
Cu@PtRu Core–Shell Nanostructured Electrocatalysts Anchored on Reduced Graphene Oxide toward Methanol Oxidation
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Reduced Graphene Oxide (rGO)
2.2. Preparation of Cu@PtRu/rGO Catalysts
2.3. Physical Characterization
2.4. Electrochemical Analyses
3. Results
3.1. Characterization
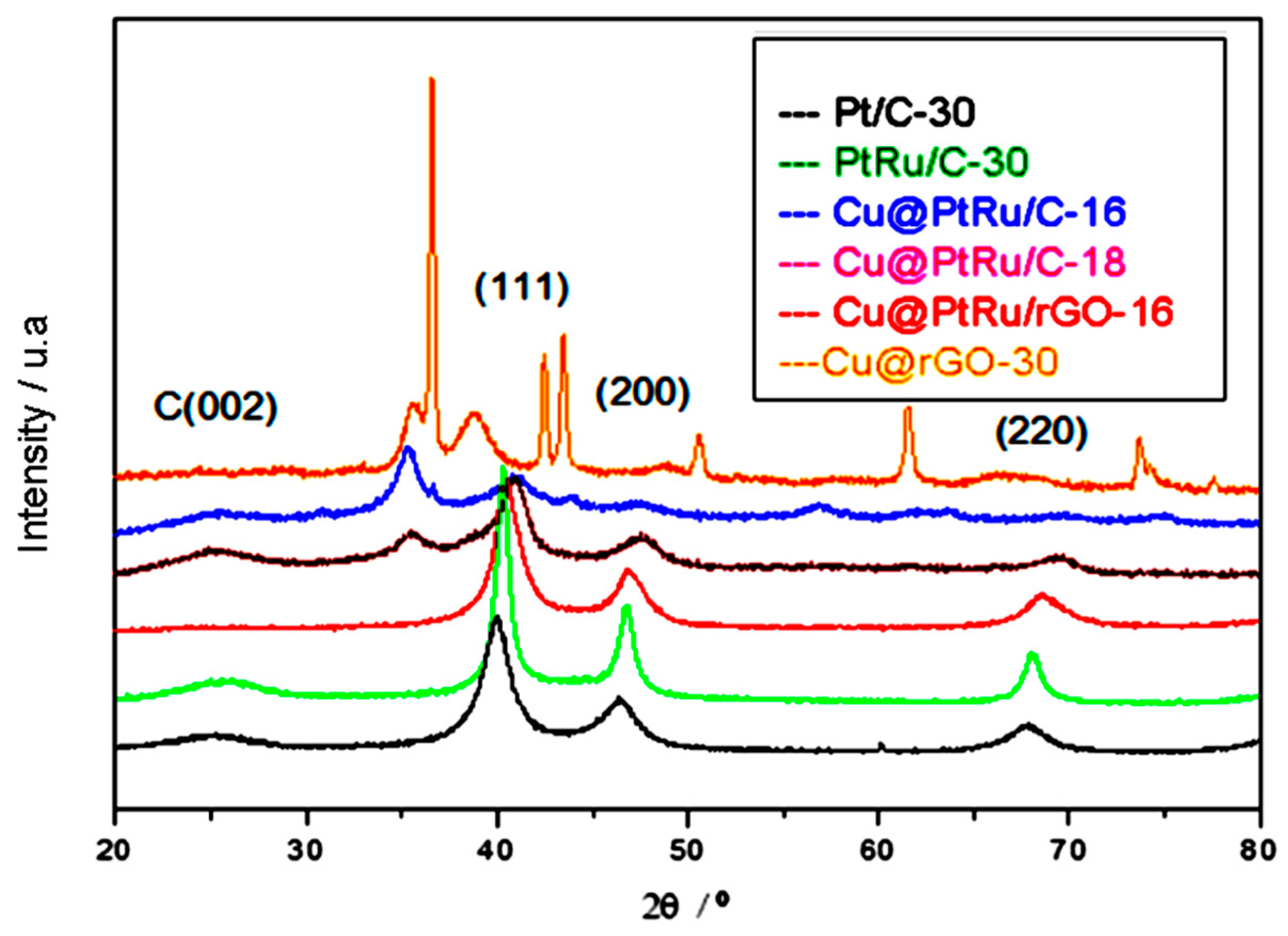
3.1.1. X-ray Analyses
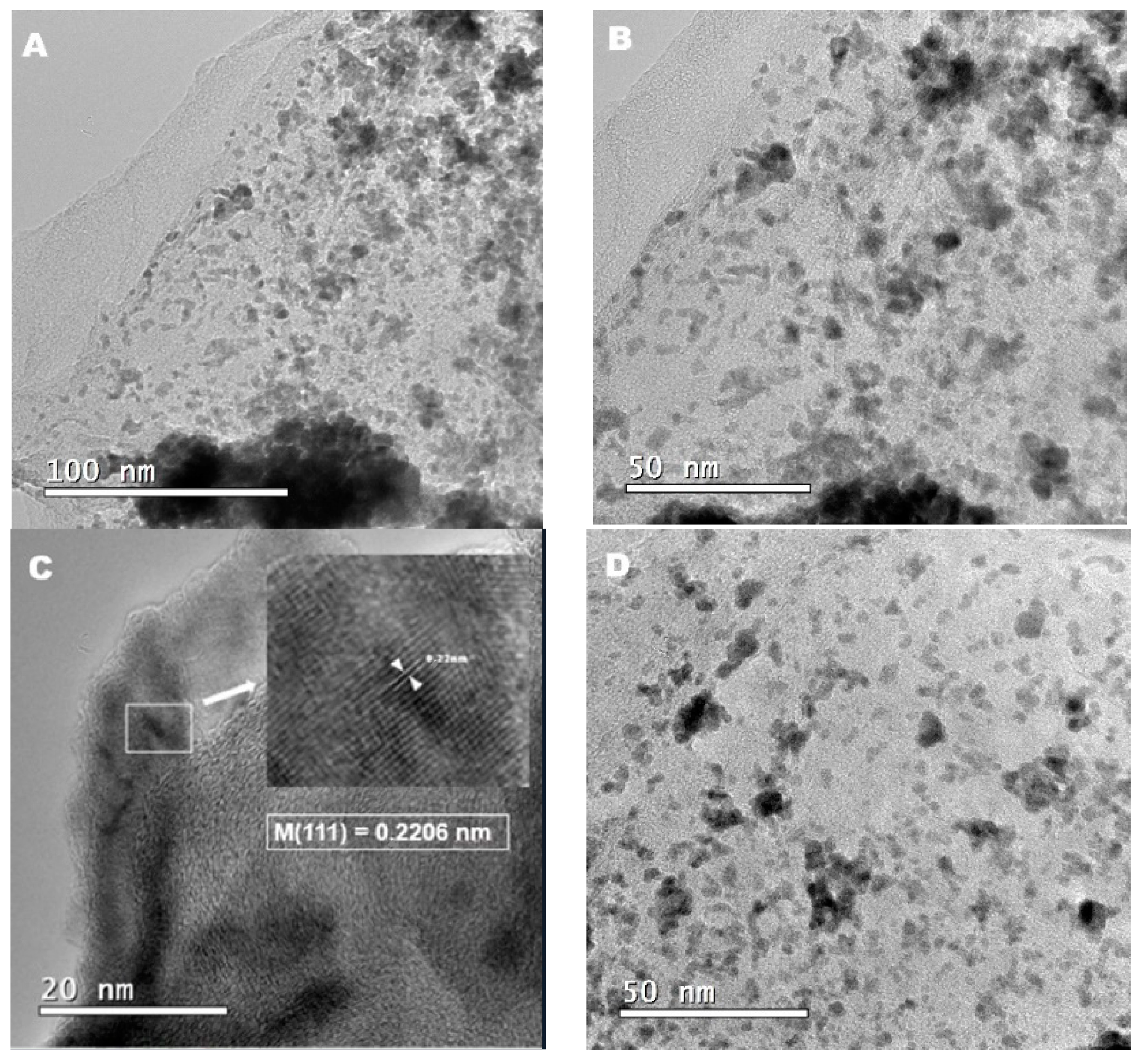
3.1.2. HRTEM Results
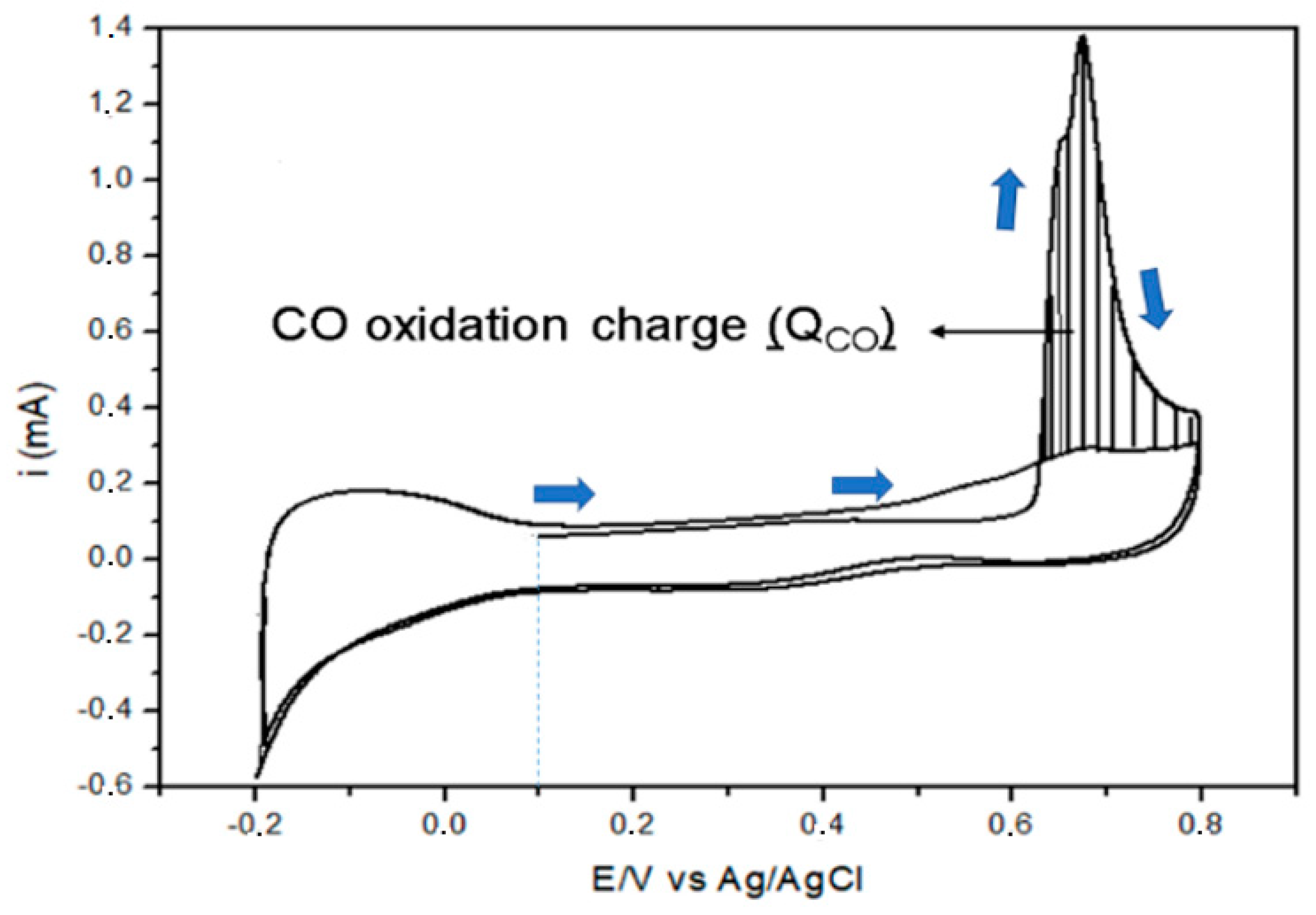
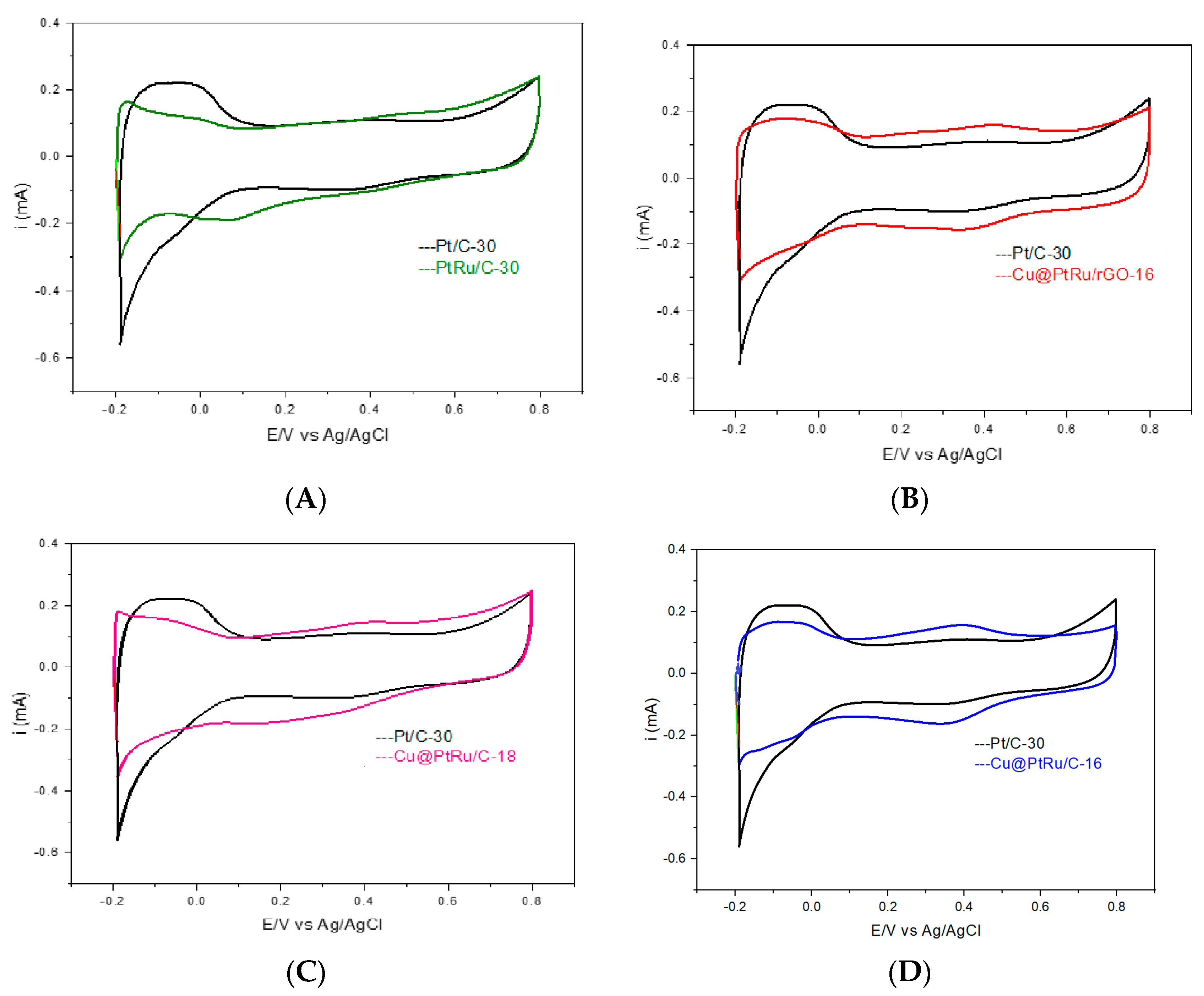
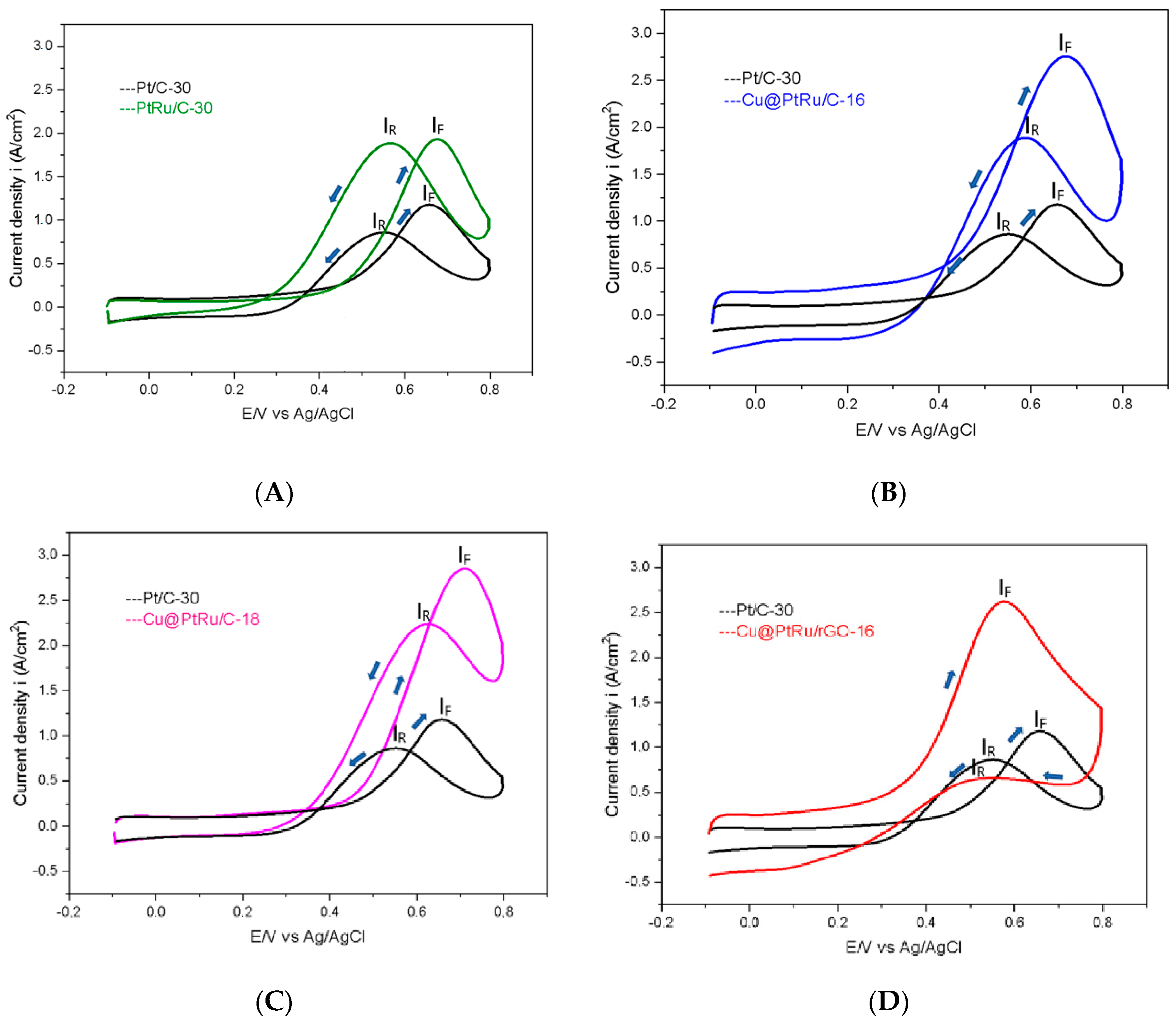
3.1.3. Cyclic Voltammetry Testing
3.1.4. Chronoamperometry Measurements for Methanol Electrooxidation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, Y.; Zhang, J.; Lu, S.; Jiang, S.P. Significantly enhanced performance of direct methanol fuel cells at elevated temperatures. J. Power Sources 2020, 450, 227620. [Google Scholar] [CrossRef]
- Sun, X.; Yang, C.; Xia, Z.; Qi, F.; Sun, H.; Sun, G. Molecular sieve as an effective barrier for methanol crossover in direct methanol fuel cells. Int. J. Hydrogen Energy 2020, 45, 8994–9003. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, M.; Xu, J.; Fang, J.; Luo, S.; Yang, C. Core–shell Pd–P@Pt–Ni nanoparticles with enhanced activity and durability as anode electrocatalyst for methanol oxidation reaction. RSC Adv. 2022, 12, 2246–2252. [Google Scholar] [CrossRef]
- Pereira, P.A.; Andrade, J.B. Fontes, reactivity and quantification of methanol and ethanol in the atmosphere. Quim. Nova 1998, 21, 744–754. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, Z.; Duan, X.; Huang, Y. Nanoscale Structure Design for High-Performance Pt-Based ORR Catalysts. Adv. Mater. 2018, 31, 1802234. [Google Scholar] [CrossRef] [PubMed]
