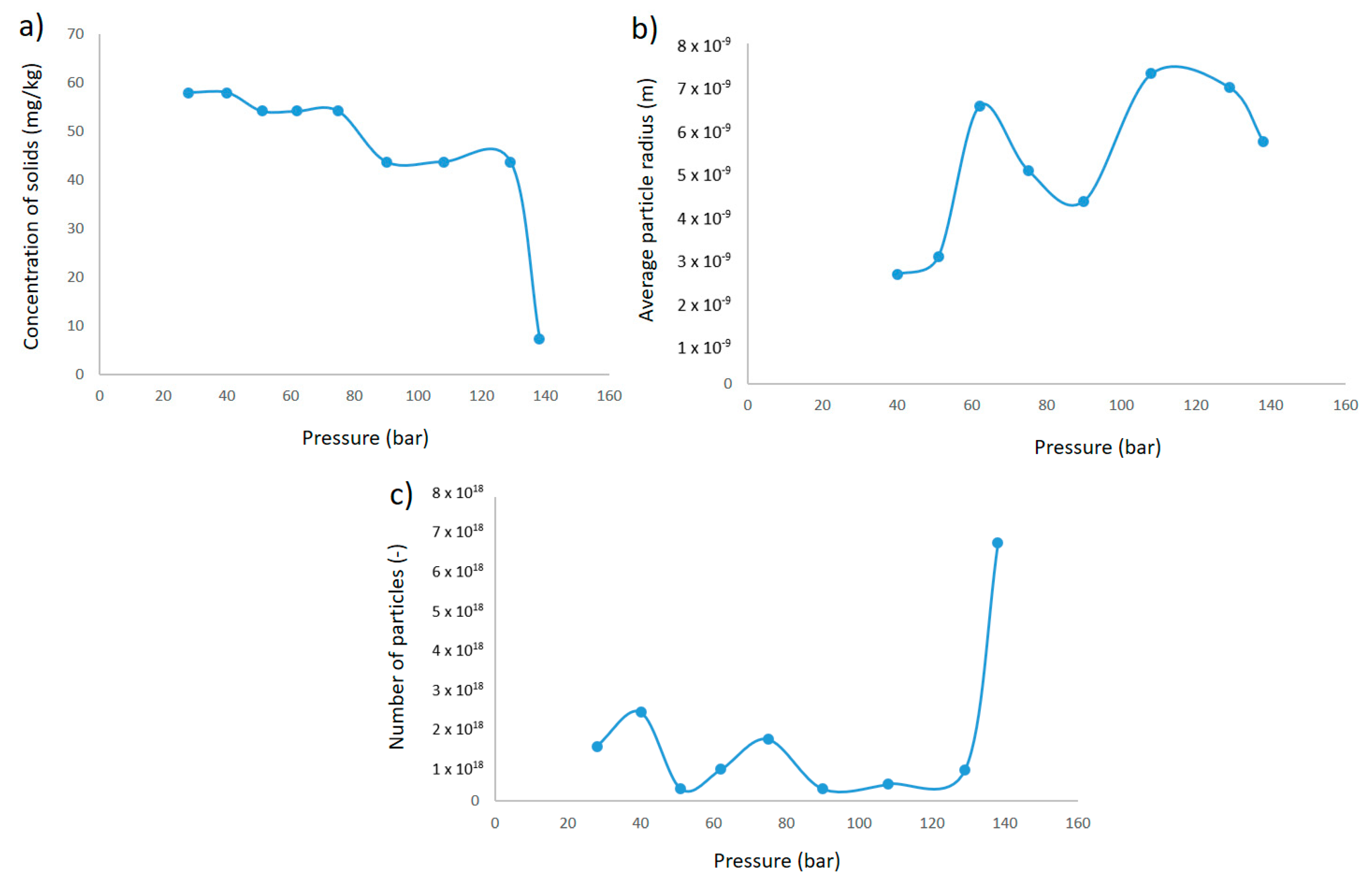
Figure 13a shows the development of the concentration of solids in the solution.
Figure 13b shows the average radius, and
Figure 13c shows the number of concentrations at chamber pressure just before entering the next orifice. In this figure, a sharp decrease in the number of particles is observed after the second pressure reduction. This corresponds to an increase in radius as more particles agglomerate than are generated in this section. After the sharp increase in solid concentration after the first pressure reduction, there are sections where supersaturation is not high enough to generate a significant amount of new particles. Here, agglomeration and growth onto existing particles occur instead, and the deposition rate decreases due to the larger particle sizes until the second deposition peak in the 62 bar pressure reduction, where supersaturation is once again high enough to generate more small particles.
In the population generated and developed by agglomeration in this time frame, the smaller-sized particles have a higher deposition rate. Heterogeneous nucleation or direct growth on existing particles could also contribute to an increase in average particle size, but a relatively high growth rate would have to be applicable for surface growth to be significant in the time span in question.
An assumption in the calculation is that there are no initial particles when the well flow enters the system. CNT (classical nucleation theory) gives the critical nucleus, which is a minimum stable particle size. Using critical nucleus size as a starting point for agglomeration will give a conservatively low particle size. Depending on concentration and supersaturation, there will be a probability of larger particles forming initially in addition to particles already present. A sensitivity analysis related to initial population balance is advisable when using the model without specifying initial particle distribution. The size of the inlet filter in the cascade experiment was 10 µm, and information regarding the percentile of concentration in solid form initially is missing. The low amount of deposition in the filter and in the first chambers in the experiment compared to the deposition on the discs after further pressure reductions indicate, however, that the initial number of particles is not high compared to the number of generated particles.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}