Sorption-Enhanced Water-Gas Shift Reaction for Synthesis Gas Production from Pure CO: Investigation of Sorption Parameters and Reactor Configurations


Abstract
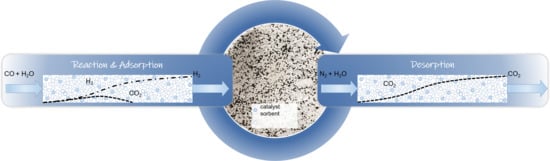
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Catalyst and Sorbent Preparation
2.1.2. Catalyst Performance Measurements
2.1.3. Sorbent Characterization and Sorption Capacity Measurements
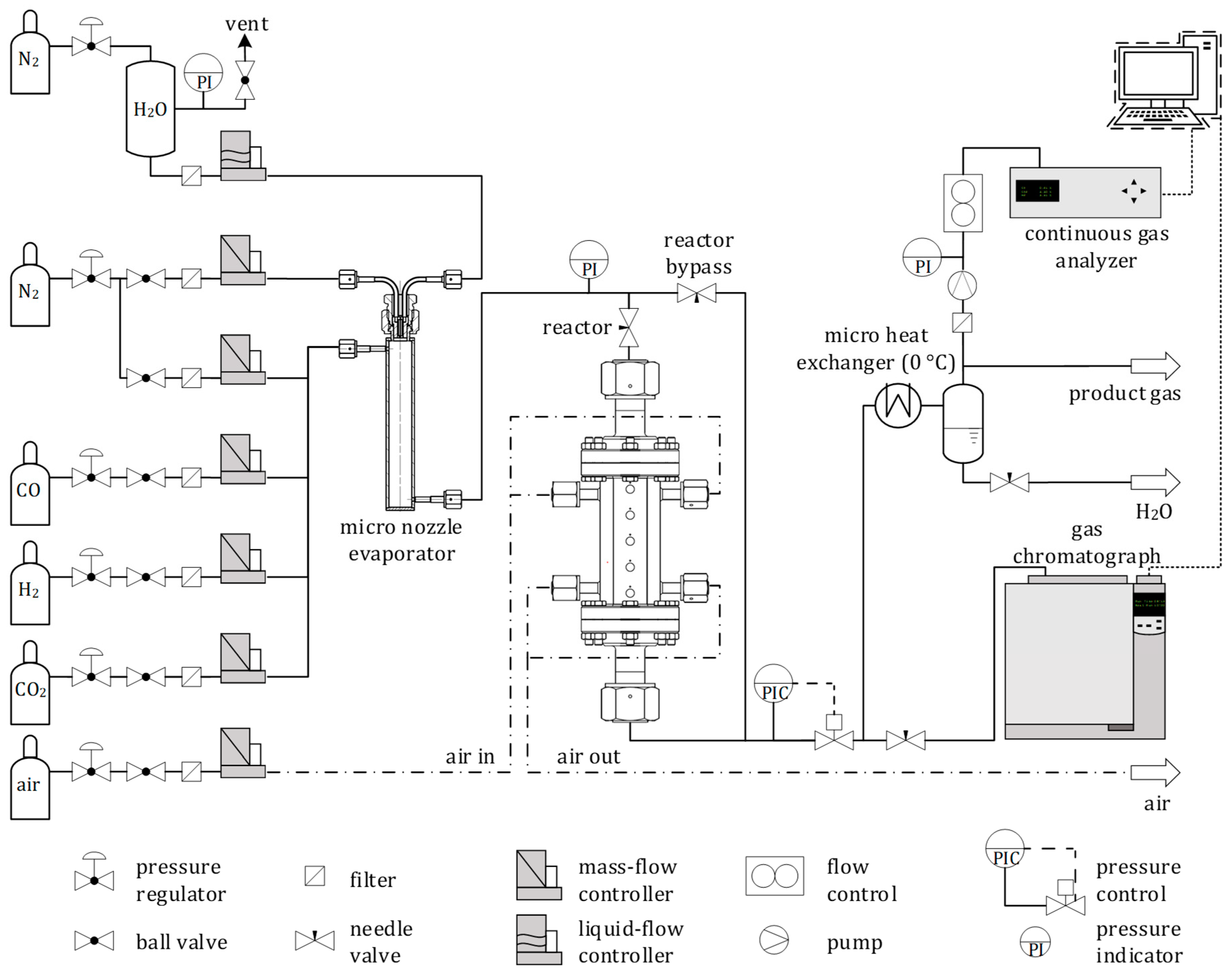
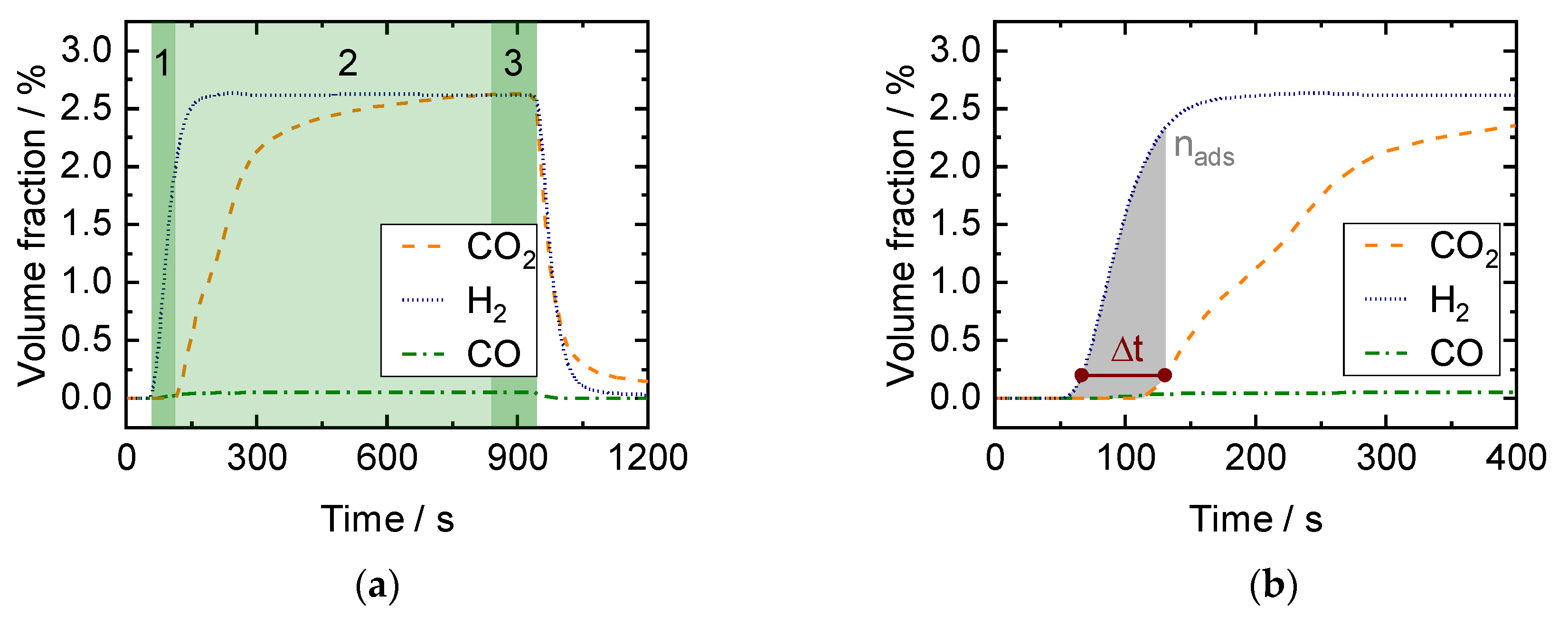
2.2. SEWGS Breakthrough Experiments
3. Results and Discussion
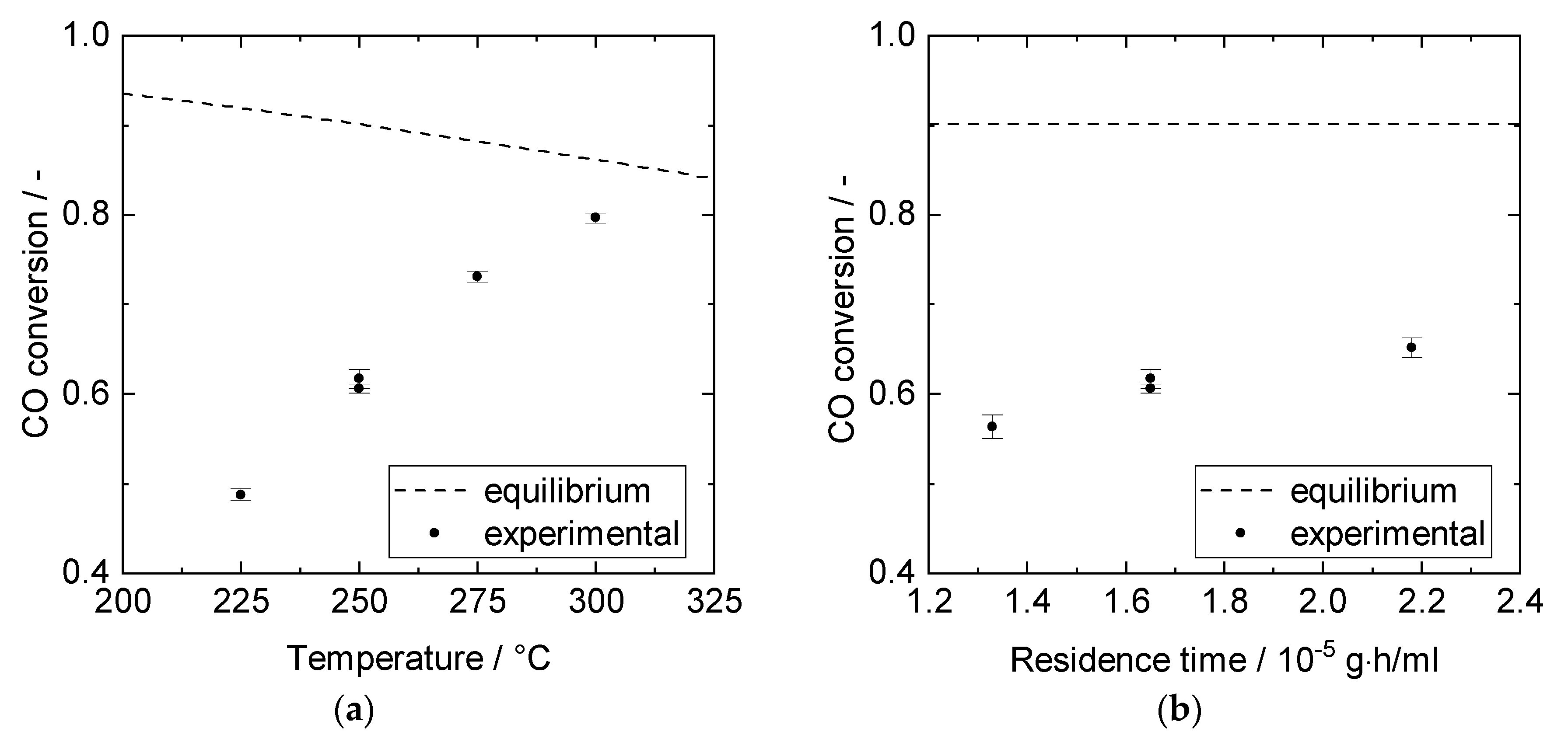
3.1. Catalyst Performance
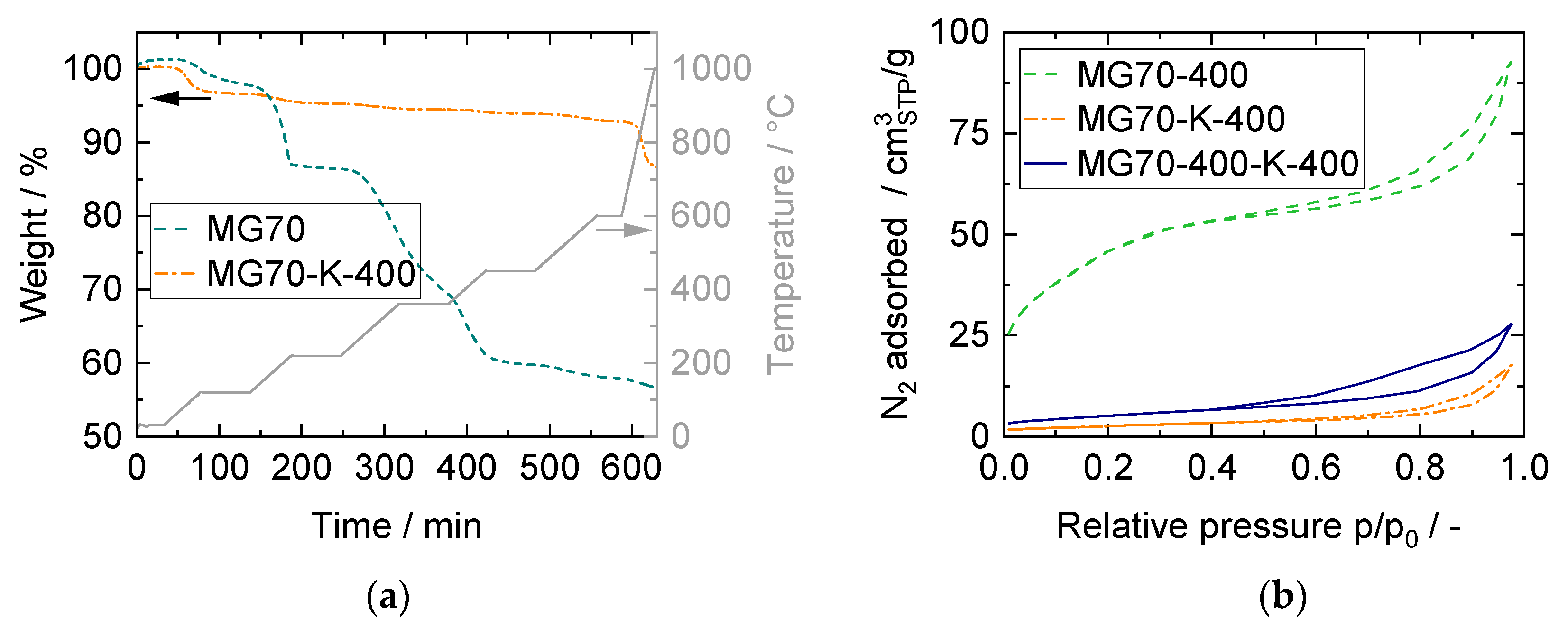
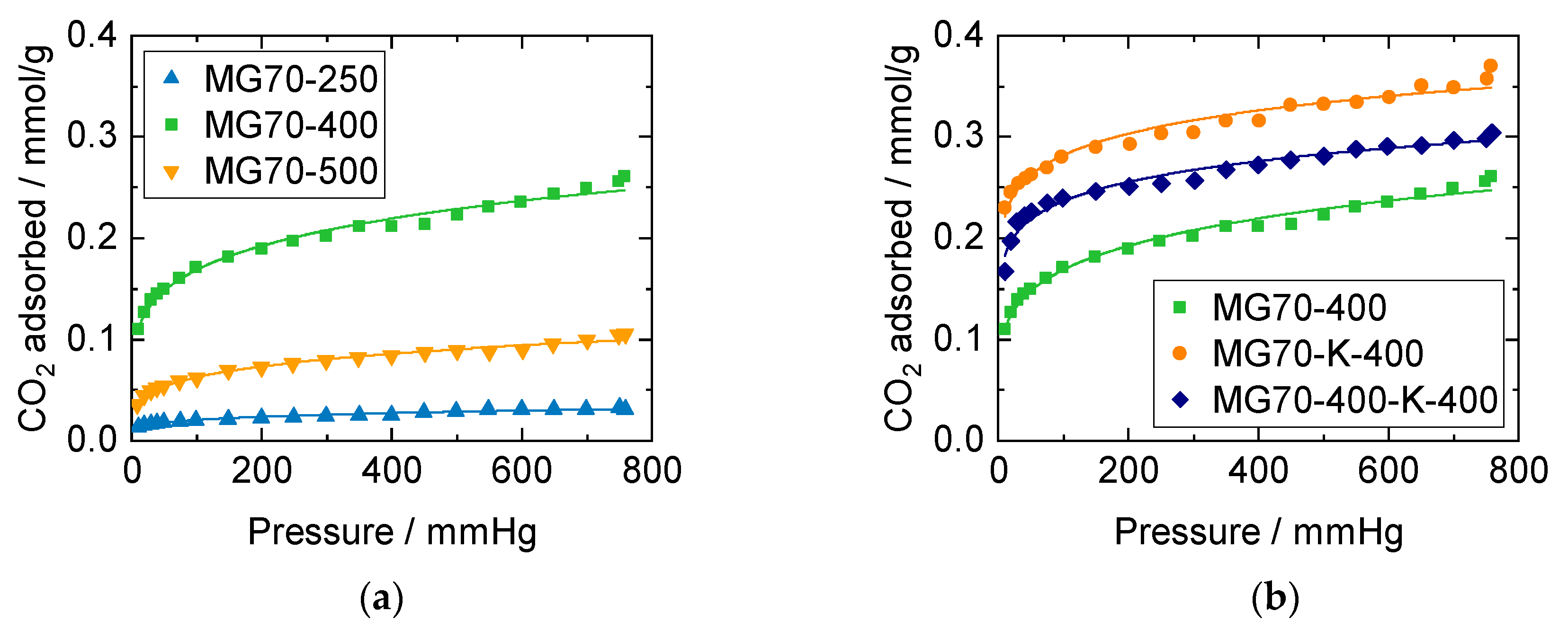
3.2. Structural Properties and CO2 Capture Performance of Sorbent
3.3. SEWGS Performance
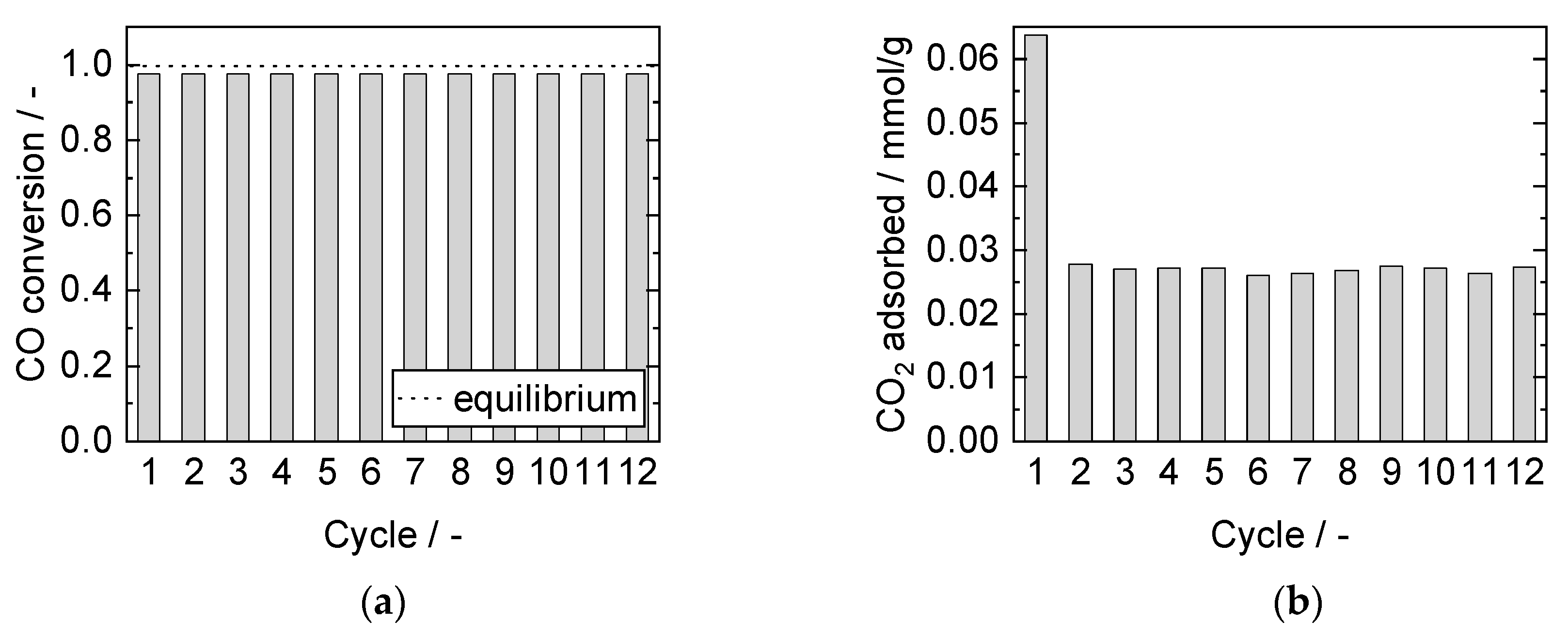
3.3.1. Cyclic Stability
3.3.2. Reproducibility
3.3.3. Variation of Adsorption Parameters
Influence of Total Pressure
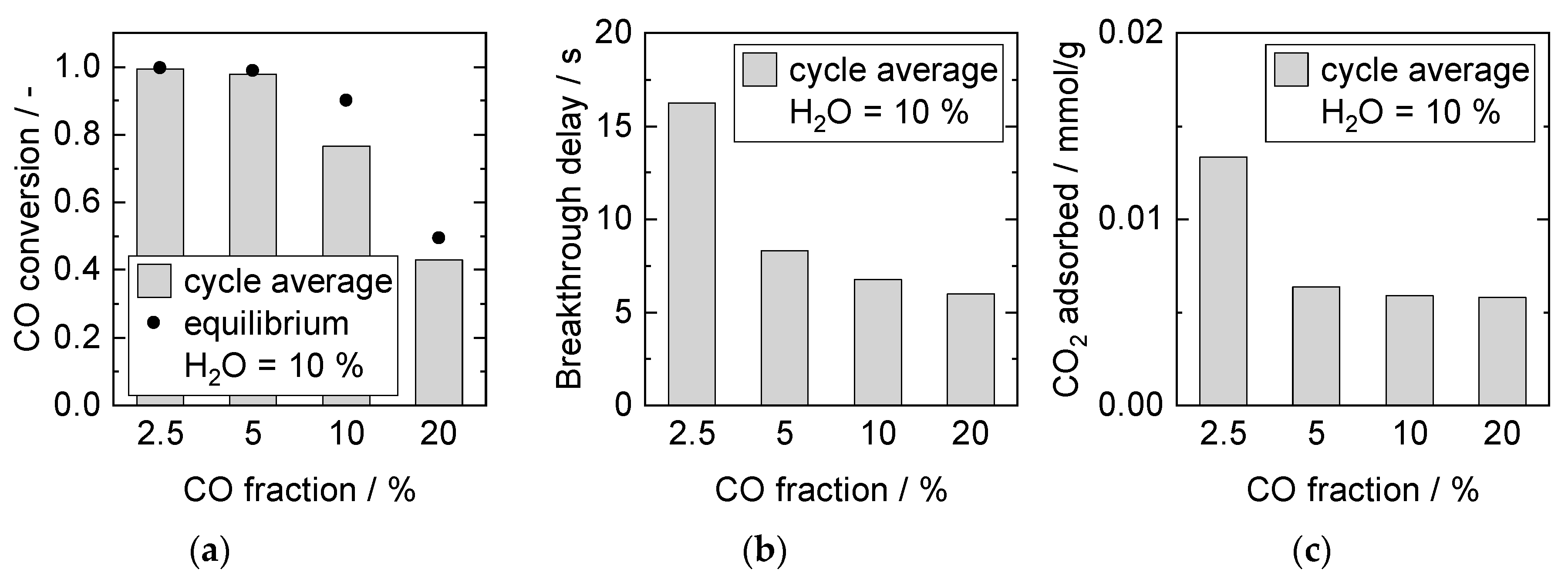
Influence of CO Partial Pressure at Constant H2O Partial Pressure
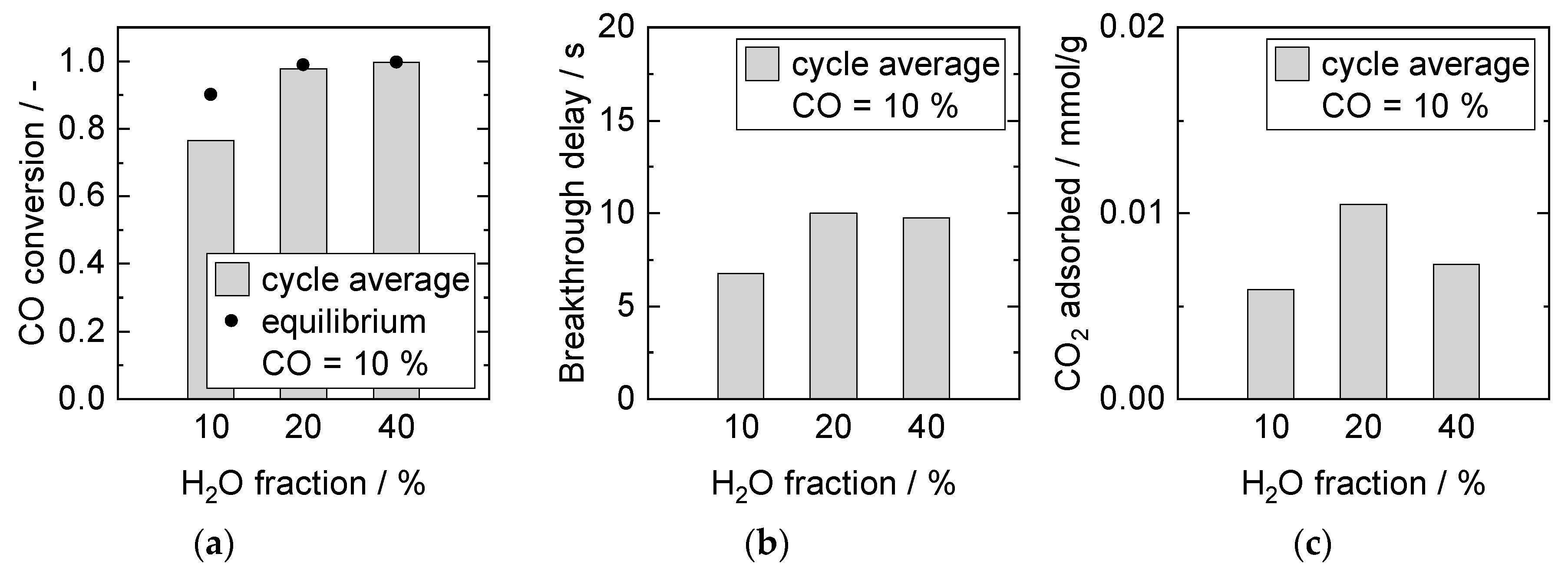
Influence of H2O Partial Pressure at Constant CO Partial Pressure
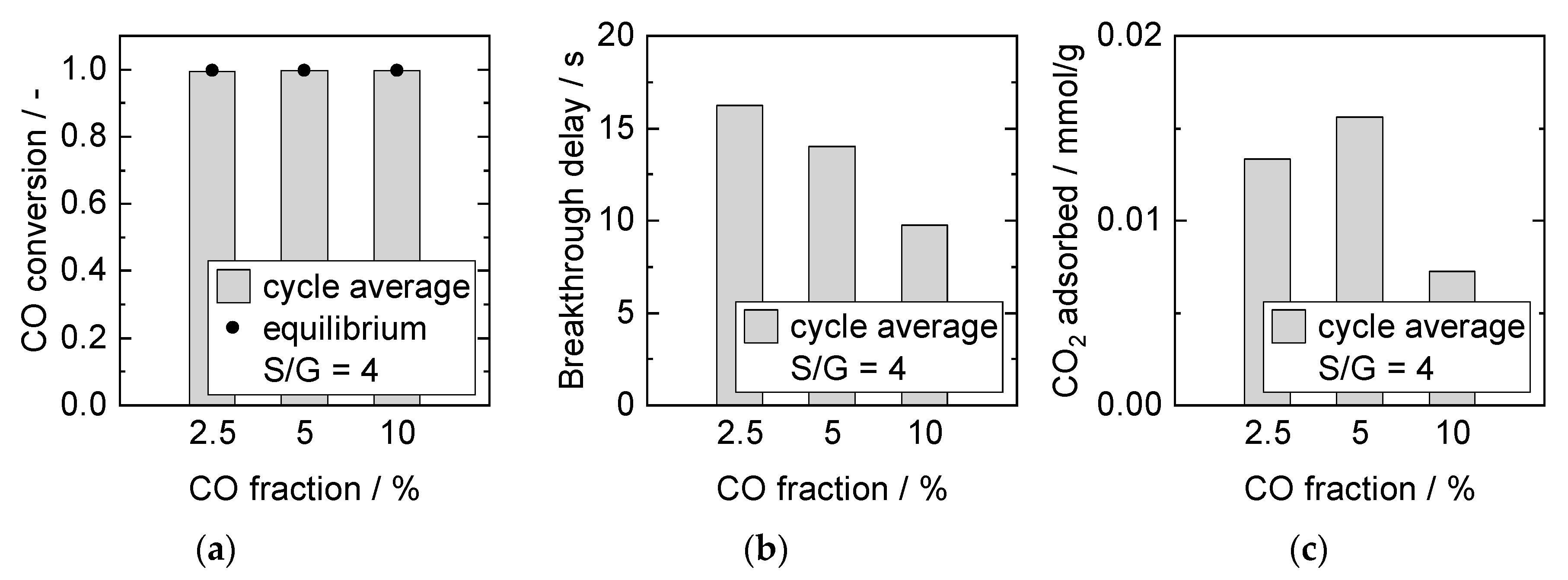
