
An Integrated Experimental and Computational Platform to Explore Gas Hydrate Promotion, Inhibition, Rheology, and Mechanical Properties at McGill University: A Review



Abstract
:1. Introduction
2. Tools and Methods
2.1. Experimental
2.1.1. Gas Hydrate Kinetics
2.1.2. Nanoparticles Additives
2.1.3. Polymer Additives
2.1.4. Rheometry
2.2. Computational Methods
2.2.1. Molecular Dynamics
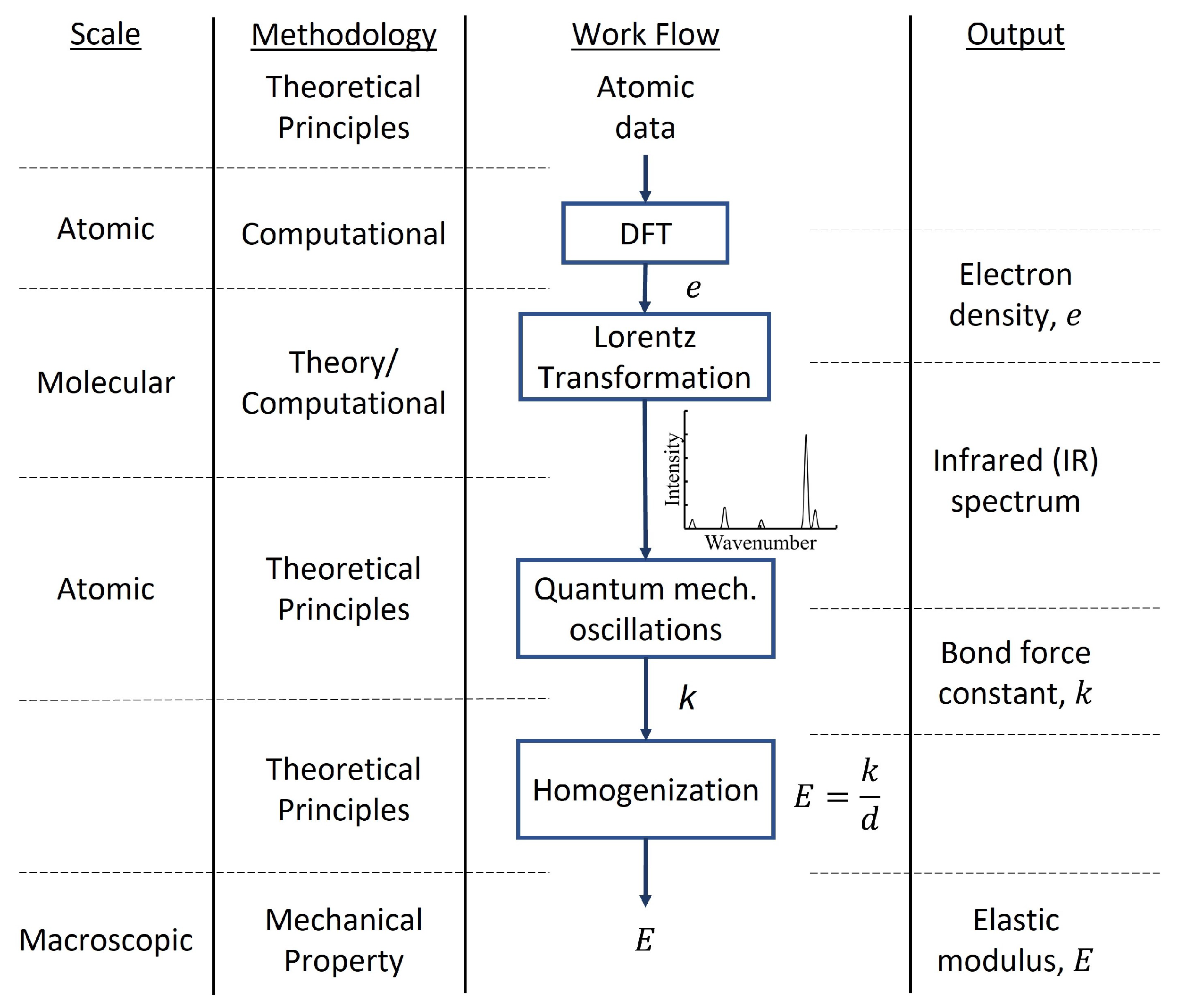
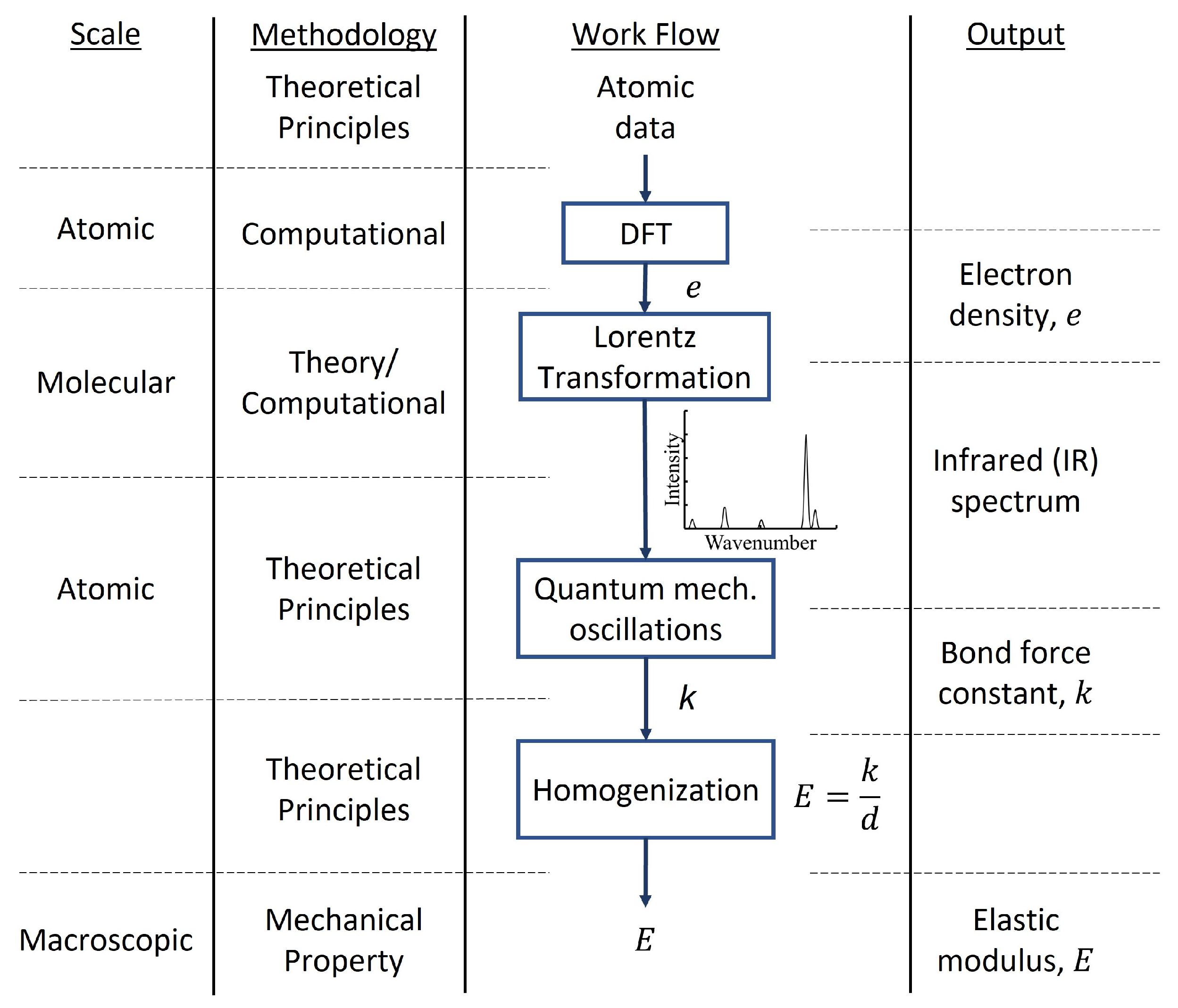
2.2.2. Density Functional Theory
3. Effects of Additives on Hydrate Growth
3.1. Multi-Walled Carbon Nanotubes
3.2. Graphene Nanoflakes
3.3. Polymer Inhibitors
4. Interfacial Effects
5. Mechanical Properties
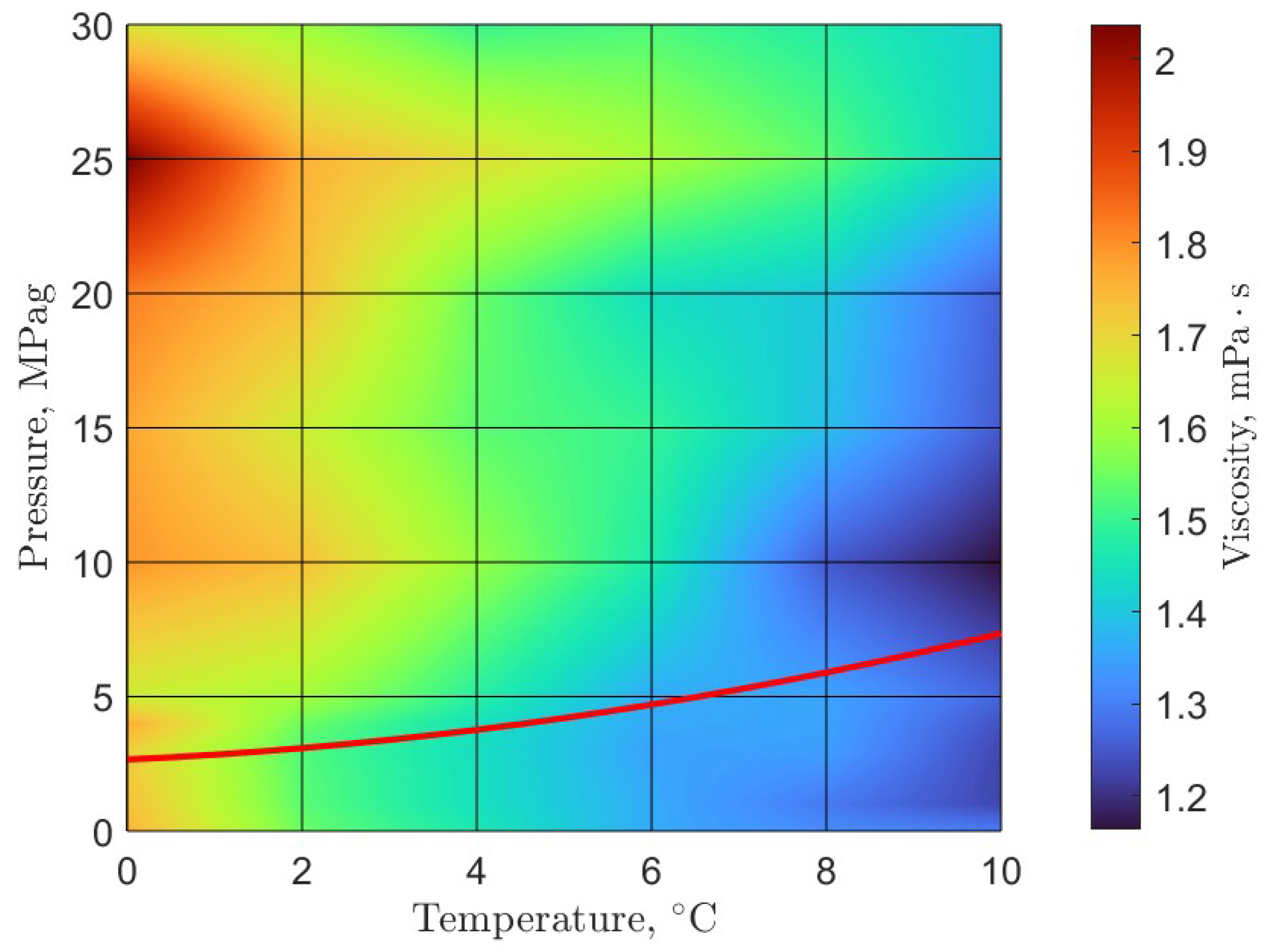
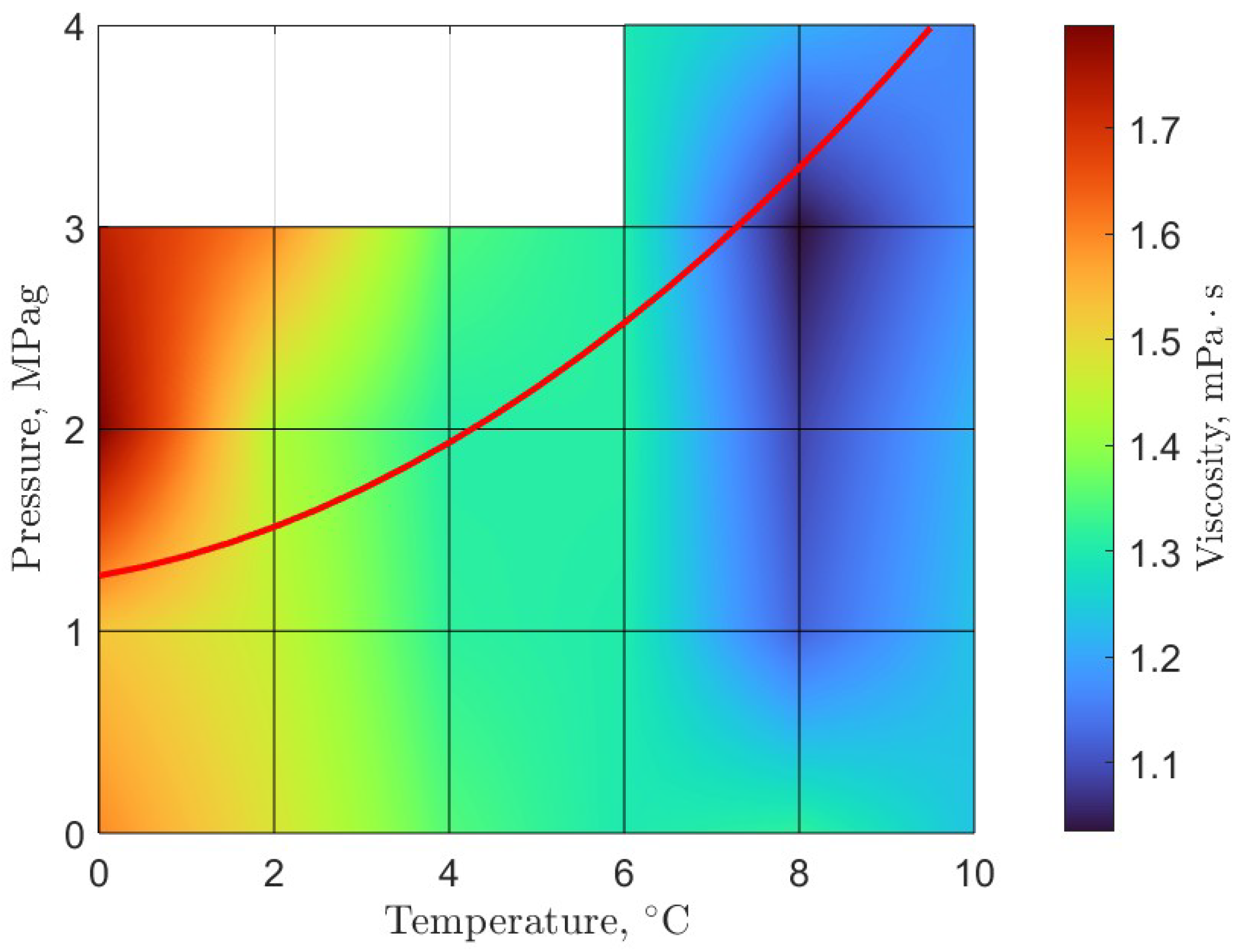
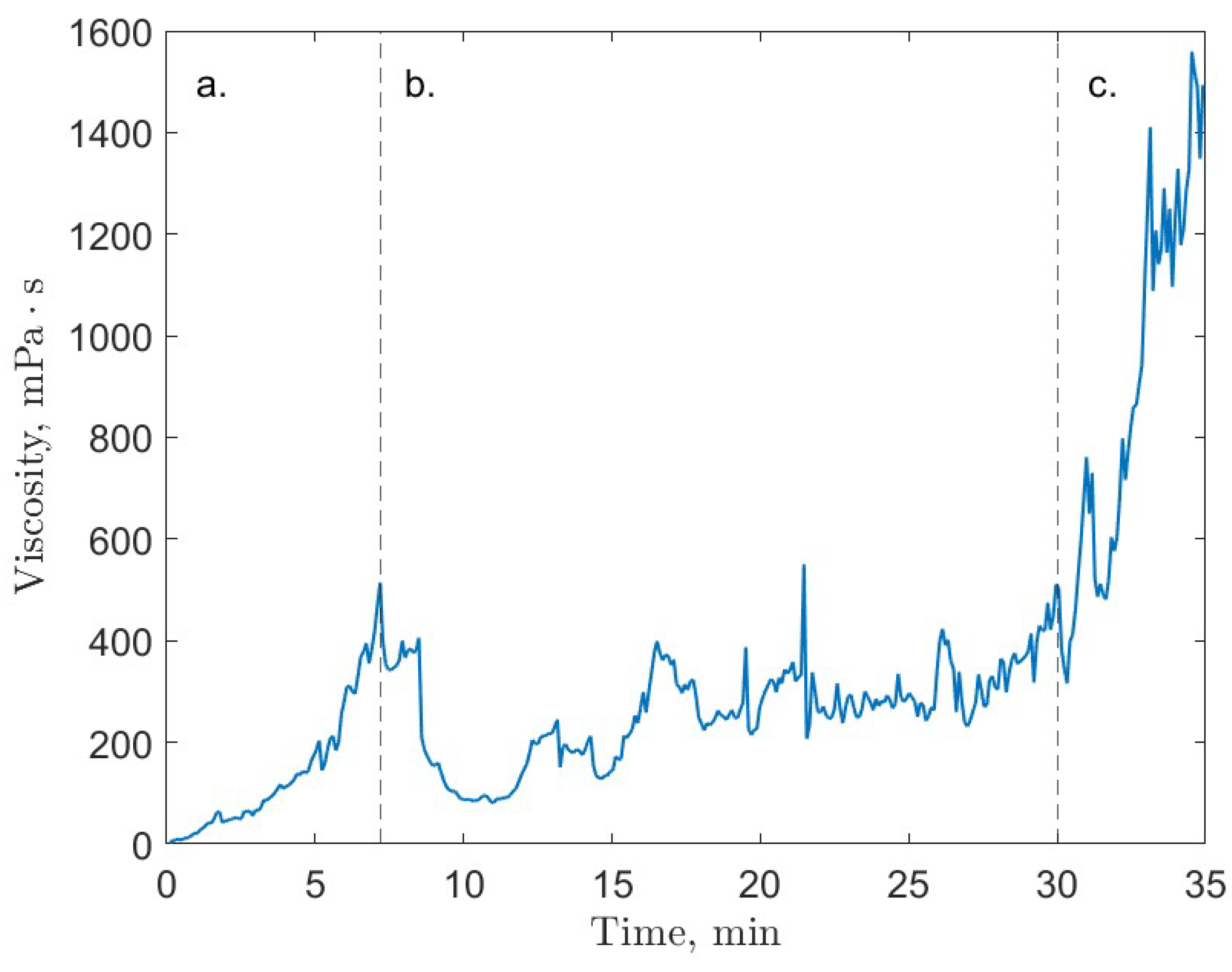
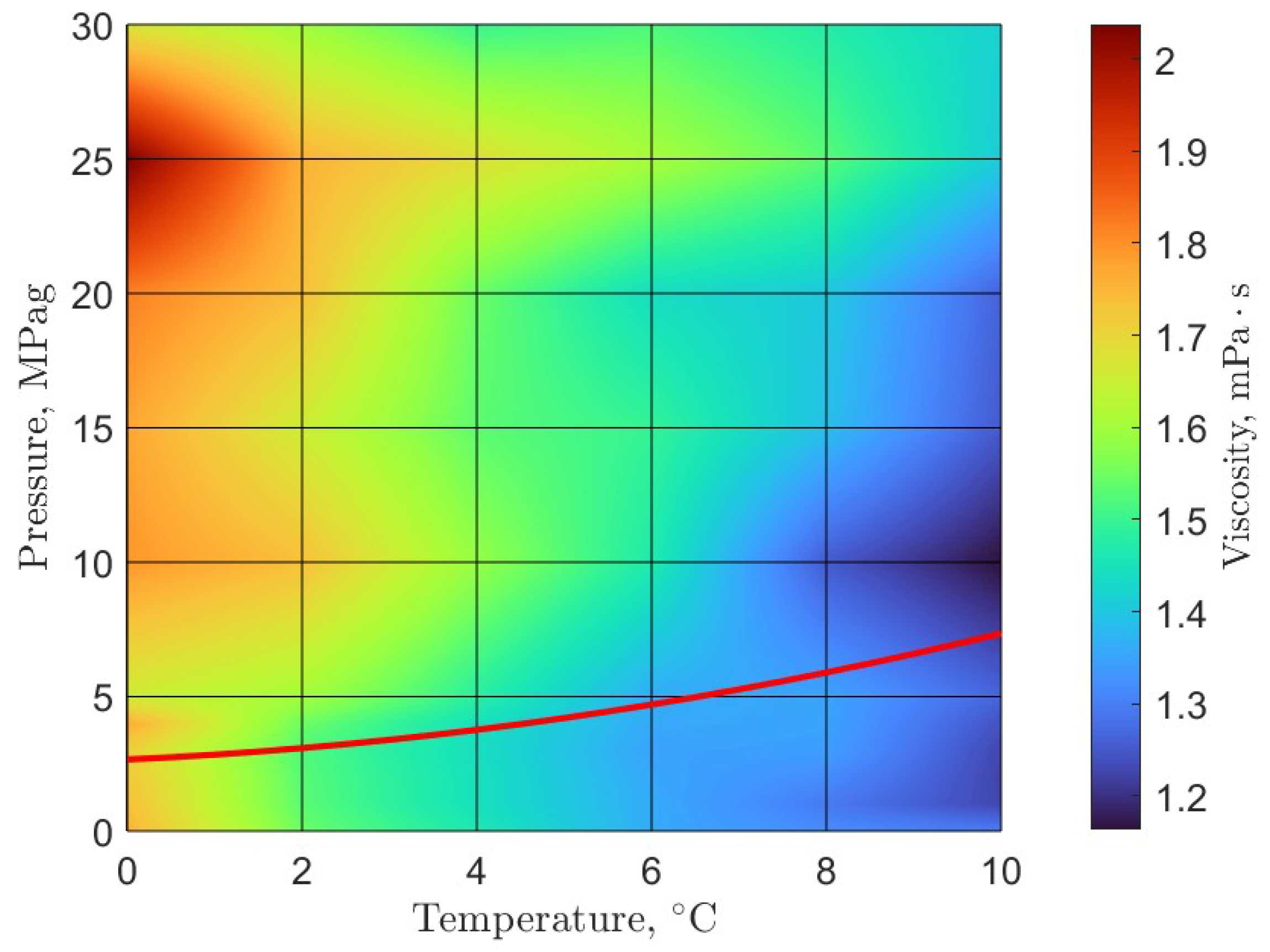
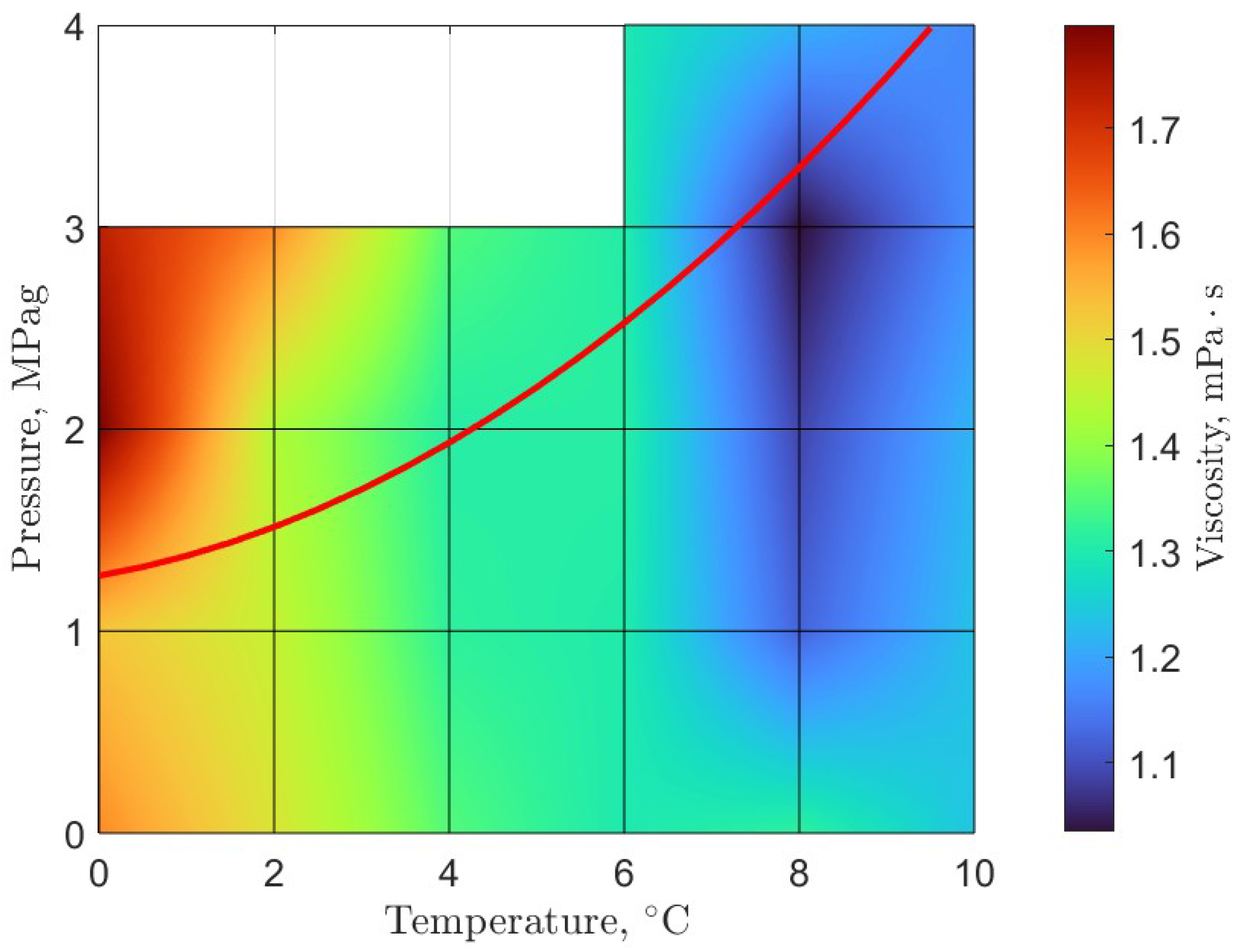
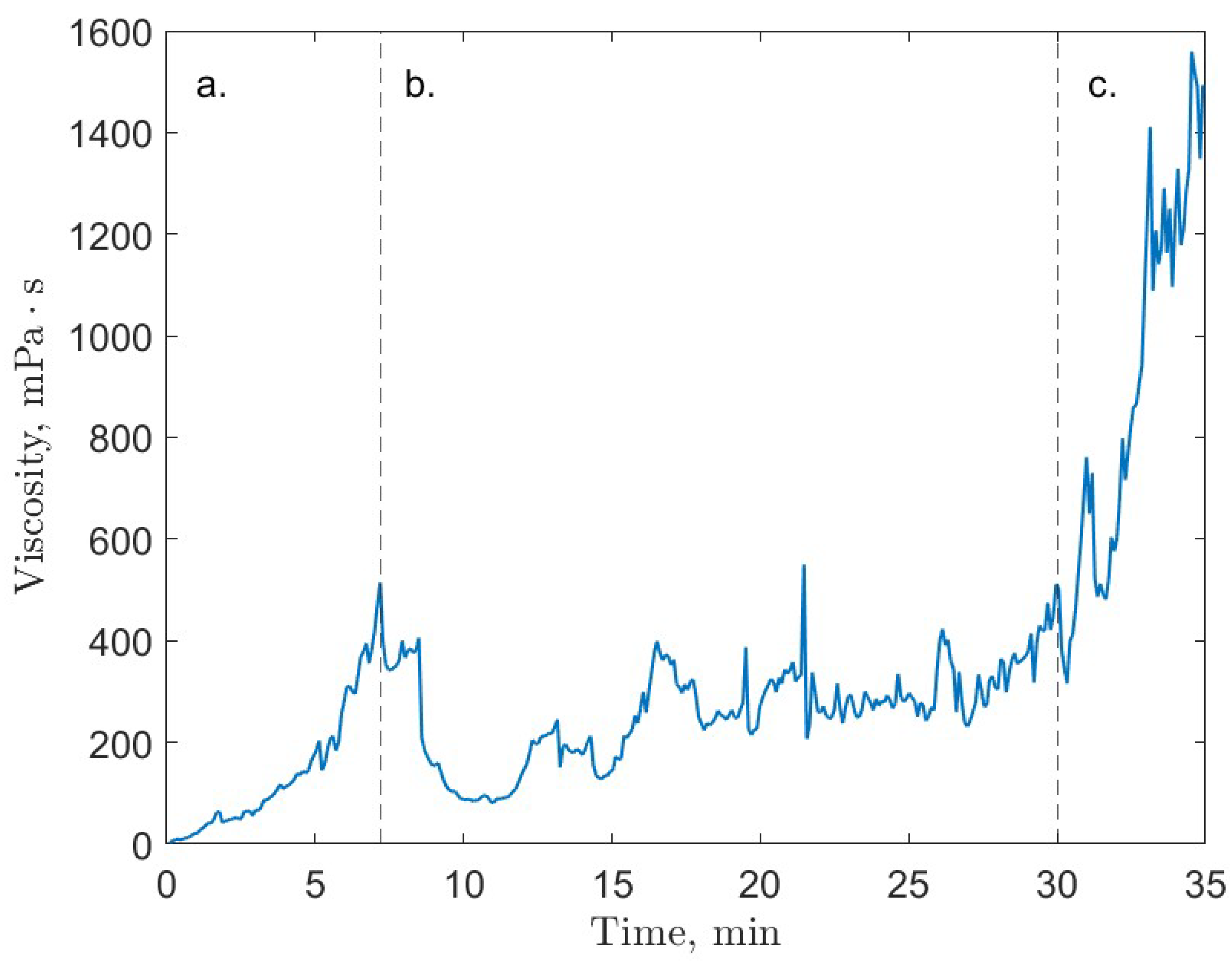
6. Rheology
7. Summary
7.1. Concluding Remarks
7.2. Future Topics to Be Explored
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMBER | Assisted model building with energy refinement |
| CHARMM | Chemistry at Harvard macromolecular mechanics |
| CMC | Critical micelle concentration |
| DFT | Density functional theory |
| GNF | Graphene nanoflakes |
| GROMOS | Groningen molecular simulation |
| IR | Infrared |
| KHI | Kinetic hydrate inhibitor |
| LAMMPS | Large-scale atomic/molecular massively parallel simulator |
| LCAO | Linear combination of atomic orbitals |
| LJ | Lennard–Jones |
| MD | Molecular dynamics |
| MWCNT | Multi-walled carbon nanotubes |
| O-GNF | Oxygen functionalized graphene nanoflakes |
| O-MWCNT | Oxygen functionalized multi-walled carbon nanotubes |
| OPLS | Optimized potentials for liquid simulations |
| PPFS | poly(pentaflourostyrene) |
| PS | poly(styrene) |
| PVA | poly(vinyl alcohol) |
| PVCap | Poly(vinylcaprolactam) |
| PVP | Poly(vinylpyrrolidone) |
| RAFT | Reversible addition-fragmentation chain transfer |
| SIESTA | Spanish initiative for electronic simulations with thousands of atoms |
| VASP | Vienna ab initio simulation package |
References
- Ripmeester, J.A.; Alavi, S. Clathrate Hydrates: Molecular Science and Characterization; Wiley-VCH: Weinheim, Germany, 2022; Available online: https://mcgill.on.worldcat.org/oclc/1301543108 (accessed on 15 June 2022). [CrossRef]
- Sloan, E.; Koh, C. Clathrate Hydrates of Natural Gases, 3rd ed.; Taylor and Francis: Boca Raton, FL, USA, 2008. [Google Scholar]
- Carroll, J. Natural Gas Hydrates: A Guide for Engineers, 3rd ed.; Gulf Professional Publishing: Calgary, AB, Canada, 2014. [Google Scholar]
- Davy, H. On some of the combinations of oxymuriatic gas andoxygene, and on the chemical relations of these principles, to inflammable bodies. Philos. Trans. R. Soc. 1811, 37, 1–35. [Google Scholar]
- Hammerschmidt, E. Formation of gas hydrates in natural gas transmission lines. Ind. Eng. Chem. 1934, 26, 851–855. [Google Scholar] [CrossRef]
- Kang, S.P.; Lee, H. Recovery of CO2 from flue gas using gas hydrate: Thermodynamic verification through phase equilibrium measurements. Environ. Sci. Technol. 2000, 34, 4397–4400. [Google Scholar] [CrossRef]
- Aaron, D.; Tsouris, C. Separation of CO2 from Flue Gas: A Review. Sep. Sci. Technol. 2005, 40, 321–348. [Google Scholar] [CrossRef]
