Abstract
Maize forms the basis of Mexican food. As a result, approximately six million tons of corncob are produced each year, which represents an environmental issue, as well as a potential feedstock for biogas production. This research aimed to analyze the taxonomic and functional shift in the microbiome of the fermenters using a whole metagenome shotgun approach. Two strategies were used to understand the microbial community at the beginning and the end of anaerobic digestion: (i) phylogenetic analysis to infer the presence and coverage of clade-specific markers to assign taxonomy and (ii) the recovery of the individual genomes from the samples using the binning of the assembled scaffolds. The results showed that anaerobic digestion brought some noticeable changes and the main microbial community was composed of Corynebacterium variable, Desulfovibrio desulfuricans, Vibrio furnissii, Shewanella spp., Actinoplanes spp., Pseudoxanthomonas spp., Saccharomonospora azurea, Agromyces spp., Serinicoccus spp., Cellulomonas spp., Pseudonocardia spp., Rhodococcus rhodochrous, Sphingobacterium spp. Methanosarcina mazei, Methanoculleus hydrogenitrophicus, Methanosphaerula spp., Methanoregula spp., Methanosaeta spp. and Methanospirillum spp. This study provides evidence of the drastic change in the microbial community structure in a short time and the functional strategy that the most representative microorganisms of the consortia used to carry out the process.
1. Introduction
Fossil fuels have played an essential role in the development of the world’s economies. However, the use of fossil fuels also has negative impacts, such as releases of carbon dioxide, methane, and nitrous oxide gases into the atmosphere [1]. All of these are harmful gases, responsible for climate change induced by global warming [2]. As a consequence of climate change, the polar ice shields are melting, the sea is rising, and extreme weather events promote the loss of crops around the world [3,4]. Therefore, a global energy transition is needed to enhance the use of renewable energies, such as that to biomass [5,6,7].
The production of biogas from the biomass of wastewater, peels, or grains and starch crops, such as corncob is a cost-effective process to generate energy and to reduce greenhouse gas emissions [8]. Biogas is promising clean energy that has some advantages over other technologies, can use the infrastructure of natural gas, use byproducts, generate organic fertilizer, and could be implemented either on small or large scales [9,10]. In order to improve biogas production, biomass is treated with several pre-treatment methods that can be chemical, physical, or biological [11,12,13]. The alkaline organosolv pre-treatment is well used in bioethanol production [14]. However, due to promising results in the increase in the hemicellulose fraction from corncob and enhanced biogas production [15], it is important to understand the microbial community involved in the process in order to improve our control and management of the process.
The biogas generated through the anaerobic digestion process involves four stages: hydrolysis, acidogenesis, acetogenesis, and methanogenesis. The microbial community related to the anaerobic process is particular in every step, and each has its specific role in the bioprocess [16]. Hydrolysis is full of bacteria, such as Syntrophomonas, Clostridium, Desulfovibrio, Sarcina and Selenomonas, which are able to break down lipids, proteins, and carbohydrates to generate simpler components [17]. During the acidogenesis and acetogenesis stages, bacteria such as Syntrophomonas, Pseudomonas, Bacillus, Clostridium and Micrococcus, take the end products of the first stage and transform them into acetate, hydrogen, carbon dioxide, formate, propionate, butyrate, and valerate. The last step is carrying out by methanogens such as Methanosarcina, Methanosaeta, Methanoregula and Methanobrevibacter, which consume the end products from the acidogenesis and acetogenesis to generate methane and carbon dioxide [16].
As noted, some microorganisms related to the bioprocess have been elucidated. However, each initial inoculum and biomass could cause unique variations in the microbial community, which will affect methane production. Furthermore, the process could be enriched with a specific genus due to the biomass used or to the variations of the physicochemical parameters [18]. Next-generation sequencing, such as the whole metagenome shotgun (WMS) approach, allows an understanding of the community’s diversity without the limitation of numerous unculturable microorganisms [19]. WMS provides a higher resolution of the microbial taxonomic profile because their approach makes the taxonomic assignation with more deoxyribonucleic acid (DNA) fragments than the amplicon technique, which uses a single marker gene and avoids other amplicon sequencing limitations as chimeric reads [20] or incorrect taxonomic association and abundance, as a result of the inefficiency of the selected primers [21]. Furthermore, using WMS, it is possible to explore the biological functions encoded in the genomes, predict the samples’ metabolic potential [19,22] and understand the biochemical interactions of the microbial population with the surrounding environment [19,22]. The metabolic potential is predicted by identifying the genes corresponding to specific metabolic pathways; however, this prediction does not prove activity or function [23]. Since 2017, 530 articles have been published in Science with the words metagenomics and biogas found in them and less than 1% of these research articles use shotgun technology to understand the microbial community. Research using WMS evidenced the microbial community’s dynamicity inside fermenters associated with the chemical changes [21]. The species of archaea found in a co-digestion experiment included Methanosarcina mazei, Methanoculleus marisnigri, Methanocorpusculum labreanum, and Methanothermobacter marburgensis [24]. Furthermore, the genes present in the anaerobic digestion associated with novel functional metabolic pathways were explored [25].
Knowledge about the ecological roles and microbial trophic networks and functions inside fermenters are still scarce [26,27,28]. A deep understanding of the microorganisms that carry out the process will help to improve the biogas generation, and consequently, the technology will be more attractive to use. The NGS has led to improve knowledge about the anaerobic digestion of different substrates [28,29,30]. In this case, interest has been shown in the corncob hydrolysate obtained in the organosolv pre-treatment because it is a highly digestible carbohydrate with previously reported efficiency in biogas production [15]. Therefore, this research aimed to observe the taxonomic and functional shift in the microbiome of the fermenters at the end of the anaerobic digestion of the hydrolysate organosolv corncob, using next-generation sequencing (NGS).
2. Materials and Methods
2.1. Substrate Sample and Characterization
Corncob was collected from agricultural land in Zapopan (Jalisco, Mexico). In order for the organosolv pre-treatment to be more effective, the corncobs were allowed to dry at room temperature (25 °C), to reduce humidity, and cut into smaller pieces. The size of the corncob is reduced to increase the surface area and for the organic solvents to better penetrate and separate the components of this raw material [14]. A particle size of 2 mm was used because it is recommended by other authors [31]. The corncob was characterized using the Van Soest fiber method, to obtain the fraction of cellulose, hemicellulose, and lignin [32].