Influence of CO and H2O Partial Pressure at Constant S/G Ratio
3.3.4. Variation of Desorption Parameters
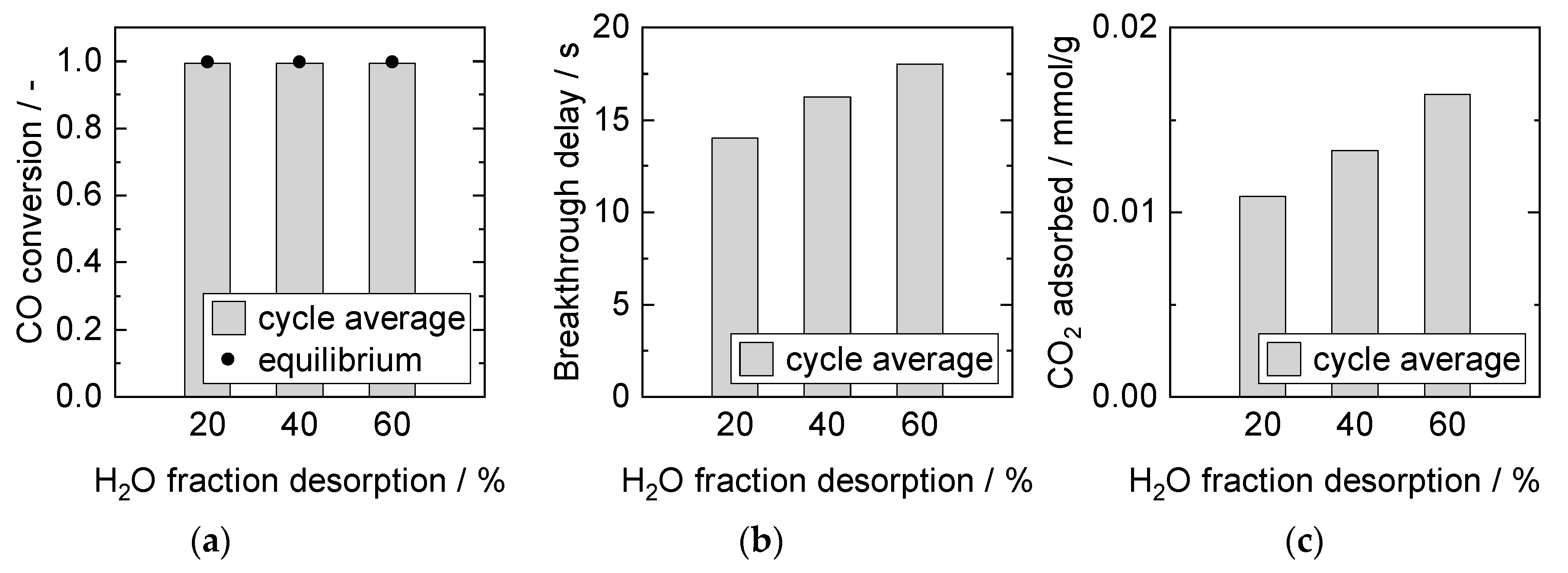
Influence of H2O Partial Pressure during Desorption
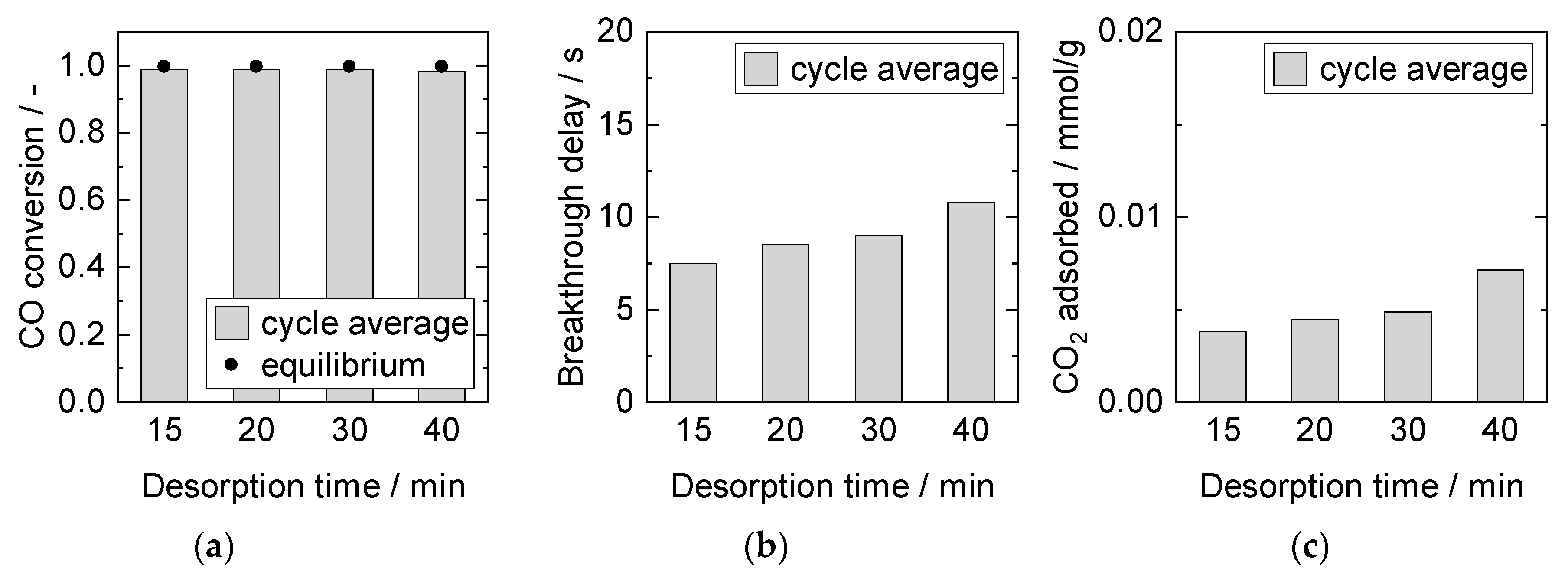
Influence of Desorption Time
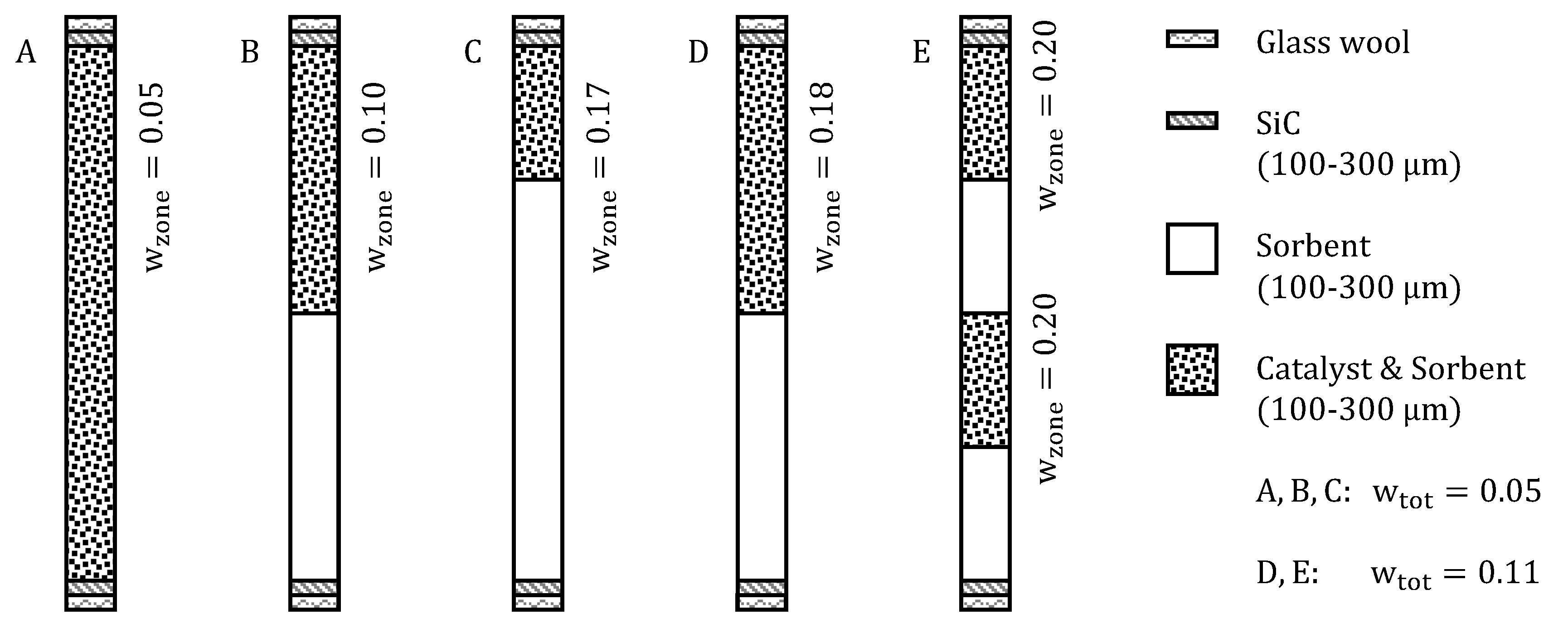
3.3.5. Variation of Reactor Configuration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| BET | Brunauer-Emmett-Teller |
| BJH | Barrett-Joyner-Halenda |
| BSE | Back-scattered electrons |
| EDS | Energy dispersive spectroscopy |
| EPMA | Electron probe micro analysis |
| Eq. | Equation |
| FID | Flame ionization detector |
| FT | Fischer-Tropsch |
| GC | Gas chromatograph |
| HTC | Hydrotalcite |
| HTdc | Hydrotalcite-derived compounds |
| LFC | Liquid flow controller |
| MFC | Mass flow controller |
| NDIR | Nondispersive infrared detector |
| PBMR | Packed bed microchannel reactor |
| Powder diffraction file | |
| PGA | Process gas analyzer |
| PtL | Power-to-Liquid |
| S/G | Steam-to-gas ratio |
| SE | Secondary electrons |
| SEWGS | Sorption-enhaned water-gas shift |
| TCD | Thermal conductivity detector |
| TGA | Thermogravimetric analysis |