- Linga, P.; Adeyemo, A.; Englezos, P. Medium-Pressure Clathrate Hydrate/Membrane Hybrid Process for Postcombustion Capture of Carbon Dioxide. Environ. Sci. Technol. 2007, 42, 315–320. [Google Scholar] [CrossRef]
- Fan, S.; Li, S.; Wang, J.; Lang, X.; Wang, Y. Efficient capture of CO2 from simulated flue gas by formation of TBAB or TBAF semiclathrate hydrates. Energy Fuels 2009, 23, 4202–4208. [Google Scholar] [CrossRef]
- Eslamimanesh, A.; Mohammadi, A.H.; Richon, D.; Naidoo, P.; Ramjugernath, D. Application of gas hydrate formation in separation processes: A review of experimental studies. J. Chem. Thermodyn. 2012, 46, 62–71. [Google Scholar] [CrossRef]
- Gudmundsson, J.; Parlaktuna, M.; Khokhar, A. Storage of natural gas as frozen hydrate. SPE Prod. Facil. 1994, 9, 69–73. [Google Scholar] [CrossRef]
- Mimachi, H.; Takahashi, M.; Takeya, S.; Gotoh, Y.; Yoneyama, A.; Hyodo, K.; Takeda, T.; Murayama, T. Effect of Long-Term Storage and Thermal History on the Gas Content of Natural Gas Hydrate Pellets under Ambient Pressure. Energy Fuels 2015, 29, 4827–4834. [Google Scholar] [CrossRef]
- Lu, Y.; Lv, X.; Li, Q.; Yang, L.; Zhang, L.; Zhao, J.; Song, Y. Molecular Behavior of Hybrid Gas Hydrate Nucleation: Separation of Soluble H2S from Mixed Gas. Phys. Chem. Chem. Phys. 2022, 24, 9509–9520. [Google Scholar] [CrossRef]
- Gaikwad, N.; Sangwai, J.; Linga, P.; Kumar, R. Separation of Coal Mine Methane Gas Mixture via sII and sH Hydrate Formation. Fuel 2021, 305, 121467. [Google Scholar] [CrossRef]
- Molaghan, P.; Jahanshahi, M.; Ahangari, M.G. H2 and H2S Separation by Adsorption Using Graphene and Zinc Oxide Sheets: Molecular Dynamic Simulations. Phys. B Condens. Matter 2021, 619, 413175. [Google Scholar] [CrossRef]
- Park, K.n.; Hong, S.Y.; Lee, J.W.; Kang, K.C.; Lee, Y.C.; Ha, M.G.; Lee, J.D. A new apparatus for seawater desalination by gas hydrate process and removal characteristics of dissolved minerals (Na+, Mg2+, Ca2+, K+, B3+). Desalination 2011, 274, 91–96. [Google Scholar] [CrossRef]
- Nallakukkala, S.; ur Rehman, A.; Zaini, D.B.; Lal, B. Gas Hydrate-Based Heavy Metal Ion Removal from Industrial Wastewater: A Review. Water 2022, 14, 1171. [Google Scholar] [CrossRef]
- Loekman, S.; Claßen, T.; Seidl, P.; Luzi, G.; Gatternig, B.; Rauh, C.; Delgado, A. Potential Application of Innovative Gas-Hydrate Technology in Fruit Juices Concentration Process. In Proceedings of the 2019 World Congress on Advances in Nano, Bio, Robotics, and Energy (ANBRE19), Jeju Island, Korea, 17–21 September 2019. [Google Scholar]
- McElligott, A.; Uddin, H.; Meunier, J.L.; Servio, P. Effects of Hydrophobic and Hydrophilic Graphene Nanoflakes on Methane Hydrate Kinetics. Energy Fuels 2019, 33, 11705–11711. [Google Scholar] [CrossRef]
- McElligott, A.; Meunier, J.L.; Servio, P. Effects of Hydrophobic and Hydrophilic Graphene Nanoflakes on Methane Dissolution Rates in Water under Vapor–Liquid–Hydrate Equilibrium Conditions. Ind. Eng. Chem. Res. 2021, 60, 2677–2685. [Google Scholar] [CrossRef]
- Guerra, A.; McElligott, A.; Yang Du, C.; Marić, M.; Rey, A.D.; Servio, P. Dynamic viscosity of methane and carbon dioxide hydrate systems from pure water at high-pressure driving forces. Chem. Eng. Sci. 2022, 252, 117282. [Google Scholar] [CrossRef]
- Vlasic, T.M.; Servio, P.D.; Rey, A.D. THF Hydrates as Model Systems for Natural Gas Hydrates: Comparing Their Mechanical and Vibrational Properties. Ind. Eng. Chem. Res. 2019, 58, 16588–16596. [Google Scholar] [CrossRef]
