
Assessing the Benthic Response to Climate-Driven Methane Hydrate Destabilisation: State of the Art and Future Modelling Perspectives


Abstract
1. Introduction
2. Marine Hydrate Destabilisation and Methane Fate within Sediments
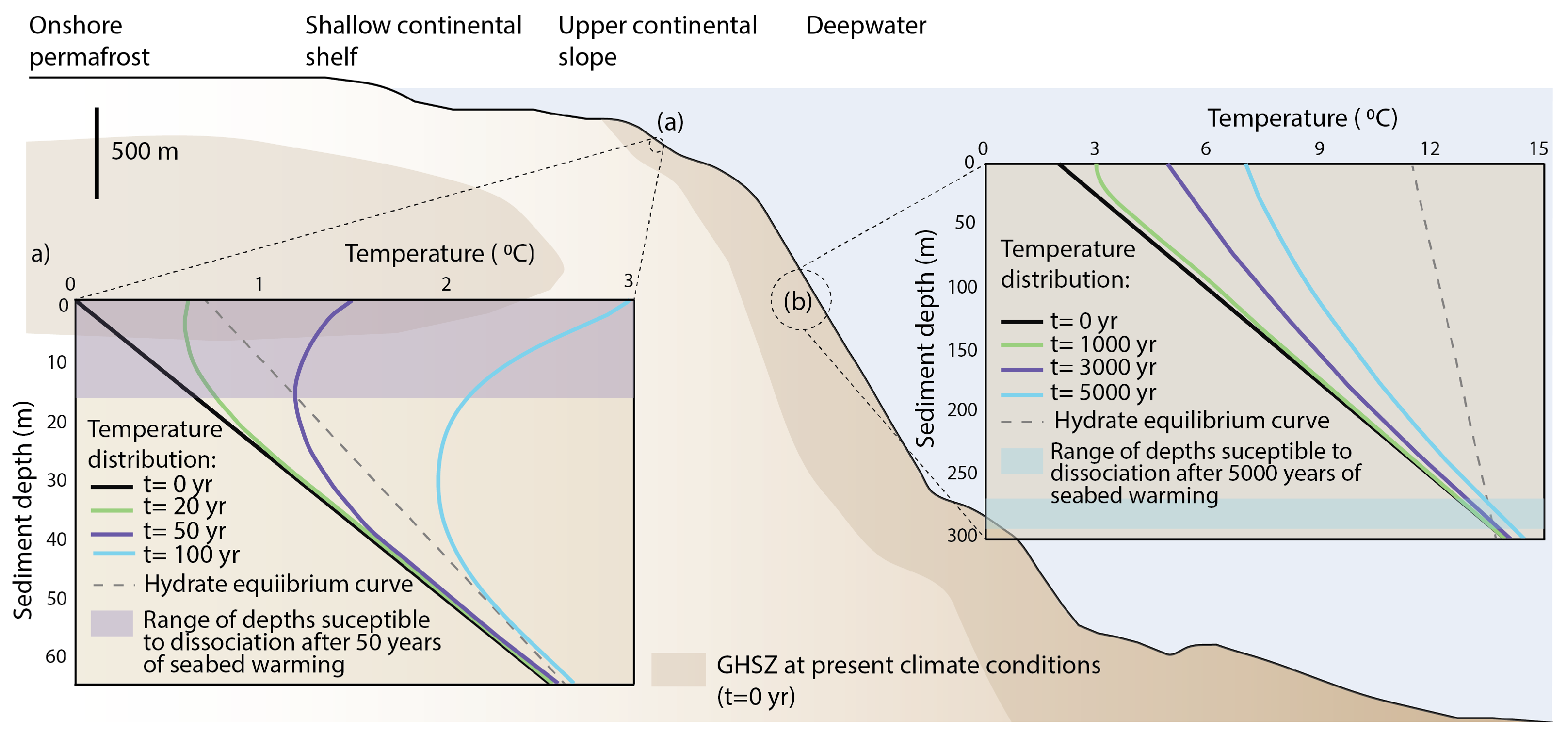
2.1. Hydrate Vulnerability to Climate Perturbations
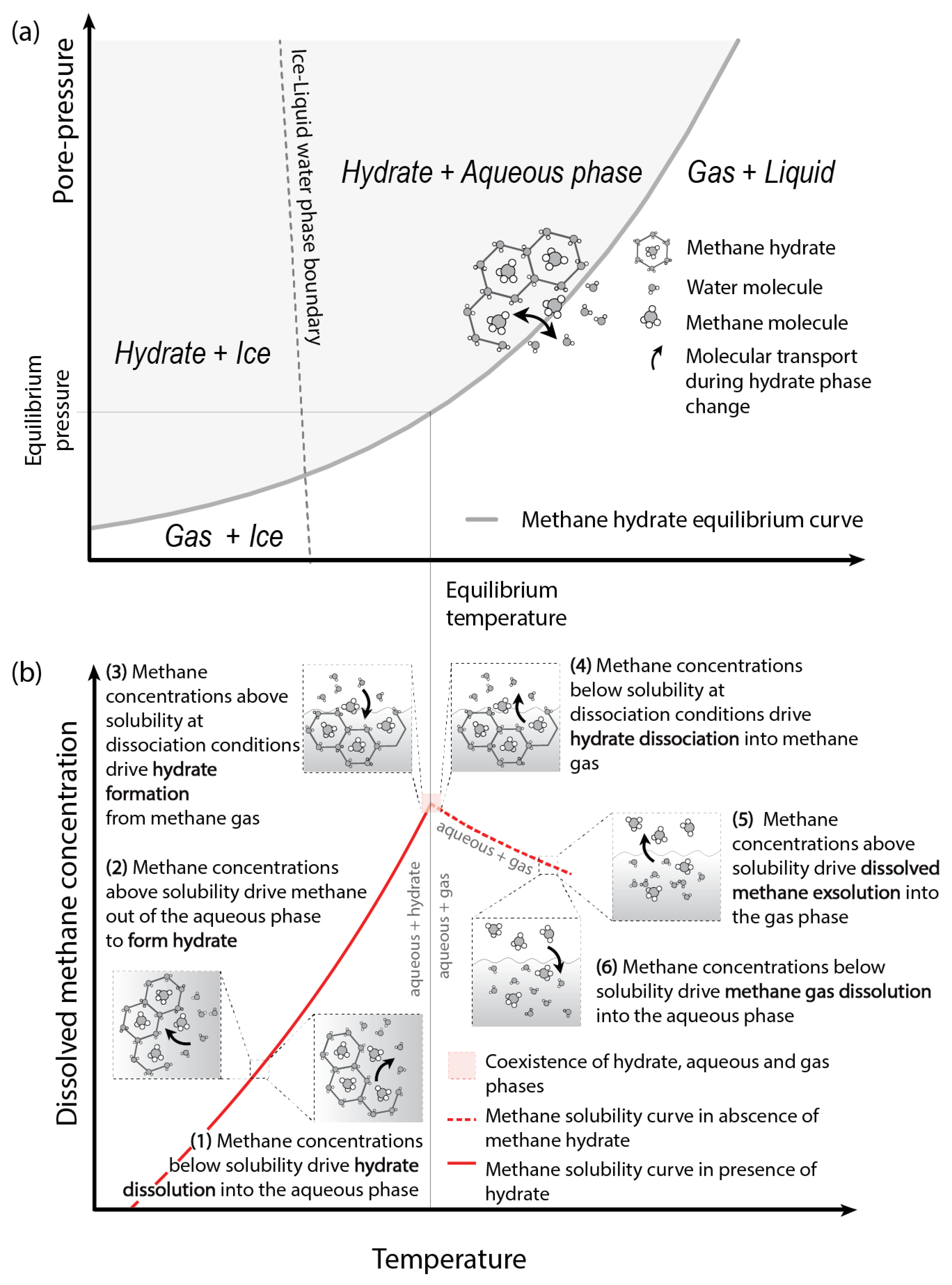
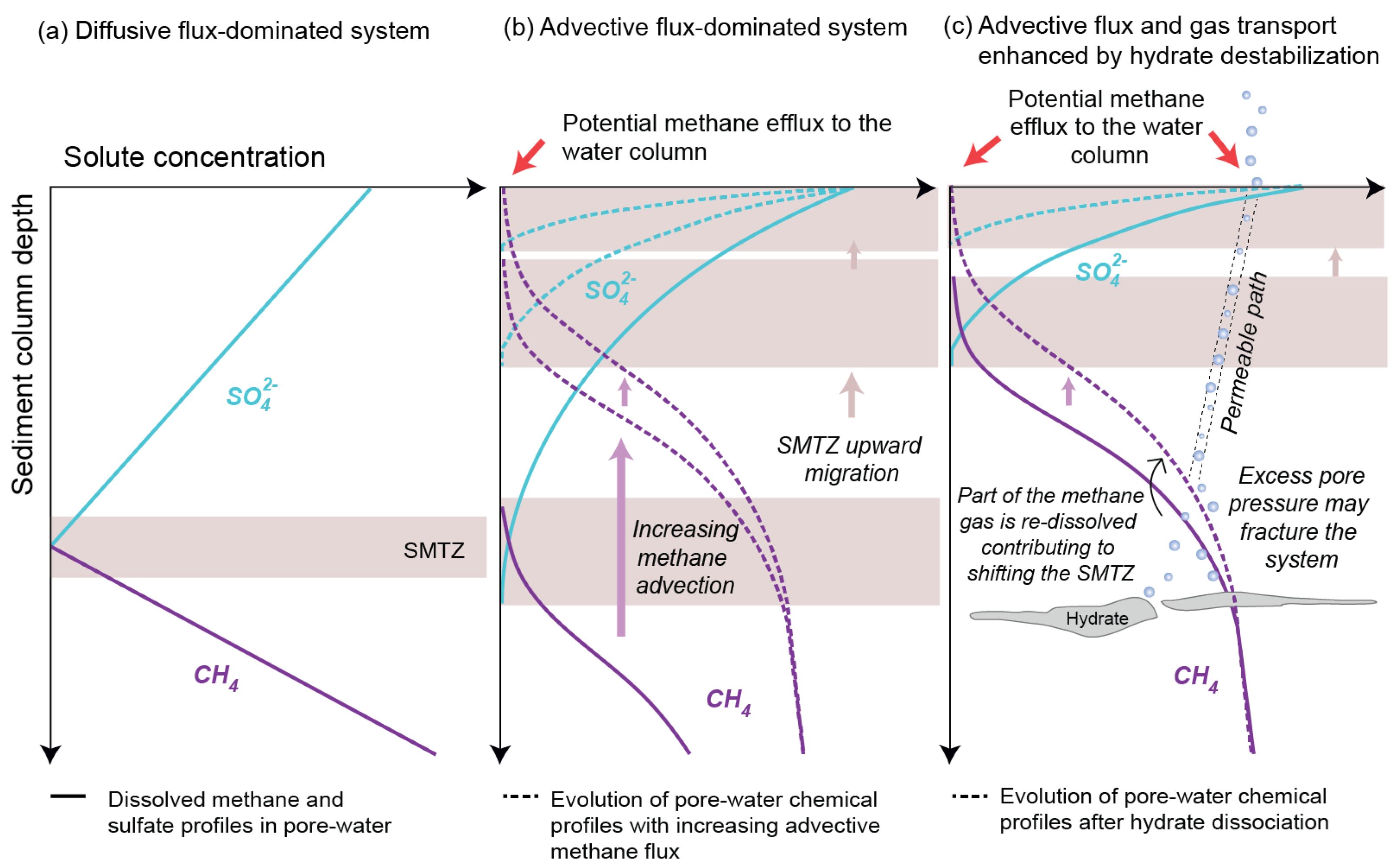
2.2. Hydrate Destabilisation and Multiphase Transport of Methane
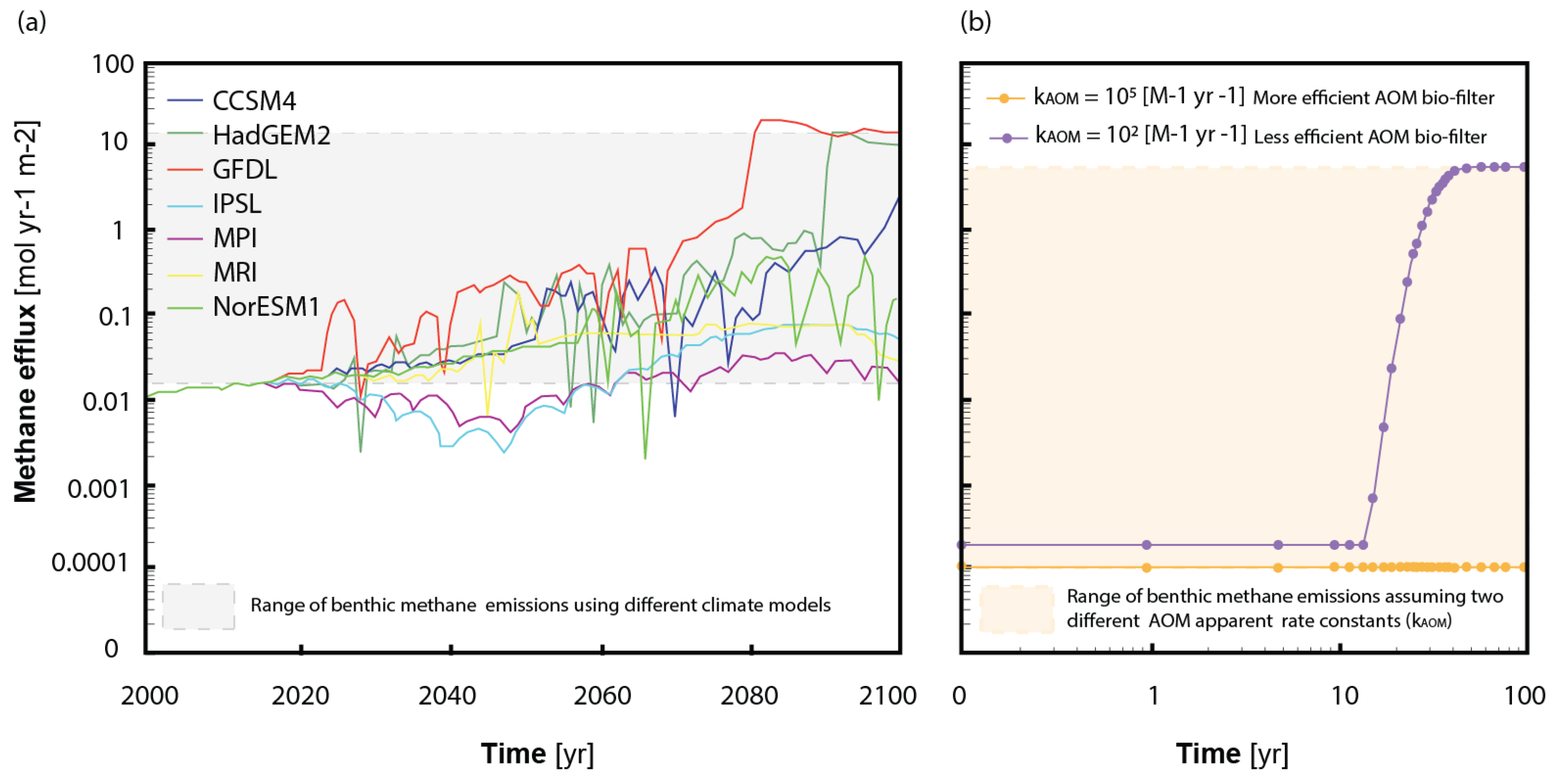
2.3. The Benthic Methane Sink: The Variable Efficiency of the AOM Bio-Filter