- Abdelkareem, M.A.; Sayed, E.T.; Nakagawa, N. Significance of diffusion layers on the performance of liquid and vapor feed passive direct methanol fuel cells. Energy 2020, 209, 118492. [Google Scholar] [CrossRef]
- Sekar, A.; Metzger, N.; Rajendran, S.; Elangovan, A.; Cao, Y.; Peng, F.; Li, X.; Li, J. PtRu Catalysts on Nitrogen-Doped Carbon Nanotubes with Conformal Hydrogenated TiO2 Shells for Methanol Oxidation. ACS Appl. Nano Mater. 2022, 5, 3275–3288. [Google Scholar] [CrossRef]
- Xu, H.; Shang, H.; Wang, C.; Du, Y. Recent Progress of Ultrathin 2D Pd-Based Nanomaterials for Fuel Cell Electrocatalysis. Small 2021, 17, 2005092. [Google Scholar] [CrossRef]
- Mohanapriya, S.; Gopi, D. Microwave assisted synthesis of core-shell Ni-Co/graphene nanosheets and their catalytic activity for methanol electro-oxidation. Mater. Today 2022, 51, 1797–1881. [Google Scholar] [CrossRef]
- Wang, X.; Li, H.; Feng, H.; Wang, H.; Lei, Z. Preparation of carbon-supported core@shell PdCu@PtRu nanoparticles for methanol oxidation. J. Power Sources 2010, 195, 1099–1102. [Google Scholar] [CrossRef]
- Lee, D.; Gok, S.; Kim, Y.; Sung, Y.E.; Lee, E.; Jang, J.H.; Lim, T. Methanol Tolerant Pt–C Core–Shell Cathode Catalyst for Direct Methanol Fuel Cells. ACS Appl. Mater. Interfaces 2020, 12, 44588–44596. [Google Scholar] [CrossRef]
- Salgado, J.R.C.; Alcaide, F.; Álvarez, G.; Calvillo, L.; Lázaro, M.J.; Pastor, E. Pt-Ru electrocatalysts supported on ordered mesoporous carbon for direct methanol fuel cell. J. Power Sources 2010, 195, 4022–4029. [Google Scholar] [CrossRef]