- Sloan, E.D. Gas Hydrates: Review of Physical/Chemical Properties. Energy Fuels 1998, 12, 191–196. [Google Scholar] [CrossRef]
- Vlasic, T.M.; Servio, P.D.; Rey, A.D. Effect of Guest Size on the Mechanical Properties and Molecular Structure of Gas Hydrates from First-Principles. Cryst. Growth Des. 2017, 17, 6407–6416. [Google Scholar] [CrossRef]
- Daghash, S.M.; Servio, P.; Rey, A.D. Structural Properties of sH Hydrate: A DFT Study of Anisotropy and Equation of State. Mol. Simul. 2019, 45, 1524–1537. [Google Scholar] [CrossRef]
- Ghafari, H.; Mohammadi-Manesh, H. The Thermal Properties of Binary Structure sI Clathrate Hydrate from Molecular Dynamics Simulation. Mol. Simul. 2019, 45, 614–622. [Google Scholar] [CrossRef]
- Mirzaeifard, S.; Servio, P.; Rey, A.D. Multiscale Modeling and Simulation of Water and Methane Hydrate Crystal Interface. Cryst. Growth Des. 2019, 19, 5142–5151. [Google Scholar] [CrossRef]
- Schicks, J.M. Gas Hydrates in Nature and in the Laboratory: Necessary Requirements for Formation and Properties of the Resulting Hydrate Phase. ChemTexts 2022, 8, 13. [Google Scholar] [CrossRef]
- Xie, Y.; Zheng, T.; Zhu, Y.J.; Zhong, J.R.; Feng, J.C.; Sun, C.Y.; Chen, G.J. Effects of H2/N2 on CO2 Hydrate Film Growth: Morphology and Microstructure. Chem. Eng. J. 2022, 431, 134004. [Google Scholar] [CrossRef]
- Daghash, S.M.; Servio, P.; Rey, A.D. Elastic Properties and Anisotropic Behavior of Structure-H (sH) Gas Hydrate from First Principles. Chem. Eng. Sci. 2020, 227, 115948. [Google Scholar] [CrossRef]
- Bagherzadeh, S.A.; Englezos, P.; Alavi, S.; Ripmeester, J.A. Influence of Hydrated Silica Surfaces on Interfacial Water in the Presence of Clathrate Hydrate Forming Gases. J. Phys. Chem. C 2012, 116, 24907–24915. [Google Scholar] [CrossRef]
- Naeiji, P.; Woo, T.K.; Alavi, S.; Varaminian, F.; Ohmura, R. Interfacial Properties of Hydrocarbon/Water Systems Predicted by Molecular Dynamic Simulations. J. Chem. Phys. 2019, 150, 114703. [Google Scholar] [CrossRef]
- Naeiji, P.; Woo, T.K.; Alavi, S.; Ohmura, R. Molecular Dynamics Simulations of Interfacial Properties of the CO2–Water and CO2–CH4–Water Systems. J. Chem. Phys. 2020, 153, 044701. [Google Scholar] [CrossRef]
- Algaba, J.; Acuña, E.; Míguez, J.M.; Mendiboure, B.; Zerón, I.M.; Blas, F.J. Simulation of the Carbon Dioxide Hydrate-Water Interfacial Energy. J. Colloid Interface Sci. 2022, 623, 354–367. [Google Scholar] [CrossRef]
- Chi, Y.; Xu, Y.; Zhao, C.; Zhang, Y.; Song, Y. In-Situ Measurement of Interfacial Tension: Further Insights into Effect of Interfacial Tension on the Kinetics of CO2 Hydrate Formation. Energy 2022, 239, 122143. [Google Scholar] [CrossRef]
- Hu, P.; Ke, W.; Chen, D. Molecular Mechanism for Methane Hydrate Nucleation on Corroded Iron Surface. Chem. Eng. Sci. 2022, 249, 117303. [Google Scholar] [CrossRef]
- He, Z.; Mi, F.; Ning, F. Molecular Insights into CO2 Hydrate Formation in the Presence of Hydrophilic and Hydrophobic Solid Surfaces. Energy 2021, 234, 121260. [Google Scholar] [CrossRef]
- Koh, C.A. Towards a fundamental understanding of natural gas hydrates. Chem. Soc. Rev. 2002, 31, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.J.; Tester, J.W.; Borghi, G.P.; Trout, B.L. Properties of inhibitors of methane hydrate formation via molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 17852–17862. [Google Scholar] [CrossRef]