2.2. Alkaline Organosolv Pre-Treatment
The procedure of the alkaline organosolv pre-treatment of the corncob was carried out in a high-pressure reactor. The dried corncob residue (400 g) was placed in the reactor with 1.9 L of water, 2 L of ethanol, and 200 mL of acetic acid for 2.5 h at 175 °C. When the reaction was complete and the reactor was cooled down to ambient temperature, the mixture in the reactor was poured out. Later, the mixture was filtered to separate the solid residue from the liquid phase. The remaining residue was dried overnight in an oven at 100 °C, before weighing [15].
2.3. Batch Anaerobic Digestion Experiments
The inoculum for the anaerobic digestion was taken from cow manure, mixed with soil and water in equal proportions, and then 10 g VSS/L was taken to inoculate the bioreactors. The inoculum was evaluated for organic composition parameters, such as the total solids (TS), volatile suspended solids (VSS) and pH [23]. Batch anaerobic digestion experiments were carried out in three bottles with a liquid volume of 300 mL, at a concentration of 10 g chemical oxygen demand (COD)/L for 15 days. The pH of the hydrolysate was adjusted to 7.5 before use and the batch reactor was flushed with nitrogen gas to guarantee the anaerobic conditions. All experiments were performed at 35 °C with constant agitation and in triplicate. Additionally, a control test without the addition of corncob hydrolysate was carried out. To analyze the microbial community changes at the end of the anaerobic digestion, the samples were taken from the batch reactor at the beginning of the process (inoculum plus hydrolysate) and at the end of the anaerobic digestion (microbial community plus digestate).
The accumulated biogas production was measured by the liquid displacement method of an alkaline solution (NaOH) for 15 days (Figure 1). The amount of biogas produced per unit of measurement of mL per day (mL/d) per unit of raw material in biogas, normalized in mL per gram of volatile solid (NmL/g VS), was quantified to determine the effectiveness of the anaerobic digestion process. The biogas samples were analyzed with an Agilent Technologies 6820 GC gas chromatograph, with a DB-35 capillary column, to determine the type and percentage of biogas generated.
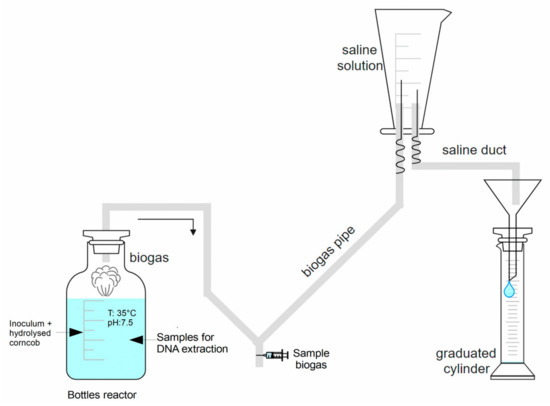
Figure 1.
Schematic diagram of the batch anaerobic digestion experiments.
2.4. Hydrolysate and Digestate Characterization
The hydrolysate and digestate were characterized in terms of: (i) total sugars by the colorimetric method of Dubois [33]; (ii) inhibitory compounds, such as furfural, by high performance liquid chromatography (HPLC) [34]; (iii) chemical oxygen demand (COD) by the colorimetric method according to Standard Methods 5220D [35]; and (iv) volatile fatty acids (VFAs) such as acetate, propionate, and butyrate by HPLC [23].
2.5. DNA Extraction and Sequencing
Samples were taken from all three bioreactors after carefully mixing and pooling them to have a composite sample and perform DNA extraction. DNA was extracted from the pooled sample using a DNeasy PowerSoil Kit (QIAGEN, Hilden, Germany). After, an Illumina library was prepared from total DNA using the TruSeq DNA PCR-Free (Illumina, Inc., San Diego, CA, USA) to obtain an average fragments size of 500 bp. NextSeq500 (Illumina, Inc., San Diego, CA, USA) was used for the sequencing of the samples with a 150 cycle configuration, generating paired-end reads with a length of 75 bp.
2.6. Bioinformatics Analysis
First, a quality control analysis using a quality control tool for high throughput sequence data (FastQC) program was carried out [36]; low-quality sequences were removed using an in-house Perl script. The phylogenetic analysis was carried out by metagenomics phylogenetic analysis (MetaPhlAn) [37]. The graphical representation of phylogenetic data was made with high-quality circular representations of taxonomic and phylogenetic trees (GraPhlan) [38]. Megahit v1.1.3 was used to assemble the raw reads [39] and the gene prediction and annotation used were Prodigal and DIAMOND [40], respectively. All samples were binned separately, using MaxBin2 [41] and MetaBAT [42], and the predictions were combined using DAS Tool [43].
The Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to search the proteins identified in the samples. The orthology numbers (KO) were set up in the KEGG Mapper website to identify the related pathways. The raw reads from the Whole Metagenome Sequencing were deposited at the National Center for Biotechnology Information database under the BioProject number PRJNA602505.
2.7. Statistical Analysis
All experiments were carried out in triplicate and the statistical package used was Statgraphics Centurion XVII software. A significance level of p = 0.05 was used for all statistical tests.
3. Results and Discussion
3.1. Anaerobic Digester Performance of Corncob
The corncob used has the following cellulosic fractions: 45% cellulose, 34% hemicellulose, 16% lignin, and 5% extractives. In contrast, the structure of the pretreated corncob had 22% cellulose, 72% hemicellulose, 5% lignin, and 2% extractives. The pre-treatment successfully gained a higher fraction of hemicellulose, a polymer consisting of monomers, including glucose, mannose, xylose, and arabinose. The hydrolysate has a higher concentration of total sugars ~158 g/L compared to the levels obtained using acid or enzymatic pre-treatments, which reached around 20 and 120 g/L, respectively [23,44]. This was followed by monosaccharides: glucose (6.23 g/L), xylose (4.12 g/L), galactose (2.42 g/L), mannose (1.66 g/L) (Table 1). Nonetheless, there is a presence of acetic (1.28 g/L), propionic (0.29 g/L), butyric (0.41 g/L), and furfural (0.02 g/L). If these compounds were not at such low concentrations, they would inhibit the anaerobic digestion [23,45].