- Christoffersen, E.; Liu, P.; Ruban, A.; Skriver, H.; Norskov, J. Anode Materials for Low-Temperature Fuel Cells: A Density Functional Theory Study. J. Catal. 2000, 199, 123–131. [Google Scholar] [CrossRef]
- Sang, Y.; Zhang, R.; Xu, B.; Yang, J.; Zhao, C.; Xu, H. Ultrafine and Highly Dispersed PtRu Alloy on Polyacrylic Acid-Grafted Carbon Nanotube@Tin Oxide Core/Shell Composites for Direct Methanol Fuel Cells. ACS Appl. Energy Mater. 2022, 5, 4179–4190. [Google Scholar] [CrossRef]
- Yola, M.L.; Eren, T.; Atar, N.; Saral, H.; Ermiş, İ. Direct-methanol Fuel Cell Based on Functionalized Graphene Oxide with Mono-metallic and Bi-metallic Nanoparticles: Electrochemical Performances of Nanomaterials for Methanol Oxidation. Electroanalysis 2015, 28, 570–579. [Google Scholar] [CrossRef]
- Li, Y.; Gao, W.; Ci, L.; Wang, C.; Ajayan, P.M. Catalytic performance of Pt nanoparticles on reduced graphene oxide for methanol electro-oxidation. Carbon 2010, 48, 1124–1130. [Google Scholar] [CrossRef]
- Lee, S.H.; Kakati, N.; Jee, S.H.; Maiti, J.; Yoon, Y.S. Hydrothermal synthesis of PtRu nanoparticles supported on graphene sheets for methanol oxidation in direct methanol fuel cell. Mater. Lett. 2011, 65, 3281–3284. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Z.; Zhu, H.; Xing, W.; Tao, P.; Shang, W.; Fu, B.; Song, C.; Hong, Y.; Dickei, M.D. Synthesis of Liquid Gallium@Reduced Graphene Oxide Core–Shell Nanoparticles with Enhanced Photoacoustic and Photothermal Performance. J. Am. Chem. Soc. 2022, 144, 6779–6790. [Google Scholar] [CrossRef]