- Perrin, A.; Musa, O.M.; Steed, J.W. The chemistry of low dosage clathrate hydrate inhibitors. Chem. Soc. Rev. 2013, 42, 1996–2015. [Google Scholar] [CrossRef] [Green Version]
- Rajput, F.; Colantuoni, A.; Bayahya, S.; Dhane, R.; Servio, P.; Maric, M. Poly(styrene/pentafluorostyrene)-block-poly(vinyl alcohol/vinylpyrrolidone) amphiphilic block copolymers for kinetic gas hydrate inhibitors: Synthesis, micellization behavior, and methane hydrate kinetic inhibition. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 2445–2457. [Google Scholar] [CrossRef]
- Rajput, F.; Maric, M.; Servio, P. Amphiphilic Block Copolymers with Vinyl Caprolactam as Kinetic Gas Hydrate Inhibitors. Energies 2021, 14, 341. [Google Scholar] [CrossRef]
- Sun, S.; Li, Y.; Gu, L.; Yang, Z.; Zhao, J. Experimental Study on Carbon Dioxide Hydrate Formation in the Presence of Static Magnetic Field. J. Chem. Thermodyn. 2022, 170, 106764. [Google Scholar] [CrossRef]
- Bazvand, M.; Madani Tehrani, D. Effect of Magnetic Field on Gas Hydrate Formation. Nat. Gas Ind. B 2022. [Google Scholar] [CrossRef]
- Ning, F.; Guo, D.; Din, S.U.; Zhang, H.; Ou, W.; Fang, B.; Liang, Y.; Zhang, L.; Lee, K.; Koh, C.A. The Kinetic Effects of Hydrate Anti-Agglomerants/Surfactants. Fuel 2022, 318, 123566. [Google Scholar] [CrossRef]
- Liu, N.; Li, T.; Liu, T.; Yang, L. Molecular Dynamics Simulations of the Effects of Metal Nanoparticles on Methane Hydrate Formation. J. Mol. Liq. 2022, 356, 118962. [Google Scholar] [CrossRef]
- Liao, Q.; Shi, B.; Li, S.; Song, S.; Chen, Y.; Zhang, J.; Yao, H.; Li, Q.; Gong, J. Molecular Dynamics Simulation of the Effect of Wax Molecules on Methane Hydrate Formation. Fuel 2021, 297, 120778. [Google Scholar] [CrossRef]
- Guo, P.; Song, G.; Ning, Y.; Li, Y.; Wang, W. Investigation on Hydrate Growth at Oil–Water Interface: In the Presence of Wax. Energy Fuels 2021, 35, 11884–11895. [Google Scholar] [CrossRef]
- Song, G.; Ning, Y.; Li, Y.; Wang, W. Investigation on Hydrate Growth at the Oil–Water Interface: In the Presence of Wax and Kinetic Hydrate Inhibitor. Langmuir 2020, 36, 14881–14891. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ning, Y.; Guo, P.; Li, Y.; Wang, W. Investigation on Hydrate Growth at the Oil–Water Interface: In the Presence of Wax and Surfactant. Langmuir 2021, 37, 6838–6845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Huang, Q.; Wang, W.; Li, H.; Zheng, H.; Li, R.; Li, W.; Kong, W. Effects of Waxes and Asphaltenes on CO2 Hydrate Nucleation and Decomposition in Oil-Dominated Systems. J. Nat. Gas Sci. Eng. 2021, 88, 103799. [Google Scholar] [CrossRef]
- Webb, E.B.; Rensing, P.J.; Koh, C.A.; Sloan, E.D.; Sum, A.K.; Liberatore, M.W. High-Pressure Rheology of Hydrate Slurries Formed from Water-in-Oil Emulsions. Energy Fuels 2012, 26, 3504–3509. [Google Scholar] [CrossRef]
- Webb, E.B.; Koh, C.A.; Liberatore, M.W. Rheological Properties of Methane Hydrate Slurries Formed From AOT + Water + Oil Microemulsions. Langmuir 2013, 29, 10997–11004. [Google Scholar] [CrossRef]
- Webb, E.B.; Koh, C.A.; Liberatore, M.W. High Pressure Rheology of Hydrate Slurries Formed from Water-in-Mineral Oil Emulsions. Ind. Eng. Chem. Res. 2014, 53, 6998–7007. [Google Scholar] [CrossRef]