Table 1.
Chemical composition of hydrolysate and digestate.
The anaerobic digestion of the hydrolysate was evaluated in triplicated for 15 days, until the methane production was exhausted. The methane production was 483 ± 12 NmLCH4/gVS. The digestate showed 85% of COD removal and 1.35 ± 0.22 g/L of the total residual sugars, as well as the following monosaccharides: glucose (1.66 ± 0.45 g/L), xylose (1.12 ± 0.19 g/L), and mannose (0.38 ± 0.051 g/L). Regarding volatile fatty acids, there was an increase in all of them and a decrease in the quantity of furfural (0.01 ± 0.006 g/L) (Table 1). The alkaline organosolv pre-treatment showed a higher methane production, compared to the control test without organosolv pre-treatment (120 ± 8 NmLCH4/g VS). The composition of the biogas produced with the anaerobic digestion of the hydrolysate was 72.80% of CH4, 26.18% of CO2 and 1.02% other gases. This confirms the results previously reported by the authors [15]. The higher level of hemicellulose may have been a stimulus to the microorganisms, due to more available carbon and energy sources [46].
3.2. The Microbial Community at the Begging and after the Anaerobic Digestion of Corn Cob Hydrolysate
The first strategy to understand the microbial community that carried out the anaerobic digestion was metagenomic phylogenetic analysis (MetaPhlAn), a method for characterizing whole metagenome shotgun samples. This computational package infers the presence and reads the coverage of clade-specific markers to assign taxonomy (~184 markers for each microorganism) [47]. The phylogenetic analysis carried out by MetaPhlAn assigned 115 different genera (Figure 2).
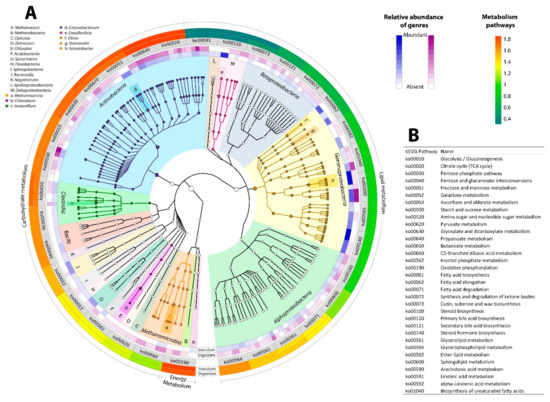
Figure 2.
Taxonomic profile: inside the circle is the inoculum microbial community, in blue, and the digestate microbial community, is purple. The intensity of the colors is related to the relative abundance of the genera listed in Table (A). The outer ring showed the abundance of the principal metabolic pathways predicted by Kyoto Encyclopedia of Genes and Genomes (KEGG) and listed in Table (B).
The second strategy for understanding the microbial community was recovering individual genomes from the samples using the assembled scaffolds’ binning. However, only genomes with a high abundance were binned, with high coverage levels of 90–99%, as shown in Table 2. Reconstructed complete genomes of individual microbes in the community allowed us to understand their individual contribution and their ecological functions in specific metabolic pathways, as well as reconstructing the community interaction networks [30,48]. The results of the individual genomes reconstruction were consistent with the data obtained using MethaPhlan2 (Table 2).
Table 2.
The genus recovered from the samples using the binning of the assembled scaffolds.
The microbial community’s composition at the beginning of the process represents the environment that it comes from, as has been demonstrated by several kinds of research [18,41,49]. Furthermore, the inoculum represents the metabolic capacity to develop the process [50]. The leading bacterial community of the inoculum was composed of the followed species: Streptomyces spp., Acinetobacter spp., Klebsiella oxytoca, Desulfovibrio desulfuricans, Klebsiella pneumoniae, Vibrio furnissii, Shewanella spp., Enterobacter cloacae, Pseudomonas stutzeri, Corynebacterium variable, Acinetobacter venetianus, Saccharomonospora azurea, Escherichia spp., Methanosarcina mazei and Methanoculleus hydrogenitrophicus (Figure 2).
As can be seen, the inoculum microbial community was mainly composed of Proteobacteria, Bacteroidetes and Actinobacteria, as has been previously reported in inoculum from manure [24], a wastewater treatment plant [51] and a mesophilic anaerobic digester plant [52]. Proteobacteria have ecologically relevant groups, capable of participating in the hydrolysis and acidogenesis/acetogenesis stage due to their metabolic capacities [29]. Members of Bacteroidetes are actively involved in the degradation of lignocellulosic biomass residues [53] and Actinobacteria, represented by genera such as Corynebacterium or Propionimicrobium are microorganisms with a heterogenic metabolic capacities which make them suitable to participate in all stages of anaerobic digestion [13,54].
The microbial community structure is determined by physicochemical parameters such as temperature, pH, and levels of fatty acids [50,55,56]. At the end of the process, the community was enriched with Corynebacterium, a variable, facultative anaerobic bacteria, which plays a vital role in the decomposition of organic material, such as cellulose, fats, and organic acids [57,58]. Other representative species founded were: Desulfovibrio desulfuricans, Vibrio furnissii, Shewanella spp., Actinoplanes spp., Pseudoxanthomonas spp., Saccharomonospora azurea, Agromyces spp., Serinicoccus spp., Cellulomonas spp., Pseudonocardia spp., Rhodococcus rhodochrous and Sphingobacterium spp. With regard to the enriched archaea community, this mainly comprised Methanosarcina mazei, followed by Methanoculleus hydrogenitrophicus, Methanosphaerula spp., Methanoregula spp., Methanosaeta spp. and Methanospirillum spp. Many studies have reported the abundance of Methanosarcina mazei in biogas production [13] as a result of their ability to use hydrogen, acetate, methanol, and any methylated C1 compounds for methanogenic metabolism [59]. Methanoculleus hydrogenitrophicus, Methanoregula spp., and Methanosphaerula spp. are hydrogenotrophic methanogens that are plentiful in several biogas reactors [28,60,61]. Methanosaeta spp. is an acetoclastic methanogen that is able to use a low concentration of acetate 7–70 µM for growth [13,62]. These archaea sets show a community structure with hydrogenotrophic members. Maintaining very low H2 levels promotes methane production and diverse acetoclastic archaea to carry out acetate oxidation, formed from other VFAs (such as propionate and butyrate), which keeps the process balanced [62,63]. The archaea structure, with a high abundance of Methanosarcina mazei, followed by Methanoculleus hydrogenitrophicus, and Methanosphaerula spp. were in line with the results showed by Hassa et al., 2018 [64] and were associated with the dominance of Methanosarcina over Methanoculleus, due the resilience of Methanosarcina to detrimental conditions and fluctuations of the bioprocess.