- Pan, J.; Li, S.; Zhang, L.; Yu, T.; Li, F.; Zhang, W.; Wang, J.; Zhang, D.; Yu, Y.; Li, X. Reduced Graphene Oxide/Ni Foam Supported ZIF-67 Derived CuCo2S4@CoS2 Core-Shell Heterostructure for Boosted Electrochemical Energy Storage. J. Energy Storage 2022, 47, 103–112. [Google Scholar] [CrossRef]
- Roh, G.; Lee, H.; Jeong, Y.; Kim, J.H. Preparation of Carbon-Supported PtRu Core-Shell Nanoparticles Using Carbonized Polydopamine and Ozone for a CO Tolerant Electrocatalyst. Int. J. Hydrogen Energy 2019, 44, 21588–21596. [Google Scholar] [CrossRef]
- Wu, Y.N.; Liao, S.J.; Liang, Z.X.; Yang, L.J.; Wang, R.F. High-Performance Core-Shell PdPt@Pt/C Catalysts via Decorating PdPt Alloy Cores with Pt. J. Power Sources 2009, 194, 805–810. [Google Scholar] [CrossRef]
- Patra, S.; Munichandraiah, N. Electrooxidation of Methanol on Pt-Modified Conductive Polymer PEDOT. Langmuir 2009, 25, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Szabó, T.; Berkesi, O.; Forgó, P.; Josepovits, K.; Sanakis, Y.; Petridis, D.; Dékány, I. Evolution of Surface Functional Groups in a Series of Progressively Oxidized Graphite Oxides. Chem. Mater. 2006, 18, 2740–2749. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, S.; Ali, A.; Chen, H.; Shen, P.K. Facile One-Pot Synthesis of a PtRh Alloy Decorated on Ag Nanocubes as a Trimetallic Core−Shell Catalyst for Boosting Methanol Oxidation Reaction. ACS Appl. Energy Mater. 2021, 4, 1085–1092. [Google Scholar] [CrossRef]