| TR | Tubular reactor |
| WGS | Water-gas shift |
| XRD | X-ray diffraction |
List of Symbols
| Reaction enthalpy | kJ/mol | |
| Equilibrium constant | - | |
| Freundlich constant | mmol/g∙bar1/n | |
| Mass | g | |
| Specific amount of CO2 adsorbed | mmol/g | |
| Molar flow | mol/s | |
| Adsorption intensity | - | |
| Pressure | bar | |
| Ideal gas constant | J/mol∙K | |
| Reaction rate | mol/g∙h | |
| Modified residence time | g∙h/mL | |
| Temperature | K | |
| Time | s | |
| Volume flow | mL/min | |
| Weight fraction | - | |
| CO conversion | - | |
| Volume fraction | - |
List of Indices
| Feed | |
| Adsorption/adsorbed | |
| Catalyst | |
| Desorption | |
| After condensation | |
| Sorbent |
References
- United Nations. Paris Agreement. Available online: https://unfccc.int/process-and-meetings (accessed on 17 November 2020).
- European Commission. A European Strategy for Low-Emission Mobility. Available online: https://eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX:52016DC0501 (accessed on 17 November 2020).
- Goede, A.P.H. CO2 neutral fuels. EPJ Web Conf. 2018, 189, 10. [Google Scholar] [CrossRef]