2.3.1. Influence of Fluid Flow Regime on the AOM
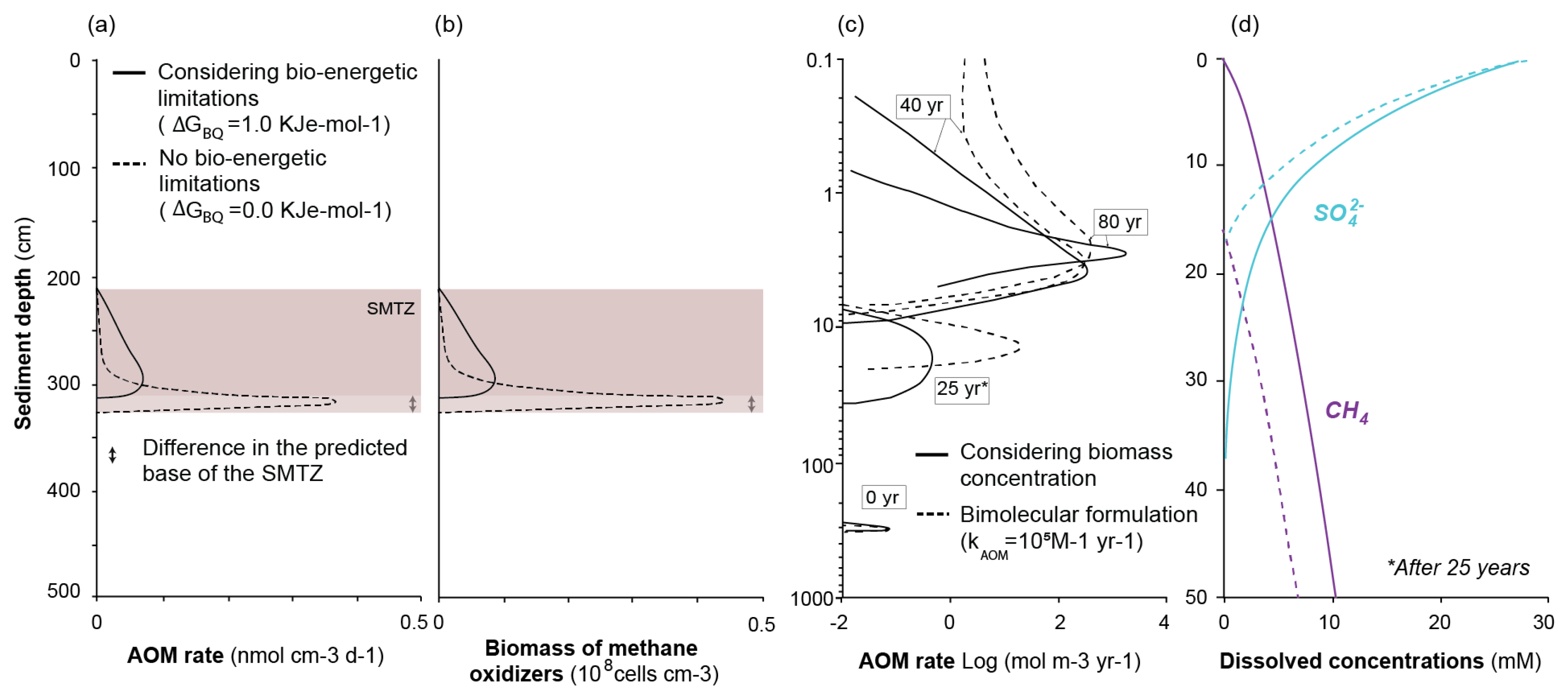
2.3.2. Kinetic and Bio-Energetic Limitations to Biomass Growth
2.3.3. Bioirrigation and Bioturbation
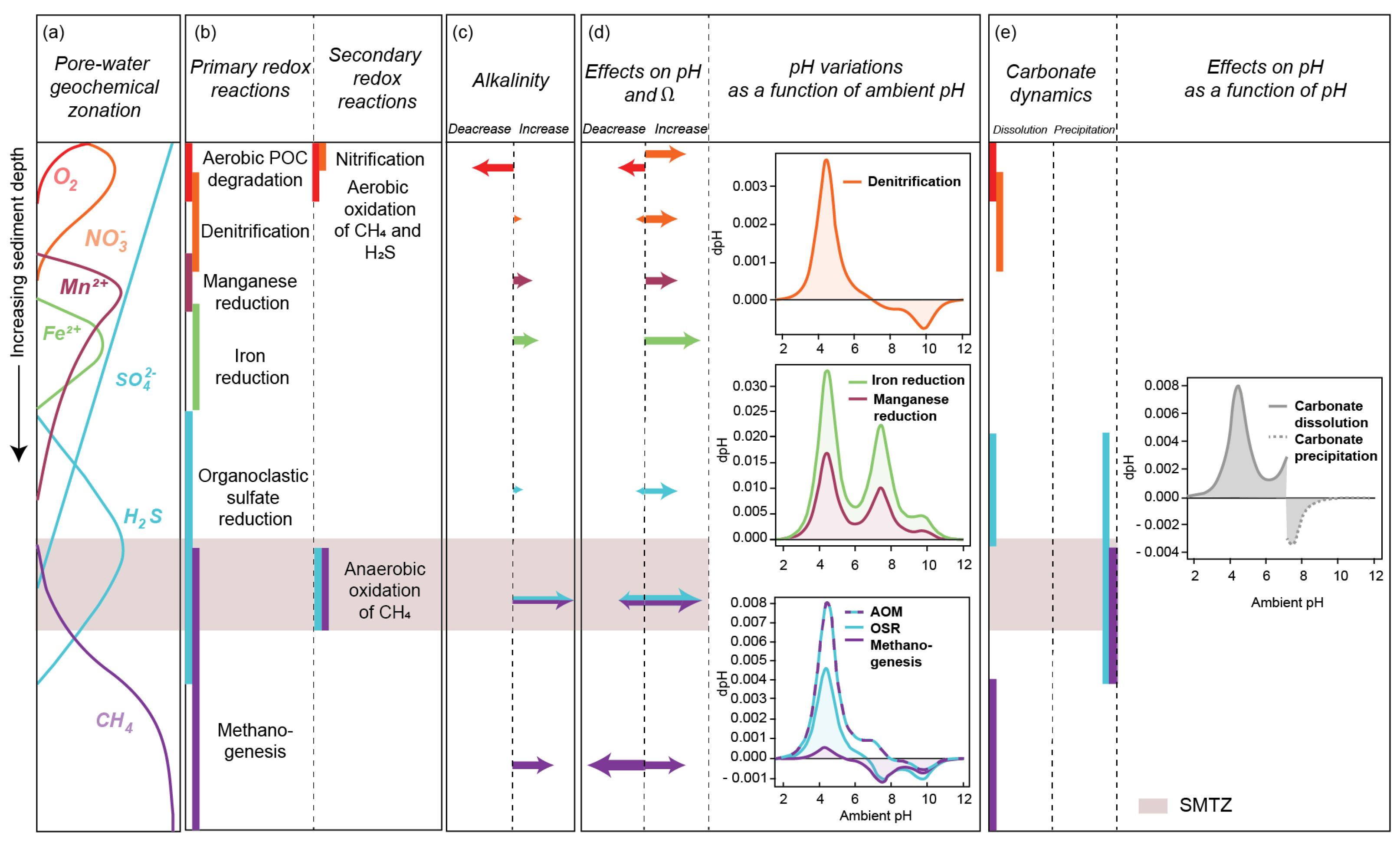
2.4. Benthic Biogeochemical Impact of Methane Consumption through the Sediment Column
3. Modelling the Benthic Response to Climate-Driven Methane Hydrate Destabilisation
3.1. What the Scientific Questions Numerical Models Should Answer
- How much methane is stored in methane hydrate and how much of it could be released in response to climate perturbations?