- Pandey, G.; Linga, P.; Sangwai, J.S. High pressure rheology of gas hydrate formed from multiphase systems using modified Couette rheometer. Rev. Sci. Instrum. 2017, 88, 025102. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Sangwai, J.S. High pressure rheological studies of methane hydrate slurries formed from water-hexane, water-heptane, and water-decane multiphase systems. J. Nat. Gas Sci. Eng. 2020, 81, 103365. [Google Scholar] [CrossRef]
- Pasieka, J.; Coulombe, S.; Servio, P. Investigating the effects of hydrophobic and hydrophilic multi-wall carbon nanotubes on methane hydrate growth kinetics. Chem. Eng. Sci. 2013, 104, 998–1002. [Google Scholar] [CrossRef]
- Pasieka, J.; Jorge, L.; Coulombe, S.; Servio, P. Effects of As-Produced and Amine-Functionalized Multi-Wall Carbon Nanotubes on Carbon Dioxide Hydrate Formation. Energy Fuels 2015, 29, 5259–5266. [Google Scholar] [CrossRef]
- Legrand, U.; Mendoza Gonzalez, N.Y.; Pascone, P.; Meunier, J.L.; Berk, D. Synthesis and in-situ oxygen functionalization of deposited graphene nanoflakes for nanofluid generation. Carbon 2016, 102, 216–223. [Google Scholar] [CrossRef]
- Hordy, N.; Coulombe, S.; Meunier, J.L. Plasma Functionalization of Carbon Nanotubes for the Synthesis of Stable Aqueous Nanofluids and Poly(vinyl alcohol) Nanocomposites. Plasma Process. Polym. 2013, 10, 110–118. [Google Scholar] [CrossRef]
- Matus Rivas, O.M.; Rey, A.D. Molecular dynamics on the self-assembly of mesogenic graphene precursors. Carbon 2016, 110, 189–199. [Google Scholar] [CrossRef]
- Matus Rivas, O.M.; Rey, A.D. Molecular dynamics of dilute binary chromonic liquid crystal mixtures. Mol. Syst. Des. Eng. 2017, 2, 223–234. [Google Scholar] [CrossRef]
- Mirzaeifard, S. Analytical and Computational Modeling of Interfacial Properties and Nucleation Process in Methane Hydrates Materials. Ph.D. Thesis, McGill University Libraries, Montreal, QC, Canada, 2020. [Google Scholar]
- Mirzaeifard, S.; Servio, P.; Rey, A.D. Characterization of Nucleation of Methane Hydrate Crystals: Interfacial Theory and Molecular Simulation. J. Colloid Interface Sci. 2019, 557, 556–567. [Google Scholar] [CrossRef]
- Mirzaeifard, S.; Servio, P.; Rey, A.D. Molecular Dynamics Characterization of Temperature and Pressure Effects on the Water-Methane Interface. Colloid Interface Sci. Commun. 2018, 24, 75–81. [Google Scholar] [CrossRef]
- Mirzaeifard, S.; Servio, P.; Rey, A.D. Molecular Dynamics Characterization of the Water-Methane, Ethane, and Propane Gas Mixture Interfaces. Chem. Eng. Sci. 2019, 208, 114769. [Google Scholar] [CrossRef]
- Rapaport, D.C. The Art of Molecular Dynamics Simulation; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell Jr, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKerell, A.D., Jr.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Schuler, L.D.; Daura, X.; van Gunsteren, W.F. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 2001, 22, 1205–1218. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Jewett, A.I.; Stelter, D.; Lambert, J.; Saladi, S.M.; Roscioni, O.M.; Ricci, M.; Autin, L.; Maritan, M.; Bashusqeh, S.M.; Keyes, T.; et al. Moltemplate: A Tool for Coarse-Grained Modeling of Complex Biological Matter and Soft Condensed Matter Physics. J. Mol. Biol. 2021, 433, 166841. [Google Scholar] [CrossRef]
- Vlasic, T.M.; Servio, P.D.; Rey, A.D. Infrared Spectra of Gas Hydrates from First-Principles. J. Phys. Chem. B 2019, 123, 936–947. [Google Scholar] [CrossRef]