- Viegas, P.; Vialetto, L.; Wolf, A.J.; Peeters, F.J.J.; Groen, P.W.C.; Righart, T.W.H.; Bongers, W.A.; van de Sanden, M.C.M.; Diomede, P. Insight into contraction dynamics of microwave plasmas for CO2 conversion from plasma chemistry modelling. Plasma Sources Sci. Technol. 2020, 29, 105014. [Google Scholar] [CrossRef]
- Soldatov, S.; Link, G.; Silberer, L.; Schmedt, C.M.; Carbone, E.; D’Isa, F.; Jelonnek, J.; Dittmeyer, R.; Navarrete, A. Time-Resolved Optical Emission Spectroscopy Reveals Nonequilibrium Conditions for CO2 Splitting in Atmospheric Plasma Sustained with Ultrafast Microwave Pulsation. ACS Energy Lett. 2020, 11, 124–130. [Google Scholar] [CrossRef]
- Kirsch, H.; Brübach, L.; Loewert, M.; Riedinger, M.; Gräfenhahn, A.; Böltken, T.; Klumpp, M.; Pfeifer, P.; Dittmeyer, R. CO2-neutrale Fischer-Tropsch-Kraftstoffe aus dezentralen modularen Anlagen: Status und Perspektiven. Chem. Ing. Tech. 2020, 92, 91–99. [Google Scholar] [CrossRef]
- Ramirez, A.; Sarathy, S.M.; Gascon, J. CO2 Derived E-Fuels: Research Trends, Misconceptions, and Future Directions. Trends Chem. 2020, 2, 785–795. [Google Scholar] [CrossRef]