- How efficient is the benthic methane sink and thus, how much, at what rate and for how long hydrate-sourced methane can be actually emitted to the ocean?
- What are the implications of methane turnover across AOM on benthic biogeochemical dynamics and fluxes?
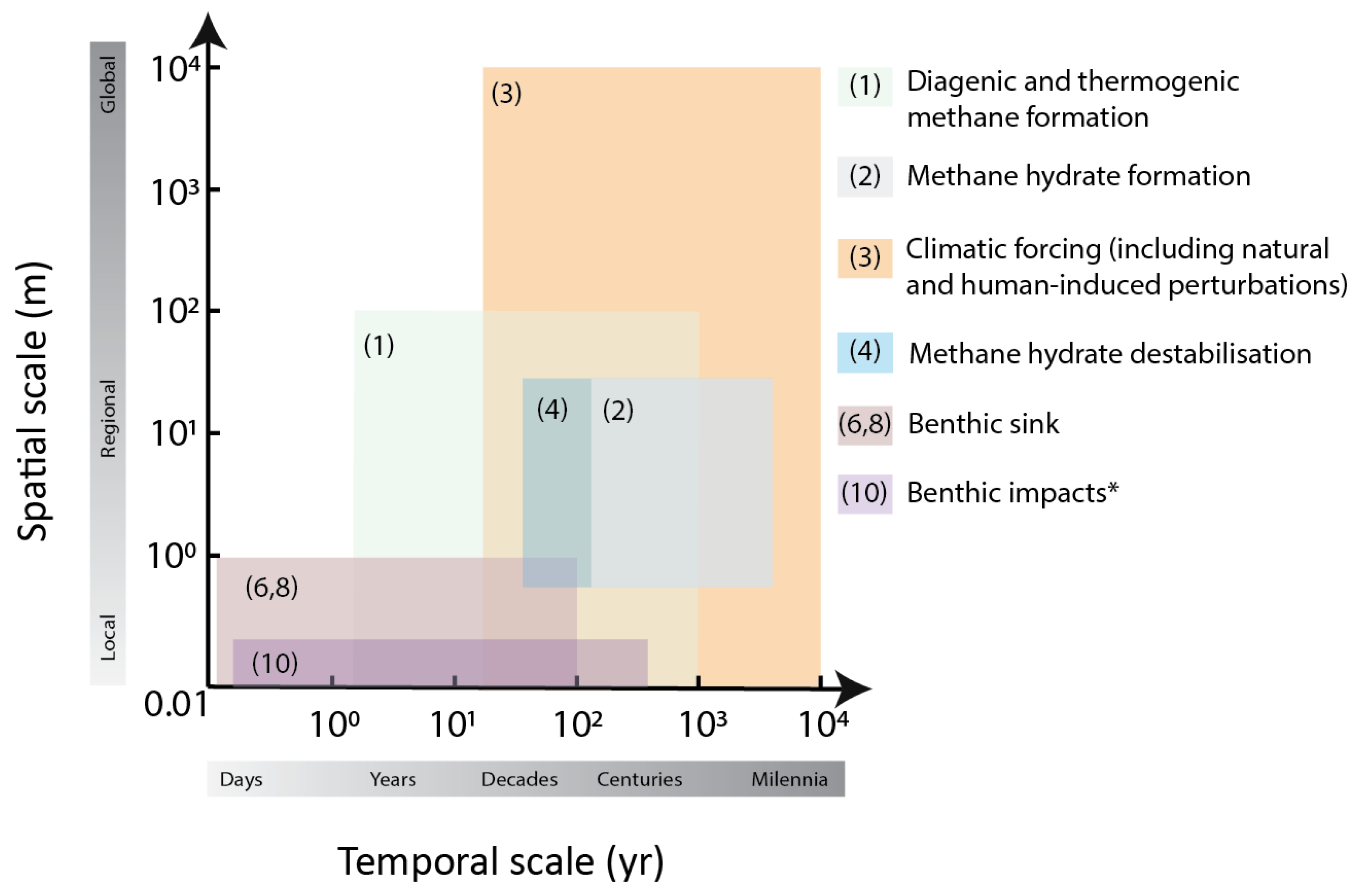
Temporal and Spatial Scales
3.2. State-of-the-Art
3.3. Future Modelling Perspectives
- Applying existing numerical models that so far have been only used to assess the hydrate inventory, to investigate the response of hydrate systems to climate perturbations. For instance, the models developed by Chatterjee et al. [119] and Tian et al. [161] could be well suited to exploring the benthic response to hydrate destabilisation but they have not been applied to examine scenarios of climate change. Chatterjee et al. [119] updated a 1D numerical model for the formation of hydrate in marine sediment [172,173] to couple it with mass balance equations for methane, sulfate, dissolved inorganic carbon, dissolved calcium () and carbon isotopes in pore water. They examined changes in these species due to methane cycling, including sediment burial, methane production from organic carbon, upward fluxes of methane, and significant loss of methane by AOM. However, they neglected hydrate and free gas phases in their calculations, only solving the dissolved methane concentrations and assuming that methane concentrations exceeding solubility do not form gas hydrate or free gas. In addition, they simplified the AOM rate to a bimolecular law. On the other hand, Tian et al. [161] coupled the TOUGH + HYDRATE model [154] with TOUGHREACT [174] to integrate reactive transport with the behaviour of hydrate-bearing systems. However, they focused on evaluating the resource of natural gas hydrate and quantifying the contribution of methane sources constrained by site-specific data.
- Extending current diagenetic formulations developed for hydrate systems to explicitly resolve AOM microbial dynamics and incorporate the full reaction-network driving the production/consumption of methane, total alkalinity, total sulfide, and dissolved inorganic carbon and controlling carbonate saturation and water pH (e.g., [160]). Burwicz et al. [160] developed a numerical model to investigate different scenarios of gas hydrate formation from both single and mixed methane sources (i.e., in situ biogenic and deep thermogenic sources). Although their model reproduces well the hydrate inventory, the AOM rate considered is too simplistic and the formulation ignores other key chemical reactions controlling benthic pore-water chemistry, for example, organoclastic sulfate consumption or authigenic carbonate precipitation/dissolution. In addition, this model has not been applied yet to explore hydrate destabilisation driven by climate perturbations. In the case of more sophisticated hydrate models (e.g., [46,47]), it would be key also to incorporate diagenetic processes for organic carbon accumulation and microbial degradation.
- Extending reactive-transport models to incorporate hydrate dynamics and consider multiphase transport. Among others reactive-transport models, the Biogeochemical Reaction Network Simulator (BRNS) [175,176,177] has been widely used to quantitatively explore the fluxes and transformations of methane carbon in the sediment column. This type of model is well suited to assessing the benthic response to hydrate destabilisation as it traces a wide range of chemical species dissolved in pore-water (e.g., [45,53]).
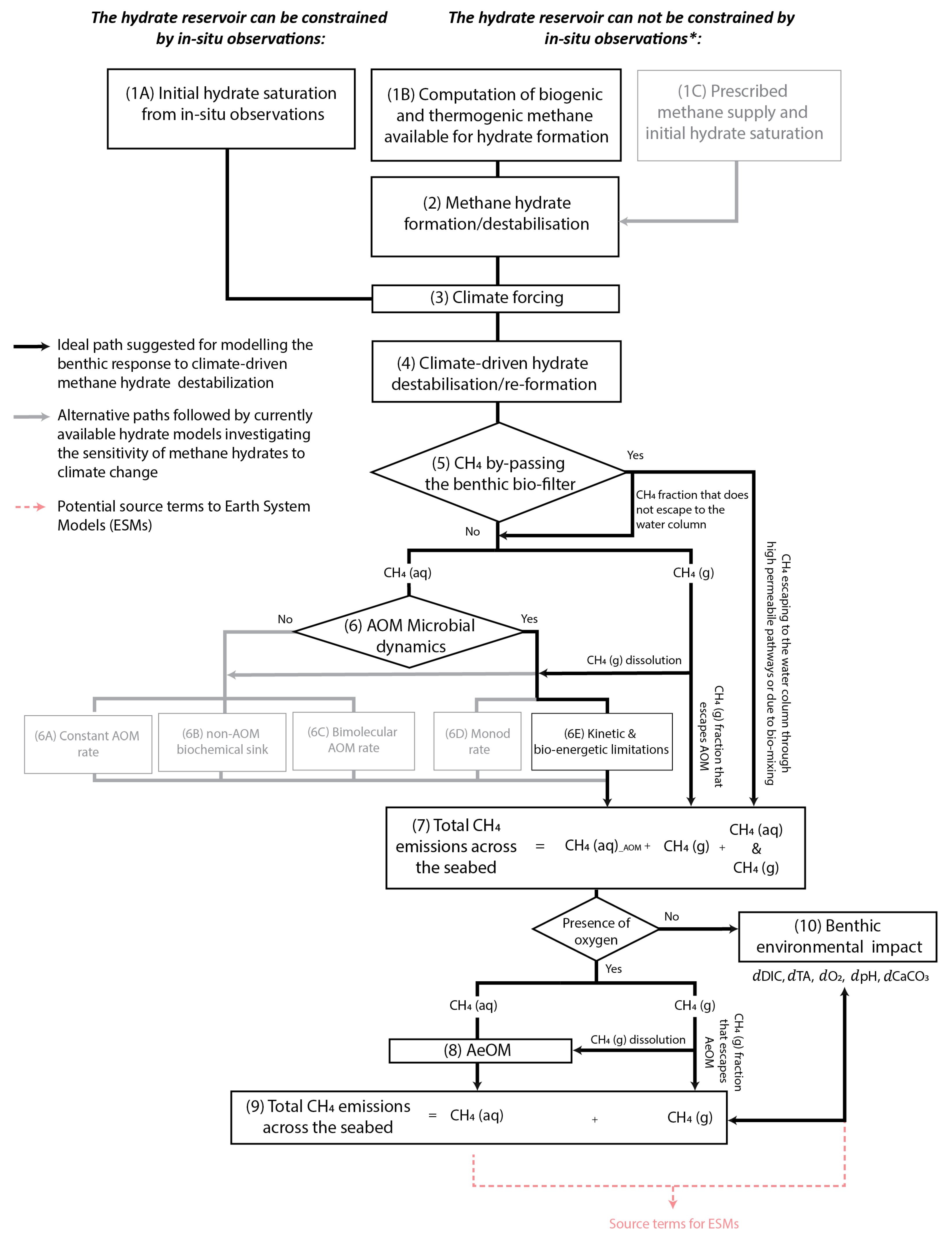
- Developing new models capable of fully coupling the dynamic interplay between hydrate thermodynamics, the different reaction and transport processes, and biomass dynamics, and which are particularly designed to assess the biogeochemical impact of methane turnover via AOM in the benthic environment (see flow path suggested in black in Figure 9). The development of new numerical schemes will avoid issues related to generalising or modifying existing formulations and can facilitate direct coupling with Earth system models (red dashed line in Figure 9, see Section 3.3.1).
- Implementing carbon isotope fractionation in fully coupled hydrate models to trace methane sources and simulate quantitative proxies of methane emissions, such as authigenic carbonates [53,178]. This will help reconstruct the activity of hydrate systems in past hyper-thermal events and establish a baseline for understanding the system response in future climate predictions.