- Daghash, S.M.; Servio, P.; Rey, A.D. From Infrared Spectra to Macroscopic Mechanical Properties of sH Gas Hydrates through Atomistic Calculations. Molecules 2020, 25, 5568. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ojamäe, L. CH-Stretching Vibrational Trends in Natural Gas Hydrates Studied by Quantum-Chemical Computations. J. Phys. Chem. C 2015, 119, 17084–17091. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ojamäe, L. C–C Stretching Raman Spectra and Stabilities of Hydrocarbon Molecules in Natural Gas Hydrates: A Quantum Chemical Study. J. Phys. Chem. A 2014, 118, 11641–11651. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA Method for Ab Initio Order-N Materials Simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Torre, D.; Escribano, R.; Archer, T.; Pruneda, J.M.; Artacho, E. First-Principles Infrared Spectrum of Nitric Acid and Nitric Acid Monohydrate Crystals. J. Phys. Chem. A 2004, 108, 10535–10541. [Google Scholar] [CrossRef]
- Liu, Y.; Ojamäe, L. Raman and IR Spectra of Ice Ih and Ice XI with an Assessment of DFT Methods. J. Phys. Chem. B 2016, 120, 11043–11051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeKock, R.; Gray, H. Chemical Structure and Bonding; University Science Books: Sausalito, CA, USA, 1989. [Google Scholar]
- Ashby, M.F.; Jones, D.R.H. Engineering Materials 1: An Introduction to Their Properties and Applications, 2nd ed.; Butterworth-Heinemann: Oxford, UK, 1996; Chapter viii; 306p. [Google Scholar]
- Vlasic, T.M.; Servio, P.; Rey, A.D. Atomistic Modeling of Structure II Gas Hydrate Mechanics: Compressibility and Equations of State. AIP Adv. 2016, 6, 085317. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Huang, C.H.; Tan, C.S. A Review of CO2 Capture by Absorption and Adsorption. Aerosol Air Qual. Res. 2012, 12, 745–769. [Google Scholar] [CrossRef] [Green Version]
- Dai, N.; Mitch, W.A. Influence of Amine Structural Characteristics on N-Nitrosamine Formation Potential Relevant to Postcombustion CO2 Capture Systems. Environ. Sci. Technol. 2013, 47, 13175–13183. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, F.A.; Yamada, H.; Higashii, T.; Goto, K.; Onoda, M. CO2 Capture by Tertiary Amine Absorbents: A Performance Comparison Study. Ind. Eng. Chem. Res. 2013, 52, 8323–8331. [Google Scholar] [CrossRef]
- Komati, S.; Suresh, A.K. Anomalous enhancement of interphase transport rates by nanoparticles: Effect of magnetic iron oxide on gas-liquid mass transfer. Ind. Eng. Chem. Res. 2010, 49, 390–405. [Google Scholar] [CrossRef]
- Zhu, H.; Shanks, B.H.; Heindel, T.J. Enhancing CO-water mass transfer by functionalized MCM41 nanoparticles. Ind. Eng. Chem. Res. 2008, 47, 7881–7887. [Google Scholar] [CrossRef] [Green Version]
- Olle, B.; Bucak, S.; Holmes, T.C.; Bromberg, L.; Hatton, T.A.; Wang, D.I. Enhancement of oxygen mass transfer using functionalized magnetic nanoparticles. Ind. Eng. Chem. Res. 2006, 45, 4355–4363. [Google Scholar] [CrossRef]
- Posteraro, D.; Verrett, J.; Maric, M.; Servio, P. New insights into the effect of polyvinylpyrrolidone (PVP) concentration on methane hydrate growth. 1. Growth rate. Chem. Eng. Sci. 2015, 126, 99–105. [Google Scholar] [CrossRef]
- Posteraro, D.; Ivall, J.; Maric, M.; Servio, P. New insights into the effect of polyvinylpyrrolidone (PVP) concentration on methane hydrate growth. 2. Liquid phase methane mole fraction. Chem. Eng. Sci. 2015, 126, 91–98. [Google Scholar] [CrossRef]