3.3. The Community Interaction Networks and Their Potential Functional Profile
The functional annotation results were summarized in Figure 2 (outer ring). The focus on pathways and lipid abundance, carbohydrate, and energy metabolism was predicted by KEGG because these gene profiles are related to cellular functions relevant to the bioprocess [50,65]. There are differences in the lipid metabolism associated with an over-representation of the glycerolipid metabolism during the anaerobic digestion, due to an increase in energy production and the need for glycerol 3-phosphate as an intermediate for the carbohydrate and lipid metabolic pathways [66]. Carbohydrate metabolism was over-represented during the anaerobic digestion, mainly in the fructose/mannose metabolism, where fructose from the hydrolysate biomass and the fructose-6-phosphate from the glycolysis were transformed to mannose, to be used as a carbon and energy source. Furthermore, at the end of the process, it was found that more genes-encoding enzymes were involved in pyruvate metabolism, an essential compound during the acidogenesis/acetogenesis stage. These genes associated pyruvate to carbohydrates via gluconeogenesis, to fatty acids such as formate, and to the pyruvic acid transformation to acetyl-CoA, by the enzyme pyruvate-ferredoxin oxidoreductase to produces acetate and adenosine triphosphate (ATP) [67].
The last over-represented pathway of carbohydrate metabolism was the butyrate metabolism, related to butyric acid production, the main fatty acid founded in some batch anaerobic digestion systems, and associated with the equilibrium of the system due to the H2 levels [68]. The result of the butyrate metabolism is in line with the high levels of butyric acid found in the chemical analysis of the digestate (Table 1). With regard to energy metabolism, which includes oxidative phosphorylation, methane, nitrogen, sulfur, photosynthesis, and the carbon fixation metabolism, we examined every pathway separately and only focused on the methane, nitrogen, and sulfur metabolism. Consistent with the statistical analysis (X2 (3, N = 200) = 5′9915, p = 0.05), the results showed the influence of anaerobic digestion over the genes founded in methane, nitrogen and sulfur metabolism. As was expected, the methane metabolism pathway showed a greater abundance of genes related to methanogenesis compared to the abundance of genes founded in inoculum.
Whereas nitrogen and sulfur metabolism pathways were under represented during the anaerobic digestion, due to a combination of two factors, first the levels of ammonia, nitrite and nitrate and sulfur and second the microbial community were not enriched in microorganisms that carry out the complete pathways of nitrogen and sulfur metabolism [63,67,69]. The key enzymes in anaerobic digestion were selected for screening, to determine the presence of those enzymes and correlate them with the microorganisms enriched during the process (Table 3). In the first stage, the microbial community began to degrade the biomass using the Cellobiose phosphorylase, which catalyzed cellulose degradation into α-D-glucose 1-phosphate and D-glucose [70]. Vibrio furnissii, Cellulomonas spp., Corynebacterium variable, and Actinoplanes spp. are able to produce this enzyme (Figure 3) [17,71].
Table 3.
Key enzymes related to the metabolic pathways of each stage of anaerobic digestion.
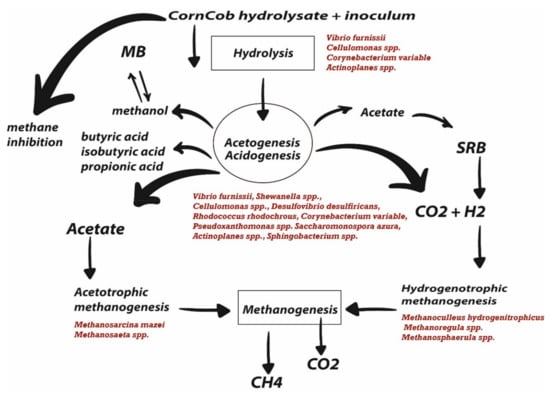
Figure 3.
General diagram of anaerobic digestion. The principal microorganisms which participate in every stage are in red.
Furthermore, the results showed the specific functional potential of each genus. For example, Corynebacterium variable, Desulfovibrio desulfuricans, Vibrio furnissii, Sphingobium spp., Rhodococcus rhodochrous, Actinoplanes spp., and Pseudoxanthomonas spp., were very active during hydrolysis with enzymes which linked them with several pathways, such as pentose phosphate (glucose 1-dehydrogenase), and fructose/mannose metabolism (mannose-6-phosphate isomerase, GDP-mannose 6-dehydrogenase, fructokinase). Vibrio furnissii, Shewanella spp., Cellulomonas spp., Desulfovibrio desulfiricans, Rhodococcus rhodochrous, Corynebacterium variable, Pseudoxanthomonas spp. Saccharomonospora azura, Actinoplanes spp., and Sphingobacterium spp. participate in starch and sucrose metabolism (glucokinase). As can be seen, there are species that only participate in a specific metabolic pathway, such as Sphingobacterium spp. However, some microorganisms participate in more than one pathway, and stages such as Corynebacterium variable. With regard to acidogenesis; the critical step is the pyruvate formation from carbohydrates, the enzyme selected to explore this step is the pyruvate kinase and the microorganism with genes related to this enzyme were Vibrio furnissii, Shewanella spp., Desulfovibrio desulfiricans, Corynebacterium variable, Rhodococcus rhodochrous, Agromyces spp., Cellulomonas spp., Saccharomonospora azura, Pseudonocardia spp., and Actinoplanes spp. Furthermore, to explore the pyruvate transformation toward the short-chain fatty acids, the enzyme selected was malate dehydrogenase, where the same microorganisms were observed as for pyruvate kinase except for Desulfovibrio desulfiricans.