3.3.1. The Need to Integrate Benthic Hydrate-Related Carbon Feed-Backs into Earth System Models (ESMs)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Westbrook, G.K.; Thatcher, K.E.; Rohling, E.J.; Piotrowski, A.M.; Pälike, H.; Osborne, A.H.; Nisbet, E.G.; Minshull, T.A.; Lanoisellé, M.; James, R.H.; et al. Escape of methane gas from the seabed along the West Spitsbergen continental margin. Geophys. Res. Lett. 2009, 36, L15608. [Google Scholar] [CrossRef]
- Ruppel, C.D.; Kessler, J.D. The interaction of climate change and methane hydrates. Rev. Geophys. 2017, 55, 126–168. [Google Scholar] [CrossRef]
- Sayedi, S.; Abbott, B.W.; Thornton, B.F.; Frederick, J.M.; Vonk, J.E.; Overduin, P.; Schädel, C.; Schuur, E.A.G.; Bourbonnais, A.; Demidov, N.; et al. Subsea permafrost carbon stocks and climate change sensitivity estimated by expert assessment. Environ. Res. Lett. 2020, 15, 124075. [Google Scholar] [CrossRef]
- McIver, R.D. Gas hydrates. In Long-Term Energy Resources; Meyer, R.F., Olson, J.C., Eds.; Pitman: Boston, MA, USA, 1981; pp. 713–726. [Google Scholar]
- Boswell, R.; Collett, T.S. Current perspectives on gas hydrate resources. Energy Environ. Sci. 2011, 4, 1206–1215. [Google Scholar] [CrossRef]
- Piñero, E.; Marquardt, M.; Hensen, C.; Haeckel, M.; Wallmann, K. Estimation of the global inventory of methane hydrates in marine sediments using transfer functions. Biogeosciences 2013, 10, 959–975. [Google Scholar] [CrossRef]
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef]
- Kennett, J.P.; Cannariato, K.G.; Hendy, I.L.; Behl, R.J. Methane Hydrates in Quaternary Climate Change: The Clathrate Gun Hypothesis; AGU: Washington, DC, USA, 2003; p. 216. [Google Scholar]
- Schmidt, G.A.; Shindell, D.T. Atmospheric composition, radiative forcing, and climate change as a consequence of a massive methane release from gas hydrates. Paleoceanography 2003, 18, 1004. [Google Scholar] [CrossRef]
- Dickens, G. Hydrocarbon-driven warming. Nature 2004, 429, 513–515. [Google Scholar] [CrossRef]
- Röhl, U.; Westerhold, T.; Bralower, T.J.; Zachos, J.C. On the duration of the Paleocene-Eocene Thermal Maximum. Geochem. Geophys. Geosyst. 2007, 8, Q12002. [Google Scholar] [CrossRef]
- Mestdagh, T.; Poort, J.; De Batist, M. The sensitivity of gas hydrate reservoirs to climate change: Perspectives from a new combined model for permafrost-related and marine settings. Earth-Sci. Rev. 2017, 169, 104–131. [Google Scholar] [CrossRef]
- Dickens, G.R.; O’Neil, J.R.; Rea, D.K.; Owen, R.M. Dissociation of oceanic methane hydrate as a cause of the carbon isotopic excursion at the end of the Paleocene. Paleoceanography 1995, 10, 965–971. [Google Scholar] [CrossRef]
- Zachos, J.; Röhl, U.; Schellenberg, S.A.; Sluijs, A.; Hodell, D.A.; Kelly, D.C.; Thomas, E.; Nicolo, M.; Raffi, I.; Lourens, L.J.; et al. Rapid acidification of the ocean during the Paleocene-Eocene thermal maximum. Science 2005, 308, 1611–1615. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, V.V.; Smith, A.M.; Brook, E.J.; Lowe, D.; Riedel, K.; Brailsford, G.; Hua, Q.; Schaefer, H.; Reeh, M.N.; Weiss, R.F.; et al. 14CH4 measurements in Greenland ice: Investigating last glacial termination CH4 sources. Science 2009, 324, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Bock, M.; Schmitt, J.; Möller, L.; Spahni, R.; Blunier, T.; Fischer, H. Hydrogen isotopes preclude marine hydrate CH4 emissions at the onset of Dansgaard-Oeschger Events. Science 2010, 328, 1686–1689. [Google Scholar] [CrossRef][Green Version]
- Gutjahr, M.; Ridgwell, A.; Sexton, P.; Anagnostou, E.; Pearson, P.N.; Pälike, H.; Norris, R.D.; Thomas, E.; Foster, G.L. Very large release of mostly volcanic carbon during the Palaeocene–Eocene Thermal Maximum. Nature 2017, 328, 573–577. [Google Scholar] [CrossRef]
- Inglis, G.N.; Rohrssen, M.; Kennedy, E.M.; Crouch, E.M.; Raine, J.I.; Strogen, D.P.; Naafs, B.D.A.; Collinson, M.E.; Pancost, R.D. Terrestrial methane cycle perturbations during the onset of the Paleocene-Eocene Thermal Maximum. Geology 2021, 45, 520–524. [Google Scholar] [CrossRef]
- Marín-Moreno, H.; Minshull, T.A.; Westbrook, G.K.; Sinha, B.; Sarkar, S. The response of methane hydrate beneath the seabed offshore Svalbard to ocean warming during the next three centuries. Geophys. Res. Lett. 2013, 40, 5163–5759. [Google Scholar] [CrossRef]
- Kretschmer, K.; Biastoch, A.; Rüpke, L.; Burwicz, E. modelling the fate of methane hydrates under global warming. Glob. Biogeochem. Cycles 2015, 29, 610–625. [Google Scholar] [CrossRef]
- Stranne, C.; O’Regan, M.; Jakobsson, M. Overestimating climate warming-induced methane gas escape from the seafloor by neglecting multiphase flow dynamics. Geophys. Res. Lett. 2016, 43, 8703–8712. [Google Scholar] [CrossRef]
- Braga, R.; Iglesias, R.S.; Romio, C.; Praeg, D.; Miller, D.J.; Viana, A.; Ketzer, J.M. Modelling methane hydrate stability changes and gas release due to seasonal oscillations in bottom water temperatures on the Rio Grande cone, offshore southern Brazil. Mar. Pet. Geol. 2020, 112, 104071. [Google Scholar] [CrossRef]
- Skarke, A.; Ruppel, C.; Kodis, M.; Brothers, D.; Lobecker, E. Modelling methane hydrate Widespread methane leakage from the sea floor on the northern US Atlantic margin. Nat. Geosci. 2014, 7, 657–661. [Google Scholar] [CrossRef]
- Paull, C.K.; Dallimore, S.R.; Caress, D.W.; Gwiazda, R.; Melling, H.; Riedel, M.; Jin, Y.K.; Hong, J.K.; Kim, Y.G.; Graves, D.; et al. Active mud volcanoes on the continental slope of the Canadian Beaufort Sea. Geochem. Geophys. Geosyst. 2015, 16, 3160–3181. [Google Scholar] [CrossRef]
- Mau, S.; Römer, M.; Torres, M.E.; Bussmann, I.; Pape, T.; Damm, E.; Geprägs, P.; Wintersteller, P.; Hsu, C.-W.; Loher, M.; et al. Widespread methane seepage along the continental margin off Svalbard—From Bjørnøya to Kongsfjorden. Sci. Rep. 2017, 7, 42997. [Google Scholar] [CrossRef] [PubMed]
- Ketzer, M.; Praeg, D.; Rodrigues, L.F.; Augustin, A.; Pivel, M.A.G.; Rahmati-Abkenar, M.; Miller, D.J.; Viana, A.R.; Cupertino, J.A. Gas hydrate dissociation linked to contemporary ocean warming in the southern hemisphere. Nat. Commun. 2020, 11, 3788. [Google Scholar] [CrossRef] [PubMed]
- Thurber, A.R.; Seabrook, S.; Welsh, R.M. Riddles in the cold: Antarctic endemism and microbial succession impact methane cycling in the Southern Ocean. Proc. R. Soc. B 2020, 287, 20201134. [Google Scholar] [CrossRef]
- Forster, P.; Ramaswamy, V.; Artaxo, P.; Berntsen, T.; Betts, R.; Fahey, D.W.; Haywood, J.; Lean, J.; Lowe, D.C.; Myhre, G.; et al. Changes in atmospheric constituents and in radiative forcing, in Climate Change 2007. In The Physical Science Basis—Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K.B., Tignor, M., Miller, H.L., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 131–234. [Google Scholar]
- Archer, D.; Buffett, B.; Brovkin, V. Ocean methane hydrates as a slow tipping point in the global carbon cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 20596–20601. [Google Scholar] [CrossRef]
- Masson-Delmotte, V.; Zhai, P.; Pirani, A.; Connors, S.L.; Péan, C.; Berger, S.; Caud, N.; Chen, Y.; Goldfarb, L.; Gomis, M.I.; et al. (Eds.) IPCC, 2021: Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2021; In press. [Google Scholar]
- Marín-Moreno, H.; Minshull, T.A.; Westbrook, G.K.; Sinha, B. Estimates of future warming-induced methane emissions from hydrate offshore west Svalbard for a range of climate models. Geochem. Geophys. Geosyst. 2015, 16, 1307–1323. [Google Scholar] [CrossRef]
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K.; et al. The Global Methane Budget 2000–2017. Earth Syst. Sci. Data 2020, 12, 1561–1623. [Google Scholar] [CrossRef]
- Beaudoin, Y.C.; Dallimore, S.R.; Boswell, R. Frozen Heat: A UNEP Global Outlook on Methane Gas Hydrates, Volume 2. 2014. Available online: https://www.unep.org/resources/report/frozen-heat-global-outlook-methane-gas-hydrates-volume-2 (accessed on 20 October 2021).