- Soria, M.A.; Tosti, S.; Mendes, A.; Madeira, L.M. Enhancing the low temperature water–gas shift reaction through a hybrid sorption-enhanced membrane reactor for high-purity hydrogen production. Fuel 2015, 159, 854–863. [Google Scholar] [CrossRef]
- Jang, H.M.; Lee, K.B.; Caram, H.S.; Sircar, S. High-purity hydrogen production through sorption enhanced water gas shift reaction using K2CO3-promoted hydrotalcite. Chem. Eng. Sci. 2012, 73, 431–438. [Google Scholar] [CrossRef]
- Moe, J.M. Design of water-gas shift reactors. Chem. Eng. Prog. 1962, 58, 3. [Google Scholar]
- Mendes, D.; Mendes, A.; Madeira, L.M.; Iulianelli, A.; Sousa, J.M.; Basile, A. The water-gas shift reaction: From conventional catalytic systems to Pd-based membrane reactors—A review. Asia Pac. J. Chem. Eng. 2010, 5, 111–137. [Google Scholar] [CrossRef]
- Smith, R.J.B.; Loganathan, M.; Shantha, M.S. A Review of the Water Gas Shift Reaction Kinetics. Int. J. Chem. React. Eng. 2010, 8, 8. [Google Scholar] [CrossRef]
- Choi, Y.; Stenger, H.G. Water gas shift reaction kinetics and reactor modeling for fuel cell grade hydrogen. J. Power Sources 2003, 124, 432–439. [Google Scholar] [CrossRef]
- Wang, S.; Shen, H.; Fan, S.; Zhao, Y.; Ma, X.; Gong, J. Enhanced CO2 adsorption capacity and stability using CaO-based adsorbents treated by hydration. AIChE J. 2013, 59, 3586–3593. [Google Scholar] [CrossRef]