- Evans, D.F.; Wennerström, H. The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet; Advances in Interfacial Engineering; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Carpena, P.; Aguiar, J.; Bernaola-Galván, P.; Carnero Ruiz, C. Problems Associated with the Treatment of Conductivity Concentration Data in Surfactant Solutions Simulations and Experiments. Langmuir 2002, 18, 6054–6058. [Google Scholar] [CrossRef]
- Bielawska, M.; Jańczuk, B.; Zdziennicka, A. Adhesion work and wettability of polytetrafluorethylene and poly(methyl methacrylate) by aqueous solutions of cetyltrimethylammonium bromide and Triton X-100 mixture with ethanol. J. Colloid Interface Sci. 2013, 404, 201–206. [Google Scholar] [CrossRef]
- Piera, E.; Erra, P.; Infante, M.R. Analysis of cationic surfactants by capillary electrophoresis. J. Chromatogr. A 1997, 757, 275–280. [Google Scholar] [CrossRef]
- Topel, Ö.; Çakır, B.A.; Budama, L.; Hoda, N. Determination of critical micelle concentration of polybutadiene-block-poly(ethyleneoxide) diblock copolymer by fluorescence spectroscopy and dynamic light scattering. J. Mol. Liq. 2013, 177, 40–43. [Google Scholar] [CrossRef]
- Zhang, J.S.; Lo, C.; Couzis, A.; Somasundaran, P.; Wu, J.; Lee, J.W. Adsorption of Kinetic Inhibitors on Clathrate Hydrates. J. Phys. Chem. C 2009, 113, 17418–17420. [Google Scholar] [CrossRef]
- Koop, T.; Murray, B.J. A Physically Constrained Classical Description of the Homogeneous Nucleation of Ice in Water. J. Chem. Phys. 2016, 145, 211915. [Google Scholar] [CrossRef] [Green Version]
- Knott, B.C.; Molinero, V.; Doherty, M.F.; Peters, B. Homogeneous Nucleation of Methane Hydrates: Unrealistic under Realistic Conditions. J. Am. Chem. Soc. 2012, 134, 19544–19547. [Google Scholar] [CrossRef]
- Bai, D.; Chen, G.; Zhang, X.; Sum, A.K.; Wang, W. How Properties of Solid Surfaces Modulate the Nucleation of Gas Hydrate. Sci. Rep. 2015, 5, 12747. [Google Scholar] [CrossRef] [Green Version]
- Koga, T.; Wong, J.; Endoh, M.K.; Mahajan, D.; Gutt, C.; Satija, S.K. Hydrate Formation at the Methane/Water Interface on the Molecular Scale. Langmuir 2010, 26, 4627–4630. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, L.; Liu, Y.; Song, Y. Microstructural Characteristics of Natural Gas Hydrates Hosted in Various Sand Sediments. Phys. Chem. Chem. Phys. PCCP 2015, 17, 22632–22641. [Google Scholar] [CrossRef] [PubMed]
- Biscay, F.; Ghoufi, A.; Malfreyt, P. Adsorption of N-Alkane Vapours at the Water Surface. Phys. Chem. Chem. Phys. 2011, 13, 11308–11316. [Google Scholar] [CrossRef]
- Biscay, F.; Ghoufi, A.; Lachet, V.; Malfreyt, P. Monte Carlo Calculation of the Methane-Water Interfacial Tension at High Pressures. J. Chem. Phys. 2009, 131, 124707. [Google Scholar] [CrossRef]
- Shuttleworth, R. The Surface Tension of Solids. Proc. Phys. Soc. Sect. A 1950, 63, 444–457. [Google Scholar] [CrossRef]
- Vázquez, U.O.M.; Shinoda, W.; Moore, P.B.; Chiu, C.c.; Nielsen, S.O. Calculating the Surface Tension between a Flat Solid and a Liquid: A Theoretical and Computer Simulation Study of Three Topologically Different Methods. J. Math. Chem. 2009, 45, 161–174. [Google Scholar] [CrossRef]
- Binks, B.P.; Clint, J.H. Solid Wettability from Surface Energy Components: Relevance to Pickering Emulsions. Langmuir 2002, 18, 1270–1273. [Google Scholar] [CrossRef]
- Ghiass, M.; Rey, A.D. Interfacial Thermodynamics of Compressible Polymer Solutions. J. Chem. Phys. 2008, 128, 071102. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, E.; Bedeaux, D.; Holcomb, C.; Zollweg, J. Tail Corrections to the Surface Tension of a Lennard-Jones Liquid-Vapour Interface. Mol. Phys. 1995, 85, 665–669. [Google Scholar] [CrossRef]