- Hinrichs, K.U.; Boetius, A. The anaerobic oxidation of methane: New insights in microbial ecology and biogeochemistry. In Ocean Margin Systems; Wefer, G., Billet, D., Hebbeln, D., Jorgensen, B.B., Schluuter, M., Weering, T.C.E.V., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 457–477. [Google Scholar]
- Reeburgh, W.S. Oceanic Methane Biogeochemistry. Chem. Rev. 2007, 107, 486–513. [Google Scholar] [CrossRef]
- Knittel, K.; Boetius, A. Anaerobic oxidation of methane: Progress with an unknown process. Ann. Rev. Microbiol. 2009, 63, 311–334. [Google Scholar] [CrossRef]
- Treude, T.; Krüger, M.; Boetius, A.; Jørgensen, B.B. Environmental control on anaerobic oxidation of methane in the gassy sediments of Eckernförde Bay (German Baltic). Limnol. Oceanogr. 2005, 50, 1771–1778. [Google Scholar] [CrossRef]
- Boetius, A.; Wenzhofer, F. Seafloor oxygen consumption fuelled by methane from cold seeps. Nat. Geosci. 2013, 6, 725–734. [Google Scholar] [CrossRef]
- Egger, M.; Riedinger, N.; Mogollón, J.M.; Jørgensen, B.B. Global diffusive fluxes of methane in marine sediments. Nat. Geosci. 2018, 11, 421–425. [Google Scholar] [CrossRef]
- Dale, A.W.; Regnier, P.; Van Cappellen, P. Bioenergetic controls on anaerobic oxidation of methane (AOM) in coastal marine sediments: A theoretical analysis. Am. J. Sci. 2006, 306, 246–294. [Google Scholar] [CrossRef]
- Dale, A.W.; Van Cappellen, P.; Aguilera, D.R.; Regnier, P. Methane efflux from marine sediments in passive and active margins: Estimations from bioenergetic reaction–transport simulations. Earth Planet. Sci. Lett. 2008, 265, 329–344. [Google Scholar] [CrossRef]
- Fu, X.; Jimenez-Martinez, J.; Nguyen, T.P.; Carey, J.W.; Viswanathan, H.; Cueto-Felgueroso, L.; Juanes, R. Crustal fingering facilitates free-gas methane migration through the hydrate stability zone. Proc. Natl. Acad. Sci. USA 2020, 117, 31660–31664. [Google Scholar] [CrossRef]
- Regnier, P.; Dale, A.W.; Arndt, S.; LaRowe, D.E.; Mogollón, M.J.; Van Cappellen, P. Quantitative analysis of anaerobic oxidation of methane (AOM) in marine sediments: A modelling perspective. Earth Sci. Rev. 2006, 106, 105–130. [Google Scholar] [CrossRef]
- Stranne, C.; O’Regan, M.; Jakobsson, M. Modelling fracture propagation and seafloor gas release during seafloor warming-induced hydrate dissociation. Geophys. Res. Lett. 2017, 44, 8510–8519. [Google Scholar] [CrossRef]
- Puglini, M.; Brovkin, V.; Regnier, P.; Arndt, S. Assessing the potential for non-turbulent methane escape from the East Siberian Arctic Shelf. Biogeosciences 2020, 17, 3247–3275. [Google Scholar] [CrossRef]
- Stranne, C.; O’Regan, M.; Jakobsson, M.; Brüchert, V.; Ketzer, M. Can anaerobic oxidation of methane prevent seafloor gas escape in a warming climate? Solid Earth 2019, 10, 1541–1554. [Google Scholar] [CrossRef]
- Reagan, T.A.M.; Moridis, G.J. Modelling of Oceanic Gas Hydrate Instability and Methane Release in Response to Climate Change. In Proceedings of the 6th International Conference on Gas Hydrates (ICGH 2008), Vancouver, BC, Canada, 6–10 July 2008. [Google Scholar]
- Elliott, S.; Reagan, M.; Moridis, G.; Smith, P.C. Geochemistry of clathrate-derived methane in Arctic ocean waters. Geophys. Res. Lett. 2010, 37, L12607. [Google Scholar] [CrossRef]
- Biastoch, A.; Treude, T.; Rüpke, L.H.; Riebesell, U.; Roth, C.; Burwicz, E.B.; Park, W.; Latif, M.; Böning, C.W.; Madec, G.; et al. Rising Arctic Ocean temperatures cause gas hydrate destabilisation and ocean acidification. Geophys. Res. Lett. 2011, 38, L08602. [Google Scholar] [CrossRef]
- Elliott, S.; Maltrud, M.; Reagan, M.; Moridis, G.; Cameron-Smith, P. Marine methane cycle simulations for the period of early global warming. J. Geophys. Res. 2011, 116, G01010. [Google Scholar]
- Hunter, S.J.; Goldobin, D.S.; Haywood, A.M.; Ridgwell, A.; Rees, J.G. Sensitivity of the global submarine hydrate inventory to scenarios of future climate change. Earth Planet. Sci. Lett. 2013, 367, 105–115. [Google Scholar] [CrossRef]
- Yamamoto, A.; Yamanaka, Y.; Oka, A.; Abe-Ouchi, A. Ocean oxygen depletion due to decomposition of submarine methane hydrate. J. Geophys. Res. Lett. 2014, 41, 5075–5083. [Google Scholar] [CrossRef]
- Blouet, J.P.; Arndt, S.; Imbert, P.; Regnier, P. Are seep carbonates quantitative proxies of CH4 leakage? modelling the influence of sulfate reduction and anaerobic oxidation of methane on pH and carbonate precipitation. Chem. Geol. 2020, 577, 120254. [Google Scholar] [CrossRef]
- Sundby, B.; Anschutz, P.; Lecroart, P.; Mucci, A. Ideas and perspectives: Sea-level change, anaerobic methane oxidation, and the glacial–interglacial phosphorus cycle. Biogeosciences 2022, 19, 1421–1434. [Google Scholar] [CrossRef]
- Akam, S.A.; Coffin, R.B.; Abdulla, H.A.; Lyons, T.W. Dissolved inorganic carbon pump in methane-charged shallow marine sediments: State of the art and new model perspectives. Front. Mar. Sci. 2020, 7, 206. [Google Scholar] [CrossRef]
- James, R.H.; Bousquet, P.; Bussmann, I.; Haeckel, M.; Kipfer, R.; Leifer, I.; Niemann, H.; Ostrovsky, I.; Piskozub, J.; Rehder, G.; et al. Effects of climate change on methane emissions from seafloor sediments in the Arctic Ocean: A review. Limnol. Oceanogr. 2016, 61, S283–S299. [Google Scholar] [CrossRef]
- Tyrrell, T.; Holligan, P.M.; Mobley, C.D. Optical impacts of oceanic coccolithophore blooms. J. Geophys. Res. Ocean. 1999, 104, 3223–3241. [Google Scholar] [CrossRef]
- Guinotte, J.M.; Fabry, V.J. Ocean acidification and its potential effects on marine ecosystems. Ann. N. Y. Acad. Sci. 2008, 1134, 320–342. [Google Scholar] [CrossRef]
- Griffith, G.P.; Fulton, E.A.; Gorton, R.; Richardson, A.J. Predicting Interactions among Fishing, Ocean Warming, and Ocean Acidification in a Marine System with Whole-Ecosystem Models. Conserv. Biol. 2012, 26, 1145–1152. [Google Scholar] [CrossRef]
- Turley, C.; Gattuso, J.P. Future biological and ecosystem impacts of ocean acidification and their socioeconomic-policy implications. Curr. Opin. Environ. Sustain. 2012, 4, 278–286. [Google Scholar] [CrossRef]
- Raven, J.; Caldeira, K.; Elderfield, H.; Hoegh-Guldberg, O.; Liss, P.S.; Riebesell, U.; Sheperd, J.; Turley, C.; Watson, A. Ocean Acidification due to Increasing Atmospheric Carbon Dioxide; The Royal Society Policy Document; The Royal Society: London, UK, 2005. [Google Scholar]
- Middelburg, J.J.; Soetaert, K.; Hagens, M. Ocean alkalinity, buffering and biogeochemical processes. Rev. Geophys. 2020, 58, e2019RG000681. [Google Scholar] [CrossRef]
- Landrigan, P.J.; Stegeman, J.J.; Fleming, L.E.; Allemand, D.; Anderson, D.M.; Backer, L.C.; Brucker-Davis, F.; Chevalier, N.; Corra, L.; Czerucka, D.; et al. Human Health and Ocean Pollution. Ann. Glob. Health 2020, 86, 151. [Google Scholar] [CrossRef]
- Krey, V.; Canadell, J.G.; Nakicenovic, N.; Abe, Y.; Andruleit, H.; Archer, D.; Grubler, A.; Hamilton, N.; Johnson, A.; Kostov, V. Gas hydrates: Entrance to a methane age or climate threat? Environ. Res. Lett. 2009, 4, 034007. [Google Scholar] [CrossRef]
- Thatcher, K.E.; Westbrook, G.K.; Sarkar, S.; Minshull, T.A. Methane release from warming-induced hydrate dissociation in the West Svalbard continental margin: Timing, rates, and geological controls. J. Geophys. Res. Solid Earth 2013, 118, 22–38. [Google Scholar] [CrossRef]
- Ruppel, C.D. Methane hydrates and contemporary climate change. Nat. Educ. Knowl. 2011, 2, 12. [Google Scholar]
- Archer, D. Methane hydrate stability and anthropogenic climate change. Biogeosciences 2007, 4, 521–544. [Google Scholar] [CrossRef]
- Wallmann, K.; Riedel, M.; Hong, W.L.; Patton, H.; Hubbard, A.; Pape, T.; Hsu, C.W.; Schmidt, C.; Johnson, J.E.; Torres, M.E.; et al. Gas hydrate dissociation off Svalbard induced by isostatic rebound rather than global warming. Nat. Commun. 2018, 9, 83. [Google Scholar] [CrossRef]
- Carson, M.; Köhl, A.; Stammer, D.; Slangen, A.B.A.; Katsman, C.A.R.; van de Wal, S.W.; Church, J.; White, N. Coastal sea level changes, observed and projected during the 20th and 21st century. Clim. Chang. 2016, 134, 269–281. [Google Scholar] [CrossRef]
- Braga, R.; Vecchia, F.D.; Iglesias, R.S. Modelling the dynamic response of shallow methane hydrates to simultaneous sea level and bottom water temperatures variations since the last glacial maximum on the Amazon Deep-Sea Fan, Brazil. Mar. Pet. Geol. 2022, 137, 105494. [Google Scholar] [CrossRef]
- Minshull, T.A.; Marín-Moreno, H.; Armstrong McKay, D.I.; Wilson, P.A. Mechanistic insights into a hydrate contribution to the Paleocene-Eocene carbon cycle perturbation from coupled thermohydraulic simulations. Geophys. Res. Lett. 2016, 43, 8637–8644. [Google Scholar] [CrossRef]
- Kvamme, B.; Clarke, M. Hydrate Phase Transition Kinetic Modeling for Nature and Industry—Where Are We and Where Do We Go? Energies 2021, 14, 4149. [Google Scholar] [CrossRef]
- De La Fuente, M.; Vaunat, J.; Marín-Moreno, H. Thermo-Hydro-Mechanical Coupled modelling of Methane Hydrate-Bearing Sediments: Formulation and Application. Energies 2019, 12, 2178. [Google Scholar] [CrossRef]