- Bhagiyalakshmi, M.; Lee, J.Y.; Jang, H.T. Synthesis of mesoporous magnesium oxide: Its application to CO2 chemisorption. Int. J. Greenh. Gas Control 2010, 4, 51–56. [Google Scholar] [CrossRef]
- Iwan, A.; Stephenson, H.; Ketchie, W.C.; Lapkin, A.A. High temperature sequestration of CO2 using lithium zirconates. Chem. Eng. J. 2009, 146, 249–258. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, W.; Yang, Y.; Qu, M.; Li, H. CO2 capture by Li4SiO4 sorbents and their applications: Current developments and new trends. Chem. Eng. J. 2019, 359, 604–625. [Google Scholar] [CrossRef]
- Yin, G.; Liu, Z.; Liu, Q.; Wu, W. The role of different properties of activated carbon in CO2 adsorption. Chem. Eng. J. 2013, 230, 133–140. [Google Scholar] [CrossRef]
- Siriwardane, R.V.; Shen, M.-S.; Fisher, E.P.; Poston, J.A. Adsorption of CO2 on Molecular Sieves and Activated Carbon. Energy Fuels 2001, 15, 279–284. [Google Scholar] [CrossRef]
- Mulloth, L.M.; Finn, J.E. Carbon Dioxide Adsorption on a 5A Zeolite Designed for CO2 Removal in Spacecraft Cabins; NASA/TM-1998-208752; NASA: Washington, DC, USA, 1998.