- Hamnett, A. Mechanism and electrocatalysis in the direct methanol fuel cell. Catal. Today 1997, 38, 445–457. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, K.; Wu, Q.; Zhang, Y.; Shi, D.; Li, H. Recent progress on reduced graphene oxide supported Pt-based catalysts and electrocatalytic oxidation performance of methanol. Int. J. Hydrogen Energy 2023, 48, 1785–1812. [Google Scholar] [CrossRef]
- Souza, J.P.I.; Queiroz, S.L.; Nart, F.C. Use of mass spectrometry in electrochemical measurements—The DEMS technique. Quim. Nova 2000, 23, 384–391. [Google Scholar] [CrossRef]
- Iwasita, T.; Nart, F.C.; Vielstich, W. An FTIR Study of the Catalytic Activity of a 85:15 Pt:Ru Alloy for Methanol Oxidation. Bunsenges. Phys. Chem. 1990, 94, 1030–1034. [Google Scholar] [CrossRef]
- Babu, P.K.; Kim, H.S.; Oldfield, E.; Wieckowski, A. Electronic Alterations Caused by Ruthenium in Pt-Ru Alloy Nanoparticles as Revealed by Electrochemical NMR. Plenum 1992, 22, 97–104. [Google Scholar] [CrossRef]
- Mukerjee, S.; Urian, R.C. Bifunctionality in Pt alloy nanocluster electrocatalysts for enhanced methanol oxidation and CO tolerance in PEM fuel cell: Electrochemical and in situ synchrotron spectroscopy. Electrochim. Acta 2002, 47, 3219–3231. [Google Scholar] [CrossRef]
- Hoster, H.; Iwasita, T.; Baumgartner, H.; Vielstich, W. Pt-Ru model catalysts for anodic methanol oxidation: Influence of structure and composition on the reactivity. Phys. Chem. Chem. Phys. 2001, 3, 337–346. [Google Scholar] [CrossRef]
- Perrozzi, F.; Prezioso, S.; Ottaviano, L. Graphene oxide: From fundamentals to applications to cite this article. J. Condens. Matter Phys. 2015, 27, 013002. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Frelink, T.; Visscher, W.; Cox, A.P.; Van Veen, J.A.R. Ellipsometry and dems study of the electrooxidation of methanol at Pt and Ru- and Sn-promoted Pt. Electrochim. Acta 1995, 40, 1537–1543. [Google Scholar] [CrossRef]