- Chapela, G.A.; Saville, G.; Thompson, S.M.; Rowlinson, J.S. Computer Simulation of a Gas–Liquid Surface. Part 1. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1977, 73, 1133–1144. [Google Scholar] [CrossRef]
- Grest, G.S.; Stevens, M.J.; Plimpton, S.J.; Woolf, T.B.; Lehoucq, R.B.; Crozier, P.S.; Ismail, A.E.; Mukherjee, R.M.; Draganescu, A.I. Substructured Multibody Molecular Dynamics; Technical Report; Sandia National Laboratories (SNL): Albuquerque, NM, USA; Livermore, CA, USA, 2006. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.Y.; Patey, G.N. Heterogeneous Ice Nucleation Induced by Electric Fields. J. Phys. Chem. Lett. 2011, 2, 2555–2559. [Google Scholar] [CrossRef]
- Yan, J.Y.; Patey, G.N. Ice Nucleation by Electric Surface Fields of Varying Range and Geometry. J. Chem. Phys. 2013, 139, 144501. [Google Scholar] [CrossRef]
- Luis, D.P.; Herrera-Hernández, E.C.; Saint-Martin, H. A Theoretical Study of the Dissociation of the sI Methane Hydrate Induced by an External Electric Field. J. Chem. Phys. 2015, 143, 204503. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PVA | PVP | PVCap | |
|---|---|---|---|
| % | % | % | |
| - | 27 | 51 | 53 |
| PS | 49 | 59 | 56 |
| PPFS | 63.5 | 76 | 73 |
| Block Copolymer | Molecular Weight | CMC |
|---|---|---|
| kg/mol | × M | |
| PS-PVA-40(0.05) | 42.8 | 2.0 |
| PPFS-PVA-40(0.05) | 44.1 | 1.5 |
| PS-PVP-20(0.10) | 21.6 | 5.0 |
| PPFS-PVP-20(0.10) | 23.1 | 4.5 |
| PS-PVCap(0.05) | 77.1 | 2.9 |
| PS-PVCap(0.10) | 42.6 | 4.0 |
| PS-PVCap(0.15) | 23.9 | 7.0 |
| PPFS-PVCap(0.05) | 90.5 | 1.3 |
| PPFS-PVCap(0.10) | 46.2 | 3.7 |
| PPFS-PVCap(0.15) | 30.0 | 4.9 |
| PPFS-PVCap(0.20) | 23.8 | 6.0 |
| PVCap-PVP(0.10) | 53.1 | 45.9 |
| PVCap-PVP(0.20) | 28.0 | 58.0 |
| Structure | Guest Molecule | k | E (IR) | E (Force Constants) | |
|---|---|---|---|---|---|
| Å | N·m | GPa | GPa | ||
| sII | Empty | 1.710 | 2.726 | 15.940 | 14.67 |
| Pr | 1.749 | 2.259 | 12.920 | 11.57 | |
| i-Bu | 1.780 | 1.886 | 10.590 | 11.33 | |
| Et-CH | 1.772 | 2.113 | 11.930 | 14.48 | |
| Pr-CH | 1.793 | 1.897 | 10.580 | 12.58 | |
| sH | Empty | 1.698 | 2.778 | 16.360 | 14.32 |
| CH-NH | 1.797 | 1.933 | 10.760 | 16.57 | |
| Xenon-NH | 1.810 | 1.933 | 10.680 | 17.00 | |
| CO-NH | 1.813 | 1.850 | 10.200 | - | |
| CO-CH-NH | 1.799 | 1.871 | 10.400 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, A.; Mathews, S.; Marić, M.; Rey, A.D.; Servio, P. An Integrated Experimental and Computational Platform to Explore Gas Hydrate Promotion, Inhibition, Rheology, and Mechanical Properties at McGill University: A Review. Energies 2022, 15, 5532. https://doi.org/10.3390/en15155532
Guerra A, Mathews S, Marić M, Rey AD, Servio P. An Integrated Experimental and Computational Platform to Explore Gas Hydrate Promotion, Inhibition, Rheology, and Mechanical Properties at McGill University: A Review. Energies. 2022; 15(15):5532. https://doi.org/10.3390/en15155532
Chicago/Turabian StyleGuerra, André, Samuel Mathews, Milan Marić, Alejandro D. Rey, and Phillip Servio. 2022. "An Integrated Experimental and Computational Platform to Explore Gas Hydrate Promotion, Inhibition, Rheology, and Mechanical Properties at McGill University: A Review" Energies 15, no. 15: 5532. https://doi.org/10.3390/en15155532