- Millward, A.R.; Yaghi, O.M. Metal-organic frameworks with exceptionally high capacity for storage of carbon dioxide at room temperature. J. Am. Chem. Soc. 2005, 127, 17998–17999. [Google Scholar] [CrossRef]
- Reijers, H.T.J.; Boon, J.; Elzinga, G.D.; Cobden, P.D.; Haije, W.G.; van den Brink, R.W. Modeling Study of the Sorption-Enhanced Reaction Process for CO2 Capture. I. Model Development and Validation. Ind. Eng. Chem. Res. 2009, 48, 6966–6974. [Google Scholar] [CrossRef]
- Van Selow, E.R.; Cobden, P.D.; Wright, A.D.; van den Brink, R.W.; Jansen, D. Improved sorbent for the sorption-enhanced water-gas shift process. Energy Procedia 2011, 4, 1090–1095. [Google Scholar] [CrossRef]
- Rives, V. Characterisation of layered double hydroxides and their decomposition products. Mater. Chem. Phys. 2002, 75, 19–25. [Google Scholar] [CrossRef]
- León, M.; Díaz, E.; Bennici, S.; Vega, A.; Ordóñez, S.; Auroux, A. Adsorption of CO2 on Hydrotalcite-Derived Mixed Oxides: Sorption Mechanisms and Consequences for Adsorption Irreversibility. Ind. Eng. Chem. Res. 2010, 49, 3663–3671. [Google Scholar] [CrossRef]
- Yong, Z.; Mata, A.V.; Rodrigues, A.E. Adsorption of Carbon Dioxide onto Hydrotalcite-like Compounds (HTlcs) at High Temperatures. Ind. Eng. Chem. Res. 2001, 40, 204–209. [Google Scholar] [CrossRef]
- Zhu, X.; Shi, Y.; Cai, N. High-pressure carbon dioxide adsorption kinetics of potassium-modified hydrotalcite at elevated temperature. Fuel 2017, 207, 579–590. [Google Scholar] [CrossRef]
- Lee, J.M.; Min, Y.J.; Lee, K.B.; Jeon, S.G.; Na, J.G.; Ryu, H.J. Enhancement of CO2 sorption uptake on hydrotalcite by impregnation with K2CO3. Langmuir 2010, 26, 18788–18797. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Williams, C.T.; Ebner, A.D.; Ritter, J.A. In Situ FTIR Spectroscopic Analysis of Carbonate Transformations during Adsorption and Desorption of CO2 in K-Promoted HTlc. Chem. Mater. 2010, 22, 3519–3526. [Google Scholar] [CrossRef]
- Belimov, M.; Metzger, D.; Pfeifer, P. On the temperature control in a microstructured packed bed reactor for methanation of CO/CO2 mixtures. AIChE J. 2017, 63, 120–129. [Google Scholar] [CrossRef]
- Lee, C.H.; Lee, K.B. Application of one-body hybrid solid pellets to sorption-enhanced water gas shift reaction for high-purity hydrogen production. Int. J. Hydrog. Energy 2014, 39, 18128–18134. [Google Scholar] [CrossRef]
- Lee, C.H.; Lee, K.B. Sorption-enhanced water gas shift reaction for high-purity hydrogen production: Application of a Na-Mg double salt-based sorbent and the divided section packing concept. Appl. Energy 2017, 205, 316–322. [Google Scholar] [CrossRef]
- Hu, Y.; Cui, H.; Cheng, Z.; Zhou, Z. Sorption-enhanced water gas shift reaction by in situ CO2 capture on an alkali metal salt-promoted MgO-CaCO3 sorbent. Chem. Eng. J. 2019, 377, 119823. [Google Scholar] [CrossRef]
- Othman, M.R.; Helwani, Z.; Martunus; Fernando, W.J.N. Synthetic hydrotalcites from different routes and their application as catalysts and gas adsorbents: A review. Appl. Organometal. Chem. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- Reijers, H.T.J.; Valster-Schiermeier, S.E.A.; Cobden, P.D.; van den Brink, R.W. Hydrotalcite as CO2 Sorbent for Sorption-Enhanced Steam Reforming of Methane. Ind. Eng. Chem. Res. 2006, 45, 2522–2530. [Google Scholar] [CrossRef]
- Yang, J.-I.; Kim, J.-N. Hydrotalcites for adsorption of CO2 at high temperature. Korean J. Chem. Eng. 2006, 23, 77–80. [Google Scholar] [CrossRef]
- Carriazo, D.; del Arco, M.; Martín, C.; Rives, V. A comparative study between chloride and calcined carbonate hydrotalcites as adsorbents for Cr(VI). Appl. Clay Sci. 2007, 37, 231–239. [Google Scholar] [CrossRef]
- Ram Reddy, M.K.; Xu, Z.P.; Lu, G.Q.; da Costa, J.C.D. Layered Double Hydroxides for CO2 Capture: Structure Evolution and Regeneration. Ind. Eng. Chem. Res. 2006, 45, 7504–7509. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Abelló, S.; van der Pers, N.M. Memory effect of activated Mg-Al hydrotalcite: In situ XRD studies during decomposition and gas-phase reconstruction. Chemistry 2007, 13, 870–878. [Google Scholar] [CrossRef]
- Yang, W.; Kim, Y.; Liu, P.K.; Sahimi, M.; Tsotsis, T.T. A study by in situ techniques of the thermal evolution of the structure of a Mg–Al–CO3 layered double hydroxide. Chem. Eng. Sci. 2002, 57, 2945–2953. [Google Scholar] [CrossRef]
- Iruretagoyena Ferrer, D. Supported Layered Double Hydroxides as CO2 Adsorbents for Sorption-Enhanced H2 Production; Springer International Publishing: Cham, Switzerland, 2016; ISBN 9783319412764. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Rocha, C.; Soria, M.A.; Madeira, L.M. Doping of hydrotalcite-based sorbents with different interlayer anions for CO2 capture. Sep. Purif. Technol. 2020, 235, 116140. [Google Scholar] [CrossRef]
- Halabi, M.H.; de Croon, M.; van der Schaaf, J.; Cobden, P.D.; Schouten, J.C. High capacity potassium-promoted hydrotalcite for CO2 capture in H2 production. Int. J. Hydrogen Energy 2012, 37, 4516–4525. [Google Scholar] [CrossRef]
- Hanif, A.; Dasgupta, S.; Divekar, S.; Arya, A.; Garg, M.O.; Nanoti, A. A study on high temperature CO2 capture by improved hydrotalcite sorbents. Chem. Eng. J. 2014, 236, 91–99. [Google Scholar] [CrossRef]
- Oliveira, E.L.; Grande, C.A.; Rodrigues, A.E. CO2 sorption on hydrotalcite and alkali-modified (K and Cs) hydrotalcites at high temperatures. Sep. Purif. Technol. 2008, 62, 137–147. [Google Scholar] [CrossRef]
- Miguel, C.V.; Trujillano, R.; Rives, V.; Vicente, M.A.; Ferreira, A.; Rodrigues, A.E.; Mendes, A.; Madeira, L.M. High temperature CO2 sorption with gallium-substituted and promoted hydrotalcites. Sep. Purif. Technol. 2014, 127, 202–211. [Google Scholar] [CrossRef]
- Ebner, A.D.; Reynolds, S.P.; Ritter, J.A. Understanding the Adsorption and Desorption Behavior of CO2 on a K-Promoted Hydrotalcite-like Compound (HTlc) through Nonequilibrium Dynamic Isotherms. Ind. Eng. Chem. Res. 2006, 45, 6387–6392. [Google Scholar] [CrossRef]