- Antolini, E.; Cardellini, F. Formation of carbon supported PtRu alloy: An XRD analysis. J. Alloys Compd. 2001, 315, 118–127. [Google Scholar] [CrossRef]
- Mcbreen, J.; Mukerjee, S. In situ X-ray Absorption Studies of a Pt-Ru Electrocatalyst. J. Electrochem. Soc. 1995, 142, 3399–3404. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Li, H.; Wang, Q.; Kang, J.; Lei, Z. Facile synthesis of carbon-supported pseudo-core@shell PdCu@Pt nanoparticles for direct methanol fuel cells. Int. J. Hydrogen Energy 2011, 36, 839–848. [Google Scholar] [CrossRef]
- Muthuswamy, N.; La Fuente, J.L.G.; Dung, T.; Walmsley, J.; Tsypkin, M.; Raaen, S.; Sunde, S.; Ronning, M.; Chen, D. Ru@Pt core-shell nanoparticles for methanol fuel cell catalyst: Control and effects of shell composition. Int. J. Hydrogen Energy 2013, 38, 16631–16641. [Google Scholar] [CrossRef]
- Wu, Q.; Huang, X.; Wang, T.; Xiang, D.; Li, X.; Wang, K.; Yuan, X.; Li, P.; Zhu, M. Enhancing Electrocatalytic Methanol Oxidation on PtCuNi Core–Shell Alloy Structures in Acid Electrolytes. Inorg. Chem. 2022, 61, 2612–2618. [Google Scholar] [CrossRef]
- Zhu, H.; Li, X.; Wang, F. Synthesis and characterization of Cu@Pt/C core-shell structured catalysts for Exchange membrane fuel cell. Int. J. Hydrogen Energy 2011, 36, 9151–9154. [Google Scholar] [CrossRef]
- Liu, H.; Sun, F.; Chen, M.; Wang, H. Reconciling the experimental and computational methanol electro-oxidation activity via potential-dependent kinetic mechanism analysis. J. Mater. Chem. A 2022, 10, 23551–23561. [Google Scholar] [CrossRef]