Acetogenesis was explored using the acetate metabolism via reactions in the citric acid cycle with the enzyme Succinyl-CoA:acetate, CoA-transferase and the microorganism related to this enzyme were Corynebacterium variable, Rhodococcus rhodochrous, and Sphingobacterium spp. In addition, the oxidation of acetate can be the result of the interaction between syntrophic acetate-oxidizing bacteria (SAOB), which oxidize acetate to produce H2/CO2 that are utilized by hydrogenotrophic methanogens which use the H2 to reduce CO2 to CH4 [72]. The marker enzymes for acetogenesis are dependent on syntrophic relations and were represented by Formyltetrahydrofolate synthetase; the microorganism with genes related to this enzyme were Vibrio furnissii and Shewanella spp.
The methanogenesis stage showed enzymes that were not present before the anaerobic digestion, such as the tetrahydromethanopterin hydro-lyase, mainly found in Methanosarcina [73]. Another key enzyme of the methanogenic process was the anaerobic carbon monoxide dehydrogenase, which is unique to archaea species and allows using carbon monoxide as a single carbon and energy-producing H2 and CO2 as well as formylmethanofuran—tetrahydromethanopterin N-formyltransferase and the NAD-reducing hydrogenase, which are enzymes that catalyze the reduction of CO2 to methane, depending on the levels of H2 in a bioreactor [74,75].
Finally, the next-generation sequencing technologies could revolutionize the diagnosis of anaerobic digestion processes. The current biogas diagnosis analyzes the methane rates, the biogas’ quality or the byproducts generated [51,65]. However, by monitoring over time, the bioreactor’s microbial community could predict some undesirable results and determine the appropriate action to be taken. For example, Methyloversatilis (0.81%) was detected at the end of our anaerobic digestion: this genus had been reported in methanol-fed heterotrophic systems as being an undesirable genus because it could reduce the rate of methane generated [76]. This finding makes sense due to the alkaline organosolv pre-treatment, which uses an organic solvent with water for lignin removal, and perhaps, these genera were enriched due to the abundance of the substrate. The solvent must be removed before fermentation. However, the remaining solvent could adhere to biomass and promote the abundance of Methyloversatilis. If the relative abundance of Methyloversatilis was more abundant, we should revise the solvent method’s removal to improve the process.
Furthermore, it is necessary to continue studying the functional potential and understand the metabolism of the microorganisms which carried out the bioprocess, to target genes, which could be changed by genetic engineering and to understand what enzymes are over represented using other substrates and what that means. It is also necessary to find maker microorganisms for the optimization and standardization of the process. The information generated during this research, such as the microbial profile at the end of the anaerobic digestion and the identified marker enzymes, will allow us to explore the possibility of generating synthetic inoculums and follow the process with the marker enzymes to improve the methane generation using corncob in future projects.
4. Conclusions
The pre-treated corncob improved the anaerobic digestion and reached a higher methane production with low concentrations of byproducts compared with the corncob without pre-treatment. The alkaline organosolv pre-treatment successfully gained a higher fraction of hemicellulose that acts as the stimulus to microorganisms during the bioprocess.
This study provides evidence of the drastic change in the microbial community structure in a short time and the functional strategy used by the most representative microorganism of the consortia to carry out the process. Gammaproteobacteria dominated the inoculum and after the anaerobic digestion, the microbial community was composed mainly of Proteobacteria, Bacteroidetes and Actinobacteria.
The functional profile allows us to support the taxonomic findings related to bioprocess and identify the main microorganism involved in the corncob digestion, such as Vibrio furnissii, Shewanella spp., Cellulomonas spp., Desulfovibrio desulfiricans, Rhodococcus rhodochrous, Corynebacterium variable, Pseudoxanthomonas spp. Saccharomonospora azura, Actinoplanes spp., and Sphingobacterium spp. Furthermore, the results allow the selection of marker enzymes for each stage of the anaerobic digestion and identify the microorganism with the genes related with those enzymes.
The metabolisms with over-representation were the lipid metabolism associated with the glycolipid pathways, the carbohydrate metabolism in the fructose/mannose pathways and the energy metabolism in the methanogenesis pathways, while the nitrogen and sulfur metabolism pathways were under represented.
Author Contributions
Methodology and experimental work: L.B.-D., J.S.A.A. and I.S.-P.; writing—review and editing: L.B.-D., K.J.G.T. and B.S.-R.; writing—original draft preparation: L.B.-D.; conceptualization and supervision: L.B.-D., K.J.G.T. and B.S.-R., funding acquisition: K.J.G.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Council of Science and Technology México (CONACyT) project number 258960 Basic Science.