- Soares, J.; Casarin, G.L.; José, H.J.; Moreira, R.D.F.P.M.; Rodrigues, A.E. Experimental and Theoretical Analysis for the CO2 Adsorption on Hydrotalcite. Adsorption 2005, 11, 237–241. [Google Scholar] [CrossRef]
- Silva, J.M.; Trujillano, R.; Rives, V.; Soria, M.A.; Madeira, L.M. High temperature CO2 sorption over modified hydrotalcites. Chem. Eng. J. 2017, 325, 25–34. [Google Scholar] [CrossRef]
- Coenen, K.; Gallucci, F.; Hensen, E.; van Sint Annaland, M. Kinetic model for adsorption and desorption of H2O and CO2 on hydrotalcite-based adsorbents. Chem. Eng. J. 2019, 355, 520–531. [Google Scholar] [CrossRef]
- Walspurger, S.; Boels, L.; Cobden, P.D.; Elzinga, G.D.; Haije, W.G.; van den Brink, R.W. The crucial role of the K+-aluminium oxide interaction in K+-promoted alumina- and hydrotalcite-based materials for CO2 sorption at high temperatures. ChemSusChem 2008, 1, 643–650. [Google Scholar] [CrossRef]
- Walspurger, S.; Cobden, P.D.; Safonova, O.V.; Wu, Y.; Anthony, E.J. High CO2 storage capacity in alkali-promoted hydrotalcite-based material: In situ detection of reversible formation of magnesium carbonate. Chemistry 2010, 16, 12694–12700. [Google Scholar] [CrossRef]
- Meis, N.N.A.H.; Bitter, J.H.; de Jong, K.P. On the Influence and Role of Alkali Metals on Supported and Unsupported Activated Hydrotalcites for CO2 Sorption. Ind. Eng. Chem. Res. 2010, 49, 8086–8093. [Google Scholar] [CrossRef]
- Hutson, N.D.; Attwood, B.C. High temperature adsorption of CO2 on various hydrotalcite-like compounds. Adsorption 2008, 14, 781–789. [Google Scholar] [CrossRef]
- Ebner, A.D.; Reynolds, S.P.; Ritter, J.A. Nonequilibrium Kinetic Model That Describes the Reversible Adsorption and Desorption Behavior of CO2 in a K-Promoted Hydrotalcite-like Compound. Ind. Eng. Chem. Res. 2007, 46, 1737–1744. [Google Scholar] [CrossRef]
- Coenen, K.; Gallucci, F.; Cobden, P.; van Dijk, E.; Hensen, E.; van Sint Annaland, M. Chemisorption working capacity and kinetics of CO2 and H2O of hydrotalcite-based adsorbents for sorption-enhanced water-gas-shift applications. Chem. Eng. J. 2016, 293, 9–23. [Google Scholar] [CrossRef]
- Boon, J.; Cobden, P.D.; van Dijk, H.; Hoogland, C.; van Selow, E.R.; van Sint Annaland, M. Isotherm model for high-temperature, high-pressure adsorption of and on K-promoted hydrotalcite. Chem. Eng. J. 2014, 248, 406–414. [Google Scholar] [CrossRef]
- Lee, K.B.; Verdooren, A.; Caram, H.S.; Sircar, S. Chemisorption of carbon dioxide on potassium-carbonate-promoted hydrotalcite. J. Colloid Interface Sci. 2007, 308, 30–39. [Google Scholar] [CrossRef]
- Ram Reddy, M.K.; Xu, Z.P.; Diniz da Costa, J.C. Influence of Water on High-Temperature CO2 Capture Using Layered Double Hydroxide Derivatives. Ind. Eng. Chem. Res. 2008, 47, 2630–2635. [Google Scholar] [CrossRef]
- Soria, M.A.; Rocha, C.; Tosti, S.; Mendes, A.; Madeira, L.M. COx free hydrogen production through water-gas shift reaction in different hybrid multifunctional reactors. Chem. Eng. J. 2019, 356, 727–736. [Google Scholar] [CrossRef]
- Coenen, K.; Gallucci, F.; Pio, G.; Cobden, P.; van Dijk, E.; Hensen, E.; van Sint Annaland, M. On the influence of steam on the CO2 chemisorption capacity of a hydrotalcite-based adsorbent for SEWGS applications. Chem. Eng. J. 2017, 314, 554–569. [Google Scholar] [CrossRef]
- Coenen, K.; Gallucci, F.; Hensen, E.; van Sint Annaland, M. CO2 and H2O chemisorption mechanism on different potassium-promoted sorbents for SEWGS processes. J. CO2 Util. 2018, 25, 180–193. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Li, P.; Yu, J.-G.; Cunha, A.F.; Rodrigues, A.E. K-Promoted Hydrotalcites for CO2 Capture in Sorption Enhanced Reactions. Chem. Eng. Technol. 2013, 36, 567–574. [Google Scholar] [CrossRef]
- Jang, H.M.; Kang, W.R.; Lee, K.B. Sorption-enhanced water gas shift reaction using multi-section column for high-purity hydrogen production. Int. J. Hydrog. Energy 2013, 38, 6065–6071. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Calcination/°C | K-Impregnation/wt% | Calcination/°C |
|---|---|---|---|
| MG70 | - | - | - |
| MG70-250 | 250 | - | - |
| MG70-400 | 400 | - | - |
| MG70-500 | 500 | - | - |
| MG70-K-400 | - | 20 | 400 |
| MG70-400-K | 400 | 20 | - |
| MG70-400-K-400 | 400 | 20 | 400 |
| Temperature | 250 | |
|---|---|---|
| Adsorption time | /min | 15 |
| Adsorption pressure | /bar | 3, 4, 8 * |
| Total adsorption volume flow (STP) | /mL/min | 2000 |
| Adsorption volume fraction CO | /% | 2.5 *, 5, 10, 20 |
| Adsorption volume fraction H2O | /% | 10 *, 20, 40 |
| Desorption time | /min | 15, 20, 30, 40 ** |
| Desorption pressure | /bar | 1 |
| Total desorption volume flow (STP) | /mL/min | 1000 |
| Desorption volume fraction H2O | /% | 20, 40 **, 60 |
| Mode A | Mode B | Mode C | Mode D | Mode E | ||
|---|---|---|---|---|---|---|
| Catalyst mass | /g | 0.58 | 0.58 | 0.58 | 1.22 | 1.22 |
| Sorbent mass | /g | 11.0 | 9.7 | 11.0 | 9.7 | 9.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stadler, T.J.; Barbig, P.; Kiehl, J.; Schulz, R.; Klövekorn, T.; Pfeifer, P. Sorption-Enhanced Water-Gas Shift Reaction for Synthesis Gas Production from Pure CO: Investigation of Sorption Parameters and Reactor Configurations. Energies 2021, 14, 355. https://doi.org/10.3390/en14020355
Stadler TJ, Barbig P, Kiehl J, Schulz R, Klövekorn T, Pfeifer P. Sorption-Enhanced Water-Gas Shift Reaction for Synthesis Gas Production from Pure CO: Investigation of Sorption Parameters and Reactor Configurations. Energies. 2021; 14(2):355. https://doi.org/10.3390/en14020355
Chicago/Turabian StyleStadler, Tabea J., Philipp Barbig, Julian Kiehl, Rafael Schulz, Thomas Klövekorn, and Peter Pfeifer. 2021. "Sorption-Enhanced Water-Gas Shift Reaction for Synthesis Gas Production from Pure CO: Investigation of Sorption Parameters and Reactor Configurations" Energies 14, no. 2: 355. https://doi.org/10.3390/en14020355
APA StyleStadler, T. J., Barbig, P., Kiehl, J., Schulz, R., Klövekorn, T., & Pfeifer, P. (2021). Sorption-Enhanced Water-Gas Shift Reaction for Synthesis Gas Production from Pure CO: Investigation of Sorption Parameters and Reactor Configurations. Energies, 14(2), 355. https://doi.org/10.3390/en14020355