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; Chemical Industries Series; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Reagan, M.; Moridis, G.J.; Collett, T.; Boswell, R.; Kurihara, M.; Reagan, M.T.; Koh, C.; Sloan, E.D. Toward production from gas hydrates: Current status, assessment of resources, and simulation-based evaluation of technology and potential. SPE Res. Eval. Eng. 2009, 12, 745–771. [Google Scholar]
- Xu, W.; Germanovich, L.N. Excess pore pressure resulting from methane hydrate dissociation in marine sediments: A theoretical approach. J. Geophys. Res. 2006, 111, B01104. [Google Scholar] [CrossRef]
- Daigle, H.; Dugan, B. Effects of multiphase methane supply on hydrate accumulation and fracture generation. Geophys. Res. Lett. 2010, 37, L20301. [Google Scholar] [CrossRef]
- Riboulot, V.; Sultan, N.; Imbert, P.; Ker, D. Initiation of gas-hydrate pockmark in deep-water Nigeria: Geo-mechanical analysis and modelling. Earth Planet. Sci. Lett. 2016, 434, 252–263. [Google Scholar] [CrossRef]
- Yang, J.; Xu, Q.; Liu, Z.; Shi, L. Pore-scale study of the multiphase methane hydrate dissociation dynamics and mechanisms in the sediment. Chem. Eng. J. 2021, 430, 132786. [Google Scholar] [CrossRef]
- Fick, A. Ueber diffusion. Ann. Phys. Chem. 1855, 94, 59–86. [Google Scholar] [CrossRef]
- Darcy, H. Les Fontaines Publiques De La Ville De Dijon; Libraire des Corps Imperiaux des Ponts et Chaussees et des Mines: Paris, France, 1856. [Google Scholar]
- Gupta, S.; Wohlmuth, B.; Haeckel, M. An All-At-Once Newton Strategy for Marine Methane Hydrate Reservoir Models. Energies 2020, 13, 503. [Google Scholar] [CrossRef]
- You, K.; Flemings, P.B. Methane hydrate formation and evolution during sedimentation. J. Geophys. Res. Solid Earth 2021, 126, e2020JB021235. [Google Scholar] [CrossRef]
- Mogollón, J.M.; L’Heureux, I.; Dale, A.W.; Regnier, P. Methane gas-phase dynamics in marine sediments: A model study. Am. J. Sci. 2009, 309, 189–220. [Google Scholar] [CrossRef]
- Waite, W.F.; Santamarina, J.C.; Cortes, D.D.; Dugan, B.; Espinoza, D.N.; Germaine, J.; Jang, J.; Jung, J.W.; Kneafsey, T.J.; Shin, H.; et al. Physical properties of hydrate-bearing sediments. Rev. Geophys. 2009, 47, RG4003. [Google Scholar] [CrossRef]
- Reeburgh, W.S. Global methane biogeochemistry. In Treatise on Geochemistry; Keeling, R.F., Ed.; Elsevier: Oxford, UK, 2003; pp. 65–89. [Google Scholar]
- Hinrichs, K.U.; Hayes, J.; Sylva, S.; Brewer, P.G.; DeLong, E.F. Methane-consuming archaebacteria in marine sediments. Nature 1999, 398, 802–805. [Google Scholar] [CrossRef]
- Boetius, A.; Ravenschlag, K.; Schubert, C.; Rickert, D.; Widdel, F.; Gieseke, A.; Amann, R.; Jørgensen, B.B.; Witte, U.; Pfannkuche, O. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 2000, 407, 623–626. [Google Scholar] [CrossRef]
- Valentine, D.L.; Blanton, D.C.; Reeburgh, W.S.; Kastner, M. Water column methane oxidation adjacent to an area of active hydrate dissociation, Eel River Basin. Geochim. Cosmochim. Acta 2001, 65, 2633–2640. [Google Scholar] [CrossRef]
- Niemann, H.; Lösekann, T.; de Beer, D.; Elvert, M.; Nadalig, T.; Knitel, K.; Amann, R.; Sauter, E.J.; Schlüter, M.; Klages, M.; et al. Novel microbial communities of the Haakon Mosby mud volcano and their role as a methane sink. Nature 2006, 443, 854–858. [Google Scholar] [CrossRef]
- Sommer, S.; Pfannkuche, O.; Linke, P.; Luff, R.; Greinert, J.; Drews, M.; Gubsch, S.; Pieper, M.; Viergutz, P.T. Efficiency of the benthic filter: Biological control of the emission of dissolved methane from sediments containing shallow gas hydrates at Hydrate Ridge. Glob. Biogeochem. Cycles 2006, 20, GB2019. [Google Scholar] [CrossRef]
- Raghoebarsing, A.A.; Pol, A.; van de Pas-Schoonen, K.T.; Smolders, A.J.P.; Ettwig, K.F.; Rijpstra, W.I.C.; Schouten, S.; Sinninghe Damsté, J.S.; Op den Camp, H.J.M.; Jetten, M.S.M.; et al. A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 2006, 44, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Beal, E.J.; House, C.H.; Orphan, V.J. Manganese- and iron-dependent marine methane oxidation. Science 2006, 325, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Winkel, M.; Mitzscherling, J.; Overduin, P.P.; Horn, F.; Winterfeld, M.; Rijkers, R.; Grigoriev, M.N.; Knoblauch, C.; Mangelsdorf, K.; Wagner, D.; et al. Anaerobic methanotrophic communities thrive in deep submarine permafrost. Sci. Rep. 2018, 8, 1291. [Google Scholar] [CrossRef] [PubMed]
- Reeburgh, W.S. Methane consumption in Cariaco Trench waters and sediments. Earth Planet Sci. Lett. 1976, 28, 337–344. [Google Scholar] [CrossRef]
- Malinverno, A.; Pohlman, J.W. modelling sulfate reduction in methane hydrate-bearing continental margin sediments: Does a sulfate-methane transition require anaerobic oxidation of methane? Geochem. Geophys. Geosyst. 2011, 12, Q07006. [Google Scholar] [CrossRef]
- Soetaert, K.; Hofmann, A.F.; Middelburg, J.J.; Meysman, F.J.R.; Greenwood, J. The effect of biogeochemical processes on pH. Mar. Chem. 2007, 105, 30–51. [Google Scholar] [CrossRef]
- Rossel Cartes, P. Microbial Communities Performing Anaerobic Oxidation of Methane: Diversity of Lipid Signatures and Habitats. Ph.D. Thesis, University of Bremen, Bremen, Germany, 2009. [Google Scholar]
- Klasek, S.A.; Hong, W.L.; Torres, M.E.; Ross, S.; Hostetler, K.; Portnov, A.; Gründger, F.; Colwell, F.S. Distinct methane-dependent biogeochemical states in Arctic seafloor gas hydrate mounds. Nat. Commun. 2021, 12, 6296. [Google Scholar] [CrossRef]
- Joye, S.B.; Boetius, A.; Orcutt, B.N.; Montoya, J.P.; Schulz, H.N.; Erickson, M.J.; Lugo, S.K. The anaerobic oxidation of methane and sulfate reduction in sediments from Gulf of Mexico cold seeps. Chem. Geol. 2004, 205, 219–238. [Google Scholar] [CrossRef]
- Orcutt, B.; Boetius, A.; Elvert, M.; Samarkin, V.; Joye, S.B. Molecular biogeochemistry of sulfate reduction, methanogenesis and the anaerobic oxidation of methane at Gulf of Mexico cold seeps. Geochim. Cosmochim. Acta 2005, 69, 4267–4281. [Google Scholar] [CrossRef]
- Jordan, S.F.A.; Treude, T.; Leifer, I.; Janßen, R.; Werner, J.; Schulz-Vogt, H.; Schmale, O. Bubble-mediated transport of benthic microorganisms into the water column: Identification of methanotrophs and implication of seepage intensity on transport efficiency. Sci. Rep. 2020, 10, 4682. [Google Scholar] [CrossRef]
- Römer, M.; Wenau, S.; Mau, S.; Veloso, M.; Greinert, J.; Schlüter, M.; Bohrmann, G. Assessing marine gas emission activity and contribution to the atmospheric methane inventory: A multidisciplinary approach from the Dutch Dogger Bank seep area (North Sea). Geochem. Geophys. Geosyst. 2017, 18, 2617–2633. [Google Scholar] [CrossRef]
- Luff, R.; Wallmann, K. Fluid flow, methane fluxes, carbonate precipitation and biogeochemical turnover in gas hydrate-bearing sediments at Hydrate Ridge, Cascadia Margin: Numerical modelling and mass balances. Geochim. Cosmochim. Acta 2003, 67, 3403–3421. [Google Scholar] [CrossRef]
- Haeckel, M.; Suess, E.; Wallmann, K.; Rickert, D. Rising methane gas bubbles form massive hydrate layers at the seafloors. Geochim. Cosmochim. Acta 2004, 68, 4335–4345. [Google Scholar] [CrossRef]
- Haeckel, M.; Boudreau, B.P.; Wallmannm, K. Bubble-induced porewater mixing: A 3-D model for deep porewater irrigation. Geochim. Cosmochim. Acta 2007, 71, 5135–5154. [Google Scholar] [CrossRef]
- Bangs, N.L.B.; Hornbach, M.J.; Berndt, C. The mechanics of intermittent methane venting at South Hydrate Ridge inferred from 4D seismic surveying. Earth Planet. Sci. Lett. 2011, 310, 105–112. [Google Scholar] [CrossRef]
- Daigle, H.; Bangs, N.L.; Dugan, B. Transient hydraulic fracturing and gas release in methane hydrate settings: A case study from southern Hydrate Ridge. Geochem. Geophys. Geosyst. 2011, 12, Q12022. [Google Scholar] [CrossRef]
- Boudreau, B.P. On the equivalence of non-local and radial-diffusion models for pore-water irrigation. J. Mar. Res. 1984, 42, 731–735. [Google Scholar] [CrossRef]
- Dale, A.W.; Regnier, P.; Knab, N.J.; Jørgensen, B.B.; Van Cappellen, P. Anaerobic oxidation of methane (AOM) in marine sediments from the Skagerrak (Denmark): II. Reaction-transport modelling. Geochim. Cosmochim. Acta 2008, 72, 2880–2894. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Findlay, A.J.; Pellerin, A. The Biogeochemical Sulfur Cycle of Marine Sediments. Front. Microbiol. 2019, 10, 849. [Google Scholar] [CrossRef]
- Fischer, D.; Sahling, H.; Nöthen, K.; Bohrmann, G.; Zabel, M.; Kasten, S. Interaction between hydrocarbon seepage, chemosynthetic communities, and bottom water redox at cold seeps of the Makran accretionary prism: Insights from habitat-specific pore water sampling and modelling. Biogeosciences 2012, 9, 2013–2031. [Google Scholar] [CrossRef]
- Cordes, E.E.; Arthur, M.A.; Shea, K.; Arvidson, R.S.; Fisher, C.R. Modelling the mutualistic interactions between tubeworms and microbial consortia. PLoS Biol. 2005, 3, e77. [Google Scholar] [CrossRef] [PubMed]
- Reagan, T.A.M.; Jones, P.W. Interrelation of Global Climate and the Response of Oceanic Hydrate Accumulations: Final Report. NETL. 2013. Available online: https://www.netl.doe.gov/node/7135 (accessed on 3 November 2021).