Acknowledgments
The authors are grateful for the financial support of student grants from CONACyT. LB-D thank CONACYT and their program CATEDRAS for supporting the Project 285. We also like to thank Unidad Universitaria de Secuenciación Masiva y Bioinformática (UUSMB) of the Universidad Nacional Autónoma de México (UNAM) for the analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gielen, D.; Boshell, F.; Saygin, D.; Bazilian, M.D.; Wagner, N.; Gorini, R. The role of renewable energy in the global energy transformation. Energy Strat. Rev. 2019, 24, 38–50. [Google Scholar] [CrossRef]
- Meunier, F. The greenhouse effect: A new source of energy. Appl. Therm. Eng. 2007, 27, 658–664. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Broughton, K.; Osanai, Y.; Anderson, I.C.; Bange, M.P.; Tissue, D.T.; Singh, B.K. Effects of elevated temperature and elevated CO2 on soil nitrification and ammonia-oxidizing microbial communities in field-grown crop. Sci. Total. Environ. 2019, 675, 81–89. [Google Scholar] [CrossRef]
- Nüsser, M.; Dame, J.; Kraus, B.; Baghel, R.; Schmidt, S. Socio-hydrology of “artificial glaciers” in Ladakh, India: Assessing adaptive strategies in a changing cryosphere. Reg. Environ. Chang. 2018, 19, 1327–1337. [Google Scholar] [CrossRef]
- Carrillo-Reyes, J.; Buitrón, G. Biohydrogen and methane production via a two-step process using an acid pretreated native microalgae consortium. Bioresour. Technol. 2016, 221, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Moraes, B.; Triolo, J.; Lecona, V.; Zaiat, M.; Sommer, S. Biogas production within the bioethanol production chain: Use of co-substrates for anaerobic digestion of sugar beet vinasse. Bioresour. Technol. 2015, 190, 227–234. [Google Scholar] [CrossRef]
- Sivagurunathan, P.; Kumar, G.; Mudhoo, A.; Rene, E.R.; Saratale, G.D.; Kobayashi, T.; Xu, K.; Kim, S.-H.; Kim, D.-H. Fermentative hydrogen production using lignocellulose biomass: An overview of pre-treatment methods, inhibitor effects and detoxification experiences. Renew. Sustain. Energy Rev. 2017, 77, 28–42. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, N.; Garcia-Bernet, D.; Domínguez, J.M. Extrusion and enzymatic hydrolysis as pretreatments on corn cob for biogas production. Renew. Energy 2017, 107, 597–603. [Google Scholar] [CrossRef]
- Aslanzadeh, S.; Rajendran, K.; Jeihanipour, A.; Taherzadeh, M.J. The Effect of Effluent Recirculation in a Semi-Continuous Two-Stage Anaerobic Digestion System. Energies 2013, 6, 2966–2981. [Google Scholar] [CrossRef]
- Kavitha, S.; Banu, J.R.; Kumar, J.V.; Rajkumar, M. Improving the biogas production performance of municipal waste activated sludge via disperser induced microwave disintegration. Bioresour. Technol. 2016, 217, 21–27. [Google Scholar] [CrossRef]
- Amnuaycheewa, P.; Hengaroonprasan, R.; Rattanaporn, K.; Kirdponpattara, S.; Cheenkachorn, K.; Sriariyanun, M. Enhancing enzymatic hydrolysis and biogas production from rice straw by pretreatment with organic acids. Ind. Crop. Prod. 2016, 87, 247–254. [Google Scholar] [CrossRef]
- Bolado, S.; Toquero, C.; Martín-Juárez, J.; Travaini, R.; García-Encina, P.A. Effect of thermal, acid, alkaline and alkaline-peroxide pretreatments on the biochemical methane potential and kinetics of the anaerobic digestion of wheat straw and sugarcane bagasse. Bioresour. Technol. 2016, 201, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, C.; Ai, S.; Wang, H.; Gao, Y.; Yan, L.; Mei, Z.; Wang, W. Biological pretreatment enhances the activity of functional microorganisms and the ability of methanogenesis during anaerobic digestion. Bioresour. Technol. 2019, 290, 121660. [Google Scholar] [CrossRef] [PubMed]
- Romaní, A.; Larramendi, A.; Yáñez, R.; Cancela, Á.; Sánchez, Á.; Teixeira, J.A.; Domingues, L. Valorization of Eucalyptus nitens bark by organosolv pretreatment for the production of advanced biofuels. Ind. Crop. Prod. 2019, 132, 327–335. [Google Scholar] [CrossRef]
- Sulbarán-Rangel, B.; Aguirre, J.S.A.; Breton-Deval, L.; Del Real-Olvera, J.; Gurubel-Tun, K.J. Improvement of Anaerobic Digestion of Hydrolysed Corncob Waste by Organosolv Pretreatment for Biogas Production. Appl. Sci. 2020, 10, 2785. [Google Scholar] [CrossRef]
- Enzmann, F.; Mayer, F.; Rother, M.; Holtmann, D. Methanogens: Biochemical background and biotechnological applications. AMB Express 2018, 8, 1. [Google Scholar] [CrossRef]
- Anderson, K.; Sallis, P.J.; Uyanik, S. Anaerobic Treatment Processes. In Handbook of Water and Waste Water Microbiology; Mara, D., Horan, N., Eds.; Academic Press: London, UK, 2003; Chapter 24; pp. 391–426. [Google Scholar] [CrossRef]
- Wojcieszak, M.; Pyzik, A.; Poszytek, K.; Krawczyk, P.S.; Sobczak, A.; Lipinski, L.; Roubinek, O.; Palige, J.; Sklodowska, A.; Drewniak, L. Adaptation of Methanogenic Inocula to Anaerobic Digestion of Maize Silage. Front. Microbiol. 2017, 8, 1881. [Google Scholar] [CrossRef]
- Techtmann, S.M.; Hazen, T.C. Metagenomic applications in environmental monitoring and bioremediation. J. Ind. Microbiol. Biotechnol. 2016, 43, 1345–1354. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef]
- Treu, L.; Campanaro, S.; Kougias, P.G.; Sartori, C.; Bassani, I.; Angelidaki, I. Hydrogen-Fueled Microbial Pathways in Biogas Upgrading Systems Revealed by Genome-Centric Metagenomics. Front. Microbiol. 2018, 9, 1079. [Google Scholar] [CrossRef] [PubMed]
- González-Toril, E.; Aguilera, Á. Chapter 14—Microbial Ecology in Extreme Acidic Environments: Use of Molecular Tools. In Microbial Diversity in the Genomic Era; Surajit, D., Dash, H.R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 227–238. [Google Scholar]