- Hsin, Y.L.; Hwang, K.C.; Yeh, C.T. Poly(vinylpyrrolidone)-Modified Graphite Carbon Nanofibers as Promising Supports for PtRu Catalysts in Direct Methanol Fuel Cells. J. Am. Chem. Soc. 2007, 129, 9999–10010. [Google Scholar] [CrossRef]
- Smith, A.S.; Lachance, A.M.; Zeng, S.; Liu, B.; Sun, L. Synthesis, properties, and applications of graphene oxide/reduced graphene oxide and their nanocomposites. Nano Mater. Sci. 2019, 1, 31–37. [Google Scholar] [CrossRef]
- Neto, A.O.; Linardi, M.; Gonzales, E.R. Electrochemical oxidation of methanol on PtRu and PtMo particles supported on high surface area carbon. Eclet. Quim. 2003, 28, 55–62. [Google Scholar] [CrossRef]
- Mekazni, S.D.; Arán-Ais, R.M.; Ferre-Vilaplana, A.; Herrero, E. Why Methanol Electro-oxidation on Platinum in Water Takes Place Only in the Presence of Adsorbed OH. ACS Catal. 2022, 12, 1965–1970. [Google Scholar] [CrossRef]
- Baomin, L.; Qiang, Z.; Yezhen, Z.; Lijuan, W.; Fenfen, L.; Haiquan, X. Core-shell Ag nanowires@Pt nanorods catalyst: Synthesis and application in direct methanol fuel cells. Mater. Lett. 2018, 233, 138–141. [Google Scholar] [CrossRef]
- Watanabe, M.; Motoo, S. Electrocatalysis by ad-atoms—Part II: Enhancement of the oxidation of carbon monoxide on platinum by ruthenium ad-atoms. J. Electroanal. Chem. Interfacial Electrochem. 1975, 60, 267–273. [Google Scholar] [CrossRef]
- Rigsby, M.A.; Zhou, W.P.; Lewera, A.; Duong, H.T.; Bagus, O.S.; Jaegermann, W.; Hunger, R.; Wieckowski, A. Experiment and Theory of Fuel Cell Catalysis: Methanol and Formic Acid Decomposition on Nanoparticle Pt/Ru. J. Phys. Chem. C 2008, 112, 15595–15601. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrocatalyst | SOP | iF | iR | iF/iR |