- Malone, M.J.; Claypool, G.; Martin, J.B.; Dickens, G.R. Variable methane fluxes in shallow marine systems over geologic time: The composition and origin of pore waters and authigenic carbonates on the New Jersey shelf. Mar. Geol. 2002, 189, 175–196. [Google Scholar] [CrossRef]
- Contreras, S.; Meister, P.; Liu, B.; Prieto-Mollar, X.; Hinrichs, K.-U.; Khalili, A.; Ferdelman, T.G.; Kuypers, M.M.; Jørgensen, B.B. Cyclic 100-ka (glacial-interglacial) migration of subseafloor redox zonation on the Peruvian shelf. Proc. Natl. Acad. Sci. USA 2013, 110, 18098–18103. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, B.P.; Luo, Y.; Meysman, F.J.R.; Middelburg, J.J.; Dickens, G.R. Gas hydrate dissociation prolongs acidification of the Anthropocene oceans. Geophys. Res. Lett. 2015, 42, 9337A–9344A. [Google Scholar] [CrossRef]
- Aloisi, G.; Wallmann, K.; Drews, M.; Bohrmann, G. Evidence for the submarine weathering of silicate minerals in Black Sea sediments: Possible implications for the marine Li and B cycles. Geochem. Geophys. Geosyst. 2004, 5, Q04007. [Google Scholar] [CrossRef]
- Chatterjee, S.; Dickens, G.R.; Bhatnagar, G.; Chapman, W.G.; Dugan, B.; Snyder, G.T.; Hirasaki, G.J. Pore water sulfate, alkalinity, and carbon isotope profiles in shallow sediment above marine gas hydrate systems: A numerical modelling perspective. J. Geophys. Res. Solid Earth 2011, 116, B09103. [Google Scholar] [CrossRef]
- Yoshinaga, M.Y.; Holler, T.; Goldhammer, T.; Wegener, G.; Pohlman, J.W.; Brunner, B.; Kuypers, M.; Hinrichs, K.; Elvert, M. Carbon isotope equilibration during sulphate-limited anaerobic oxidation of methane. Nat. Geosci. 2014, 7, 190. [Google Scholar] [CrossRef]
- Munhoven, G. Mathematics of the total alkalinity–pH equation–Pathway to robust and universal solution algorithms: The SolveSAPHE package v1.0.1. Geosci. Model Dev. 2013, 6, 1367–1388. [Google Scholar] [CrossRef]
- Krumins, V.; Gehlen, M.; Arndt, S.; Van Cappellen, P.; Regnier, P. Dissolved inorganic carbon and alkalinity fluxes from coastal marine sediments: Model estimates for different shelf environments and sensitivity to global change. Biogeosciences 2013, 10, 371–398. [Google Scholar] [CrossRef]
- Orphan, V.J.; Ussler, W.; Naehr, T.H.; House, C.H.; Hinrichs, K.; Paull, C.K. Geological, geochemical, and microbiological heterogeneity of the seafloor around methane vents in the Eel River Basin, offshore California. Chem. Geol. 2004, 205, 265–289. [Google Scholar] [CrossRef]
- Aloisi, G.; Bouloubassi, I.; Heijs, S.K.; Pancost, R.D.; Pierre, C.; Sinninghe, J.S.; Gottschal, D.C.; Forney, L.J.; Rouchye, J. CH4-consuming microorganisms and the formation of carbonate crusts at cold seeps. Earth Planet. Sci. Lett. 2002, 203, 195–203. [Google Scholar]
- Portnov, A.; Vadakkepuliyambatta, S.; Mienert, J.; Hubbard, A. Ice-sheet-driven methane storage and release in the Arctic. Nat. Commun. 2016, 7, 10314. [Google Scholar] [CrossRef] [PubMed]
- Prouty, N.G.; Sahy, D.; Ruppel, C.D.; Roark, E.B.; Condon, D.; Brooke, S.; Ross, S.W.; Demopoulos, A.W.J. Insights into methane dynamics from analysis of authigenic carbonates and chemosynthetic mussels at newly-discovered Atlantic Margin seeps. Earth Planet. Sci. Lett. 2016, 499, 332–344. [Google Scholar] [CrossRef]
- Luff, R.; Wallmann, K.; Aloisi, G. Numerical modelling of carbonate crust formation at cold vent sites: Significance for fluid and methane budgets and chemosynthetic biological communities. Earth Planet. Sci. Lett. 2004, 221, 337–353. [Google Scholar] [CrossRef]
- Karaca, D.; Hensen, C.; Wallmann, K. Controls on authigenic carbonate precipitation at cold seeps along the convergent margin off Costa Rica. Geochem. Geophys. Geosyst. 2010, 11, Q08S27. [Google Scholar] [CrossRef]
- Himmler, T.; Brinkmann, F.; Bohrmann, G.; Peckmann, J. Corrosion patterns of seep-carbonates from the eastern Mediterranean Sea. Terra Nova 2011, 23, 206–212. [Google Scholar] [CrossRef]
- Stachnik, L.; Majchrowska, E.; Yde, J.C.; Nawrot, A.P.; Cichała-Kamrowska, K.; Ignatiuk, D.; Piechota, A. Chemical denudation and the role of sulfide oxidation at Werenskioldbreen, Svalbard. J. Hydrol. 2016, 538, 177–193. [Google Scholar] [CrossRef]
- Torres, M.E.; Hong, W.L.; Solomon, E.A.; Milliken, K.; Kim, J.; Sample, J.C.; Teichert, B.M.A.; Wallmann, K. Silicate weathering in anoxic marine sediment as a requirement for authigenic carbonate burial. Earth-Sci. Rev. 2020, 200, 102960. [Google Scholar] [CrossRef]
- Caesar, K.H.; Kyle, J.R.; Lyons, T.W.; Tripati, A.; Loyd, S.J. Carbonate formation in salt dome cap rocks by microbial anaerobic oxidation of methane. Nat Commun 2019, 10, 808. [Google Scholar] [CrossRef]
- Singurindy, O.; Berkowitz, B. Flow, dissolution, and precipitation in dolomite. Water Resour. Res. 2003, 39, 1143. [Google Scholar] [CrossRef]
- Luff, R.; Greinert, J.; Wallmann, K.; Klaucke, I.; Suess, E. Simulation of long-term feed-backs from authigenic carbonate crust formation at cold vent sites. Chem. Geo. 2004, 216, 157–174. [Google Scholar] [CrossRef]
- Marcon, Y.; Ondreás, H.; Sahling, H.; Bohrmann, G.; Olu, K. Fluid flow regimes and growth of a giant pockmark. Geology 2014, 42, 63–66. [Google Scholar] [CrossRef]
- Garcia-Tigreros, F.; Leonte, M.; Ruppel, C.D.; Ruiz-Angulo, A.; Joung, D.J.; Young, B.; Kessler, J.D. Estimating the impact of seep methane oxidation on ocean pH and dissolved inorganic radiocarbon along the U.S. Mid-Atlantic Bight. J. Geophys. Res. Biogeosci. 2021, 126, e2019JG005621. [Google Scholar] [CrossRef]
- Giustiniani, M.; Tinivella, U.; Jakobsson, M.; Rebesco, M. Arctic Ocean Gas Hydrate Stability in a Changing Climate. J. Geo. Res. 2013, 1687–8833. [Google Scholar] [CrossRef]
- Crémiere, A.; Lepland, A.; Chand, S.; Sahy, D.; Condon, D.J.; Noble, S.R.; Martma, T.; Thorsnes, T.; Sauer, S.; Brunstad, H. Timescales of methane seepage on the Norwegian margin following collapse of the Scandinavian Ice Sheet. Nat. Commun. 2016, 7, 11509. [Google Scholar] [CrossRef]
- Xu, W.; Lowell, R.P.; Peltzer, E.T. Effect of seafloor temperature and pressure variations on methane flux from a gas hydrate layer: Comparison between current and late Paleocene climate conditions. J. Geophys. Res. 2001, 106, 26413–26423. [Google Scholar] [CrossRef]
- Reagan, T.A.M.; Moridis, G.J.; Elliott, S.M.; Maltrud, M. Contribution of oceanic gas hydrate dissociation to the formation of Arctic Ocean methane plumes. J. Geophys. Res. 2011, 116, C09014. [Google Scholar] [CrossRef]
- Darnell, K.N.; Flemings, P.B. Transient seafloor venting on continental slopes from warming-induced methane hydrate dissociation. Geophys. Res. Lett. 2015, 42, 10765–10772. [Google Scholar] [CrossRef]
- Hong, W.L.; Torres, M.; Carroll, J.; Crémière, A.; Panieri, G.; Yao, H.; Serov, P. Seepage from an arctic shallow marine gas hydrate reservoir is insensitive to momentary ocean warming. Nat. Commun. 2017, 8, 15745. [Google Scholar] [CrossRef]
- Holder, G.D.; Angert, P.F.; John, V.T.; Yen, S. A thermodynamic evaluation of thermal recovery of gas from hydrates in the Earth. J. Pet. Geol. 1982, 34, 1127–1132. [Google Scholar]
- Xu, W.; Ruppel, C.D. Predicting the occurrence, distribution, and evolution of methane gas hydrate in porous marine sediments. J. Geophys. Res. 1999, 104, 5081–5095. [Google Scholar] [CrossRef]
- Moridis, G.J.; Kowalsky, M.B.; Pruess, K. TOUGH-Fx/HYDRATE v1.0 User’s Manual; Report LBNL-58950; Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2005. [Google Scholar]
- Luff, R.; Wallmann, K.; Grandel, S.; Schluter, M. Numerical modelling of benthic processes in the deep Arabian Sea. Deep. Sea Res. II 2000, 47, 3039–3072. [Google Scholar] [CrossRef]
- Boudreau, B.P. A method-of-lines code for carbon and nutrient diagenesis in aquatic sediments. Comput. Geosci. 1996, 22, 479–496. [Google Scholar] [CrossRef]
- Davie, M.K.; Buffett, B.A. A numerical model for the formation of gas hydrate below the seafloor. J. Geophys. Res. 2001, 106, 497–514. [Google Scholar] [CrossRef]
- Moridis, G.J.; Kowalsky, M.B.; Pruess, K. TOUGH+HYDRATE v1.0 User’s Manual: A Code for the Simulation of System Behaviour in Hydrate-Bearing Geological Medial; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2008. [Google Scholar]
- Tishchenko, P.; Hensen, C.; Wallmann, K.; Wong, C.S. Calculation of the stability and solubility of methane hydrate in seawater. Chem. Geol. 2005, 219, 37–52. [Google Scholar] [CrossRef]
- Liu, X.L.; Flemings, P.B. Dynamic multiphase flow model of hydrate formation in marine sediments. J. Geophys. Res.-Solid Earth 2007, 112, B03101. [Google Scholar] [CrossRef]
- Wallmann, K.; Pinero, E.; Burwicz, E.; Haeckel, M.; Hensen, C.; Dale, A.; Rupke, L. The Global Inventory of Methane Hydrate in Marine Sediments: A Theoretical Approach. Energies 2012, 5, 2449–2498. [Google Scholar] [CrossRef]
- Moridis, G.J.; Kowalsky, M.B.; Pruess, K. TOUGH1HYDRATE v1.2 User’s Manual: A Code for the Simulation of System Behavior Inhydrate-Bearing Geological Media; Per. LBNL-0149E; Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2012. [Google Scholar]
- Moridis, G.J. User’s Manual for the Hydrate v1.5 Option of TOUGH+V1.5: A Code for the Simulation of System Behaviour in Hydrate-Bearing Geologic Media; Earth Sciences Division, Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2014. [Google Scholar]
- White, M.D.; Kneafsey, T.J.; Seol, Y.; Waite, W.F.; Uchida, S.; Lin, J.S.; Myshakin, E.M.; Gai, X.; Gupta, S.; Reagan, T.A.M.; et al. An international code comparison study on coupled thermal, hydrologic and geomechanical processes of natural gas hydrate-bearing sediments. Mar. Pet. Geol. 2020, 120, 104566. [Google Scholar] [CrossRef]
- Soetaert, K.; Herman, P.M.J.; Middelburg, J.J. A model of early diagenetic processes from the shelf to abyssal depths. Geochim. Cosmochim. Acta 1996, 60, 1019–1040. [Google Scholar] [CrossRef]
- Soetaert, K.; Herman, P.M.J.; Middelburg, J.J. Dynamic response of deep-sea sediments to seasonal variations: A model. Limnol. Oceanogr. 1996, 41, 1651–1668. [Google Scholar] [CrossRef]