- Breton-Deval, L.; Méndez-Acosta, H.O.; González-Álvarez, V.; Snell-Castro, R.; Gutiérrez-Sánchez, D.; Arreola-Vargas, J. Agave tequilana bagasse for methane production in batch and sequencing batch reactors: Acid catalyst effect, batch optimization and stability of the semi-continuous process. J. Environ. Manag. 2018, 224, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Tsapekos, P.; Kougias, P.; Treu, L.; Campanaro, S.; Angelidaki, I. Process performance and comparative metagenomic analysis during co-digestion of manure and lignocellulosic biomass for biogas production. Appl. Energy 2017, 185, 126–135. [Google Scholar] [CrossRef]
- Abendroth, C.; Pérez, A.L.; Porcar, M.; Simeonov, C.; Luschnig, O.; Vilanova, C.; Pascual, J. Shedding light on biogas: Phototrophic biofilms in anaerobic digesters hold potential for improved biogas production. Syst. Appl. Microbiol. 2020, 43, 126024. [Google Scholar] [CrossRef] [PubMed]
- Elnaker, N.A.; Elektorowicz, M.; Naddeo, V.; Hasan, S.W.; Yousef, A.F. Assessment of Microbial Community Structure and Function in Serially Passaged Wastewater Electro-Bioreactor Sludge: An Approach to Enhance Sludge Settleability. Sci. Rep. 2018, 8, 7013. [Google Scholar] [CrossRef] [PubMed]
- Liguori, R.; Ventorino, V.; Pepe, O.; Faraco, V. Bioreactors for lignocellulose conversion into fermentable sugars for production of high added value products. Appl. Microbiol. Biotechnol. 2016, 100, 597–611. [Google Scholar] [CrossRef]
- Shin, S.G.; Han, G.; Lee, J.; Shin, J.; Hwang, S. A snapshot of microbial community structures in 20 different field-scale anaerobic bioreactors treating food waste. J. Environ. Manag. 2019, 248, 109297. [Google Scholar] [CrossRef]
- Guo, J.; Peng, Y.; Ni, B.-J.; Han, X.; Fan, L.; Yuan, Z. Dissecting microbial community structure and methane-producing pathways of a full-scale anaerobic reactor digesting activated sludge from wastewater treatment by metagenomic sequencing. Microb. Cell Factories 2015, 14, 33. [Google Scholar] [CrossRef]
- Song, K.; Ren, J.; Sun, F. Reads Binning Improves Alignment-Free Metagenome Comparison. Front. Genet. 2019, 10, 1156. [Google Scholar] [CrossRef]
- Qing, Q.; Zhou, L.; Guo, Q.; Gao, X.; Zhang, Y.; He, Y.; Zhang, Y. Mild alkaline presoaking and organosolv pretreatment of corn stover and their impacts on corn stover composition, structure, and digestibility. Bioresour. Technol. 2017, 233, 284–290. [Google Scholar] [CrossRef]
- Van Soest, P.V.; Robertson, J.B.; Lewis, B.A. Methods for Dietary Fiber, Neutral Detergent Fiber, and Nonstarch Polysaccharides in Relation to Animal Nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Dubois, M.Y.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F.G. A Colorimetric Method for the Determination of Sugars. Nat. Cell Biol. 2006, 168, 167. [Google Scholar] [CrossRef] [PubMed]
- Lins, P.; Malin, C.; Wagner, A.O.; Illmer, P. Reduction of accumulated volatile fatty acids by an acetate-degrading enrichment culture. FEMS Microbiol. Ecol. 2010, 71, 469–478. [Google Scholar] [CrossRef]
- Baird, R.; Eaton, A.; Rice, E. Standard Methods for the Examination of Water and Wastewater, 23 ed.; American Public Health Association: Washington, DC, USA, 2017. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Asnicar, F.; Weingart, G.; Tickle, T.L.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Ecai, M.; Ewilkins, D.; Echen, J.; Eng, S.K.; Elu, H.; Ejia, Y.; Lee, P.K.H. Metagenomic Reconstruction of Key Anaerobic Digestion Pathways in Municipal Sludge and Industrial Wastewater Biogas-Producing Systems. Front. Microbiol. 2016, 7, 778. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef]
- Velázquez-Valadez, U.; Farías-Sánchez, J.C.; Vargas-Santillán, A.; Castro-Montoya, A.J. Tequilana weber Agave Bagasse Enzymatic Hydrolysis for the Production of Fermentable Sugars: Oxidative-Alkaline Pretreatment and Kinetic Modeling. BioEnergy Res. 2016, 9, 998–1004. [Google Scholar] [CrossRef]
- Siegert, I.; Banks, C. The effect of volatile fatty acid additions on the anaerobic digestion of cellulose and glucose in batch reactors. Process Biochem. 2005, 40, 3412–3418. [Google Scholar] [CrossRef]
- Naraian, R.; Gautam, R.L. Chapter 6—Penicillium Enzymes for the Saccharification of Lignocellulosic Feedstocks. In New and Future Developments in Microbial Biotechnology and Bioengineering; Gupta, V.K., Rodriguez-Couto, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 121–136. [Google Scholar]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, D.; Huang, R.; Cao, X.; Zeng, J.; Yu, Z.; Hooker, K.V.; Hambright, K.D.; Wu, Q. Contrasting Network Features between Free-Living and Particle-Attached Bacterial Communities in Taihu Lake. Microb. Ecol. 2018, 76, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Guo, B.; Zhang, L.; Zhang, Y.; Liu, Y. Microbial community dynamics in anaerobic digesters treating conventional and vacuum toilet flushed blackwater. Water Res. 2019, 160, 249–258. [Google Scholar] [CrossRef]
- Tapadia-Maheshwari, S.; Pore, S.; Engineer, A.; Shetty, D.; Dagar, S.S.; Dhakephalkar, P.K. Illustration of the microbial community selected by optimized process and nutritional parameters resulting in enhanced biomethanation of rice straw without thermo-chemical pretreatment. Bioresour. Technol. 2019, 289, 121639. [Google Scholar] [CrossRef]
- Liu, C.; Li, H.; Zhang, Y.; Liu, C. Improve biogas production from low-organic-content sludge through high-solids anaerobic co-digestion with food waste. Bioresour. Technol. 2016, 219, 252–260. [Google Scholar] [CrossRef]
- Abendroth, C.; Vilanova, C.; Günther, T.; Luschnig, O.; Porcar, M. Eubacteria and archaea communities in seven mesophile anaerobic digester plants in Germany. Biotechnol. Biofuels 2015, 8, 87. [Google Scholar] [CrossRef]
- Azman, S.; Khadem, A.F.; Van Lier, J.B.; Zeeman, G.; Plugge, C.M. Presence and Role of Anaerobic Hydrolytic Microbes in Conversion of Lignocellulosic Biomass for Biogas Production. Crit. Rev. Environ. Sci. Technol. 2015, 45, 2523–2564. [Google Scholar] [CrossRef]