|---|---|---|---|---|
| (V) | (A/cm2) | (A/cm2) | ||
| Pt/C-30 | 0.437 | 1.21 | 0.99 | 1.22 |
| PtRu/C-30 | 0.425 | 1.96 | 1.88 | 1.04 |
| Cu@PtRu/C-16 | 0.300 | 2.79 | 2.02 | 1.38 |
| Cu@PtRu/C-18 | 0.391 | 2.89 | 2.17 | 1.33 |
| Cu@PtRu/rGO-16 | 0.250 | 2.68 | 1.85 | 1.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, W.d.S.; Noce, R.d.; de Matos, T.d.S.; Andrade, F.V.; Molfetta, F.A.; Iúdice de Souza, J.P. Cu@PtRu Core–Shell Nanostructured Electrocatalysts Anchored on Reduced Graphene Oxide toward Methanol Oxidation. Energies 2023, 16, 6508. https://doi.org/10.3390/en16186508
Gomes WdS, Noce Rd, de Matos TdS, Andrade FV, Molfetta FA, Iúdice de Souza JP. Cu@PtRu Core–Shell Nanostructured Electrocatalysts Anchored on Reduced Graphene Oxide toward Methanol Oxidation. Energies. 2023; 16(18):6508. https://doi.org/10.3390/en16186508
Chicago/Turabian StyleGomes, Walber dos Santos, Rodrigo della Noce, Tamires de Sousa de Matos, Flávio Vargas Andrade, Fábio Alberto Molfetta, and José Pio Iúdice de Souza. 2023. "Cu@PtRu Core–Shell Nanostructured Electrocatalysts Anchored on Reduced Graphene Oxide toward Methanol Oxidation" Energies 16, no. 18: 6508. https://doi.org/10.3390/en16186508
APA StyleGomes, W. d. S., Noce, R. d., de Matos, T. d. S., Andrade, F. V., Molfetta, F. A., & Iúdice de Souza, J. P. (2023). Cu@PtRu Core–Shell Nanostructured Electrocatalysts Anchored on Reduced Graphene Oxide toward Methanol Oxidation. Energies, 16(18), 6508. https://doi.org/10.3390/en16186508