- Garg, S.K.; Pritchett, J.W.; Katoh, A.; Baba, K.; Fujii, T. A mathematical model for the formation and dissociation of methane hydrates in the marine environment. J. Geophys. Res. 2008, 113, B01201. [Google Scholar] [CrossRef]
- Wallmann, K.; Aloisi, G.; Haeckel, M.; Obzhirov, A.; Pavlova, G.; Tishchenko, P. Kinetics of organic matter degradation, microbial methane generation, and gas hydrate formation in anoxic marine sediments. Geochim. Cosmochim. Acta 2006, 15, 3905–3927. [Google Scholar] [CrossRef]
- Burwicz, E.; Rupke, L. Thermal State of the Blake Ridge Gas Hydrate Stability Zone (GHSZ)—Insights on Gas Hydrate Dynamics from a New Multi-Phase Numerical Model. Energies 2019, 12, 3403. [Google Scholar] [CrossRef]
- Tian, H.; Yu, C.; Xu, T.; Liu, C.; Jia, W.; Li, Y.; Shang, S. Combining reactive transport modelling with geochemical observations to estimate the natural gas hydrate accumulation. Appl. Energy 2020, 275, 115362. [Google Scholar] [CrossRef]
- Zeebe, R.E. Modelling CO2 chemistry, δ13C, and oxidation of organic carbon and methane in sediment pore-water: Implications for paleo-proxies in benthic foraminifera. Geochim. Cosmochim. Acta 2007, 71, 3238–3256. [Google Scholar] [CrossRef]
- Berner, R.A. Early Diagenesis: A Theoretical Approach; Princeton University Press: Princeton, NJ, USA, 1980. [Google Scholar]
- Boudreau, B.P. Diagenetic Models and Their Implementation; Springer: Berlin/Heidelberg, Germany, 1997; Volume 505. [Google Scholar]
- Treude, T.; Boetius, A.; Knittel, K.; Wallmann, K.; Jørgensen, B.B. Anaerobic oxidation of methane above gas hydrates at Hydrate Ridge, NE Pacific Ocean. Mar. Ecol. Prog. Ser. 2003, 264, 1–14. [Google Scholar] [CrossRef]
- Caldwell, S.L.; Laidler, J.R.; Brewer, E.A.; Eberly, J.O.; Sandborgh, S.C.; Colwell, F.S. Anaerobic oxidation of methane: Mechanisms, bioenergetics, and the ecology of associated microorganisms. Environ. Sci. Technol. 2008, 42, 6791–6799. [Google Scholar] [CrossRef] [PubMed]
- Nauhaus, K.; Albrecht, M.; Elvert, M.; Boetius, A.; Widdel, F. In vitro cell growth of marine archaeal–bacterial consortia during anaerobic oxidation of methane with sulfate. Environ. Microbiol. 2007, 9, 187–196. [Google Scholar] [CrossRef]
- Girguis, P.R.; Cozen, A.E.; DeLong, E.F. Growth and population dynamics of anaerobic methane-oxidizing archaea and sulfate-reducing bacteria in a continuous-flow bioreactor. Appl. Environ. Microbiol. 2005, 71, 3725–3733. [Google Scholar] [CrossRef]
- Regnier, P.; Dale, A.W.; Pallud, C.; van Lith, Y.; Bonneville, S.; Hyacinthe, C.; Thullner, M.; Laverman, A.; Van Cappellen, P. Incorporating geo-microbial processes in subsurface reactive transport models. In Reactive Transport in Soil and Groundwater: Processes and Models; Nuetzmann, G., Viotti, P., Agaard, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 107–126. [Google Scholar]
- Jin, Q.; Bethke, C.M. Predicting the rate of microbial respiration in geochemical environments. Geochim. Cosmochim. Acta 2005, 69, 1133–1143. [Google Scholar] [CrossRef]
- Cavicchioli, R.; Ripple, W.J.; Timmis, K.N.; Azam, F.; Bakken, L.R.; Baylis, M.; Behrenfeld, M.J.; Boetius, A.; Boyd, P.W.; Classen, A.T.; et al. Scientists’ warning to humanity: Microorganisms and climate change. Nat. Rev. Microbiol. 2019, 17, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, G.; Chapman, W.G.; Dickens, G.R.; Dugan, B.; Hirasaki, G.J. Generalization of gas hydrate distribution and saturation in marine sediments by scaling of thermodynamic and transport processes. Am. J. Sci. 2007, 307, 861–900. [Google Scholar] [CrossRef]
- Bhatnagar, G.; Chatterjee, S.; Chapman, W.G.; Dugan, B.; Dickens, G.R.; Hirasaki, G.J. Analytical theory relating the depth of the sulfate-methane transition to gas hydrate distribution and saturation. Geochem. Geophys. Geosyst. 2011, 12, Q03003. [Google Scholar] [CrossRef]
- Xu, T.; Spycher, N.; Sonnenthal, E.; Zheng, L.; Pruess, K. TOUGHREACT User’s Guide: A Simulation Program for Non-Isothermal Multiphase Reactive Transport in Variably Saturated Geologic Media; Version 2.0; Earth Sciences Division; National Laboratory: Berkeley, CA, USA, 2012. [Google Scholar]
- Regnier, P.; O’kane, J.; Steefel, C.; Vanderborght, J.P. modelling complex multi-component reactive-transport systems: Towards a simulation environment based on the concept of a Knowledge Base. Appl. Math. Model. 2002, 26, 913–927. [Google Scholar] [CrossRef]
- Aguilera, D.; Jourabchi, P.; Spiteri, C.; Regnier, P. A knowledge-based reactive transport approach for the simulation of biogeochemical dynamics in Earth systems. Geochem. Geophys. Geosyst. 2005, 6, Q07012. [Google Scholar] [CrossRef]
- Centler, F.; Shao, H.; De Biase, C.; Park, C.-H.; Regnier, P.; Kolditz, O.; Thullner, M. GeoSysBRNS—A flexible multidimensional reactive transport model for simulating biogeochemical subsurface processes. Comput. Geosci. 2010, 36, 397–405. [Google Scholar] [CrossRef]
- Berndt, C.; Feseker, T.; Treude, T.; Krastel, S.; Liebetrau, V.; Niemann, H.; Bertics, V.J.; Dumke, I.; Dünnbier, K.; Ferré, B.; et al. Temporal constraints on hydrate-controlled methane seepage off Svalbard. Science 2014, 343, 284–287. [Google Scholar] [CrossRef]
- Dean, J.F.; Middelburg, J.J.; Röckmann, T.; Aerts, R.; Blauw, L.G.; Egger, M.; Jetten, M.S.M.; de Jong, A.E.E.; Meisel, O.H.; Rasigraf, O.; et al. Methane feedbacks to the global climate system in a warmer world. Rev. Geophys. 2018, 13, 841–863. [Google Scholar] [CrossRef]
- Fu, X.; Waite, W.F.; Ruppel, C.D. Hydrate formation on marine seep bubbles and the implications for water column methane dissolution. J. Geophys. Res. Ocean. 2021, 126, e2021JC017363. [Google Scholar] [CrossRef]
- Brenner, H.; Braeckman, U.; Le Guitton, M.; Meysman, F.J. Fate of rising methane bubbles in stratified waters: How much methane reaches the atmosphere? Biogeosciences 2016, 111, C09007. [Google Scholar]
- Kim, B.; Zhang, Y.G. Methane hydrate dissociation across the Oligocene–Miocene boundary. Nat. Geosci. 2022, 15, 203–209. [Google Scholar] [CrossRef]
- Heavens, N.G.; Ward, D.S.; Natalie, M.M. Studying and Projecting Climate Change with Earth System Models. Nat. Educ. Knowl. 2013, 4, 4. [Google Scholar]
- O’Connor, F.M.; Boucher, O.; Gedney, N.; Jones, C.D.; Folberth, G.A.; Coppell, R.; Friedlingstein, P.; Collins, W.J.; Chappellaz, J.; Ridley, J.; et al. Possible role of wetlands, permafrost, and methane hydrates in the methane cycle under future climate change: A review. Rev. Geophys. 2010, 48, RG4005. [Google Scholar] [CrossRef]
- Moore, J.K.; Doney, S.; Lindsay, K. Upper ocean ecosystem dynamics and iron cycling in a global 3D model. Glob. Biogeochem. Cycles. 2004, 18, GB4028. [Google Scholar] [CrossRef]
- Elliott, S. Dependence of DMS global sea-air flux distribution on transfer velocity and concentration field type. J. Geophys. Res. 2009, 114, G02001. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sulfate Reduction | |
| Catabolic reaction: | |
| Biomass growth: | |
| Methanogenesis | |
| Catabolic reaction: | |
| Biomass growth: | |
| AOM | |
| Catabolic reaction: | |
| Biomass growth: | |
| Secondary Redox Reactions | |
| R1: Nitrification | |
| R2: Sulfide Oxidation | |
| R3: AeOM | |
| R4: AOM | |
| Precipitation Reactions | |
| R5: Carbonate precipitation | |
| R6: Sulfide precipitation | |
| Numerical Study | Model Reference | Scientific Question 1 | Scientific Question 2 | Scientific Question 3 | ||||||||||||||
| (1,2) Estimation of the Hydrate Inventory | (3,4) Climate-Driven Hydrate Destabilisation/Re-Formation | (5) Methane by-Passing AOM | (6) AOM Rate | (8, 10) Benthic Environmental Impact | ||||||||||||||
|
(1C) Prescribed Methane Supply and Initial Hydrate Saturation |
(1B) Diagenetic Approach for Methane Sources |
Changes in GHSZ Thickness |
Hydrate Thermodynamics |
Gas Phase Escapes the AOM |
Hydraulic Fractures or Permeable Paths | Bio-Mixing |
(6A) Constant
Rate |
(6B) Non- Biochemical Sink |
(6C) Bimolecular |
(6D) Monod Rate | (6E) Kinetic & Bio-Energetic limitations | DIC | TA | pH | ||||
| Xu et al. [139] | Xu and Ruppel [144] | x | x | x | ||||||||||||||
| Reagan and Moridis [47] | TOUGH+Hydrate [145] C.CANDI [146,147] | x | x | x | x | x | x | |||||||||||
| Biastoch [49] | Biastoch [49] | x | x | |||||||||||||||
| Archer et al. [29] | Davie and Buffet [148] | x | x | x | x | |||||||||||||
| Reagan et al. [140] | TOUGH+Hydrate [149] | x | x | |||||||||||||||
| Hunter et al. [51] | Tishchenko et al. [150] | x | x | x | ||||||||||||||
| Thatcher et al. [65] | TOUGH+Hydrate [149]. | x | x | |||||||||||||||
| Darnell and Flemings [141] | Liu and Flemings [151] | x | x | |||||||||||||||
| Kretschmer et al. [20] | Wallmann et al. [152] | x | x | |||||||||||||||
| Marin-Moreno et al. [31] | TOUGH+Hydrate [153] | x | x | |||||||||||||||
| Mestdagh et al. [12] | Xu and Ruppel [144] | x | x | |||||||||||||||
| Wallmann et al. [68] | Wallmann et al. [68] | x | x | x | x | |||||||||||||
| Stranne et al. [46] | TOUGH+Hydrate -GeoMech [44,154] | x | x | x | x | |||||||||||||
| Braga et al. [22] | TOUGH+Hydrate [154] | x | x | |||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De La Fuente, M.; Arndt, S.; Marín-Moreno, H.; Minshull, T.A. Assessing the Benthic Response to Climate-Driven Methane Hydrate Destabilisation: State of the Art and Future Modelling Perspectives. Energies 2022, 15, 3307. https://doi.org/10.3390/en15093307
De La Fuente M, Arndt S, Marín-Moreno H, Minshull TA. Assessing the Benthic Response to Climate-Driven Methane Hydrate Destabilisation: State of the Art and Future Modelling Perspectives. Energies. 2022; 15(9):3307. https://doi.org/10.3390/en15093307
Chicago/Turabian StyleDe La Fuente, Maria, Sandra Arndt, Héctor Marín-Moreno, and Tim A. Minshull. 2022. "Assessing the Benthic Response to Climate-Driven Methane Hydrate Destabilisation: State of the Art and Future Modelling Perspectives" Energies 15, no. 9: 3307. https://doi.org/10.3390/en15093307
APA StyleDe La Fuente, M., Arndt, S., Marín-Moreno, H., & Minshull, T. A. (2022). Assessing the Benthic Response to Climate-Driven Methane Hydrate Destabilisation: State of the Art and Future Modelling Perspectives. Energies, 15(9), 3307. https://doi.org/10.3390/en15093307