- Bae, H.-S.; Moe, W.M.; Yan, J.; Tiago, I.; Da Costa, M.S.; Rainey, F.A. Brooklawnia cerclae gen. nov., sp. nov., a propionate-forming bacterium isolated from chlorosolvent-contaminated groundwater. Int. J. Syst. Evol. Microbiol. 2006, 56, 1977–1983. [Google Scholar] [CrossRef]
- Ziels, R.M.; Karlsson, A.; Beck, D.A.; Ejlertsson, J.; Yekta, S.S.; Bjorn, A.; Stensel, H.D.; Svensson, B. Microbial community adaptation influences long-chain fatty acid conversion during anaerobic codigestion of fats, oils, and grease with municipal sludge. Water Res. 2016, 103, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Shayegan, S.S.; Ng, W.J.; He, J. Development and characteristics of rapidly formed hydrogen-producing granules in an acidic anaerobic sequencing batch reactor (AnSBR). Biochem. Eng. J. 2010, 49, 119–125. [Google Scholar] [CrossRef]
- Alvarez, A.; Saez, J.M.; Costa, J.S.D.; Colin, V.L.; Fuentes, M.S.; Cuozzo, S.A.; Benimeli, C.S.; Polti, M.A.; Amoroso, M.J. Actinobacteria: Current research and perspectives for bioremediation of pesticides and heavy metals. Chemosphere 2017, 166, 41–62. [Google Scholar] [CrossRef]
- Anandan, R.; Dharumadurai, D.; Manogaran, G.P. An Introduction to Actinobacteria. In Actinobacteria—Basics and Biotechnological Applications; IntechOpen: Tamil Nadu, India, 2016. [Google Scholar]
- Welte, C.U.; Deppenmeier, U. Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 1130–1147. [Google Scholar] [CrossRef] [PubMed]
- Bräuer, S.L.; Cadillo-Quiroz, H.; Ward, R.J.; Yavitt, J.B.; Zinder, S.H. Methanoregula boonei gen. nov., sp. nov., an acidiphilic methanogen isolated from an acidic peat bog. Int. J. Syst. Evol. Microbiol. 2011, 61, 45–52. [Google Scholar] [CrossRef]
- Kougias, P.G.; Campanaro, S.; Treu, L.; Zhu, X.; Angelidaki, I. A novel archaeal species belonging to Methanoculleus genus identified via de-novo assembly and metagenomic binning process in biogas reactors. Anaerobe 2017, 46, 23–32. [Google Scholar] [CrossRef]
- Lv, L.; Mbadinga, S.M.; Wang, L.-Y.; Liu, J.-F.; Gu, J.-D.; Mu, B.-Z.; Yang, S.-Z. Acetoclastic methanogenesis is likely the dominant biochemical pathway of palmitate degradation in the presence of sulfate. Appl. Microbiol. Biotechnol. 2015, 99, 7757–7769. [Google Scholar] [CrossRef]
- Tian, G.; Xi, J.; Yeung, M.; Ren, G. Characteristics and mechanisms of H2S production in anaerobic digestion of food waste. Sci. Total. Environ. 2020, 724, 137977. [Google Scholar] [CrossRef]
- Hassa, J.; Maus, I.; Off, S.; Pühler, A.; Scherer, P.; Klocke, M.; Schlüter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef]
- Joo, J.-Y.; Park, C.-H.; Han, G.-B. Optimization of two-phased anaerobic sludge digestion using the pressurized ultra filtration membrane with a mesh screen (MS-PUFM). Chem. Eng. J. 2016, 300, 20–28. [Google Scholar] [CrossRef]
- Doi, Y. Glycerol metabolism and its regulation in lactic acid bacteria. Appl. Microbiol. Biotechnol. 2019, 103, 5079–5093. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, B.; Khanna, N.; Das, D. Chapter 4—Dark-Fermentative Biohydrogen Production, in Biohydrogen, 2nd ed.; Ashok Pandey, S., Mohan, V., Chang, J., Patrick, C., Larroche, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 79–122. [Google Scholar]
- Esquivel-Elizondo, S.; Ilhan, Z.E.; Garcia-Peña, E.I.; Krajmalnik-Brown, R. Insights into Butyrate Production in a Controlled Fermentation System via Gene Predictions. mSystems 2017, 2, e00051-17. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.W.; Trottier, T.M. Effect of sulfur-containing compounds on anaerobic degradation of cellulose to methane by mixed cultures obtained from sewage sludge. Appl. Environ. Microbiol. 1978, 35, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Chomvong, K.; Kordić, V.; Li, X.; Bauer, S.; Gillespie, A.E.; Ha, S.-J.; Oh, E.J.; Galazka, J.; Jin, Y.-S.; Cate, J.H.D. Overcoming inefficient cellobiose fermentation by cellobiose phosphorylase in the presence of xylose. Biotechnol. Biofuels 2014, 7, 85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liaquat, R.; Jamal, A.; Tauseef, I.; Qureshi, Z.; Farooq, U.; Imran, M.; Ali, M. Characterizing Bacterial Consortia from an Anaerobic Digester Treating Organic Waste for Biogas Production. Pol. J. Environ. Stud. 2017, 26, 709–716. [Google Scholar] [CrossRef]
- Schnürer, A.; Zellner, G.; Svensson, B.H. Mesophilic syntrophic acetate oxidation during methane formation in biogas reactors. FEMS Microbiol. Ecol. 1999, 29, 249–261. [Google Scholar] [CrossRef]
- Goenrich, M.; Thauer, R.K.; Yurimoto, H.; Kato, N. Formaldehyde activating enzyme (Fae) and hexulose-6-phosphate synthase (Hps) in Methanosarcina barkeri: A possible function in ribose-5-phosphate biosynthesis. Arch. Microbiol. 2005, 184, 41–48. [Google Scholar] [CrossRef]
- Nölling, J.; Pihl, T.D.; Reeve, J.N. Cloning, sequencing, and growth phase-dependent transcription of the coenzyme F420-dependent N5, N10-methylenetetrahydromethanopterin reductase-encoding genes from Methanobacterium thermoautotrophicum delta H and Methanopyrus kandleri. J. Bacteriol. 1995, 177, 7238–7244. [Google Scholar] [CrossRef]
- Schwartz, E.; Henne, A.; Cramm, R.; Eitinger, T.; Friedrich, B.; Gottschalk, G. Complete Nucleotide Sequence of pHG1: A Ralstonia eutropha H16 Megaplasmid Encoding Key Enzymes of H2-based Lithoautotrophy and Anaerobiosis. J. Mol. Biol. 2003, 332, 369–383. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Hu, C.; Liu, H.; Bai, Y.; Qu, J. Sulfur-based mixotrophic denitrification corresponding to different electron donors and microbial profiling in anoxic fluidized-bed membrane bioreactors. Water Res. 2015, 85, 422–431. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).