Lumped Kinetics for Homogeneous Reactions of n-Hexadecane and n-Decene as Model Compounds for PE Pyrolysis Primary Tars



Abstract
1. Introduction
2. Experimental and Methods
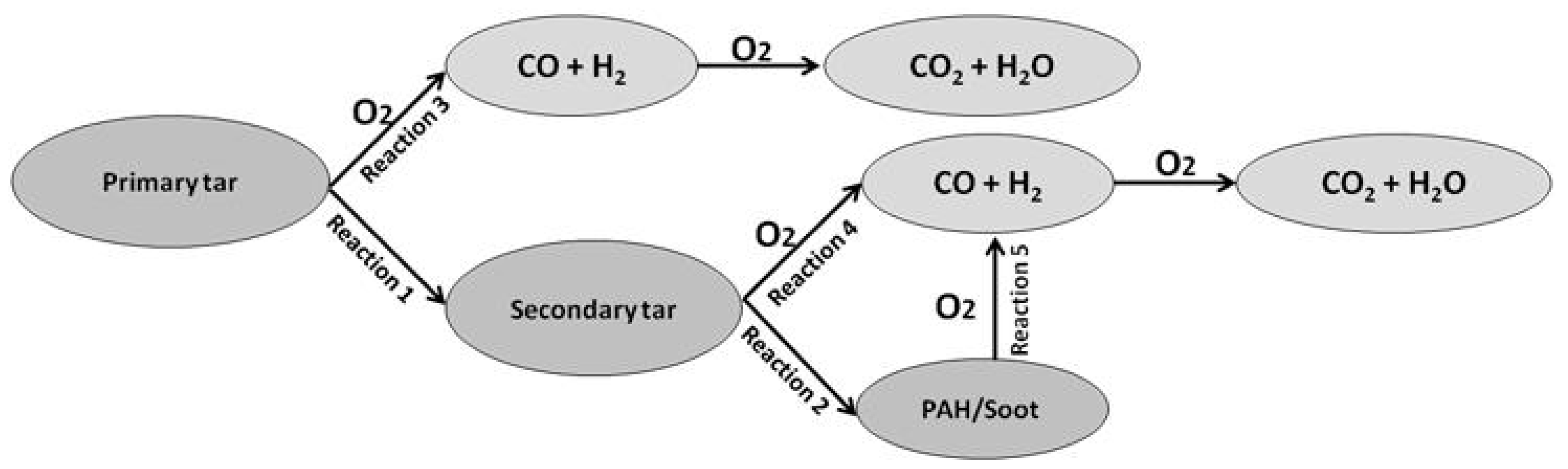
2.1. Computational Experiments
- Reaction of primary tar under inert conditions
- Reaction of secondary tar under inert conditions
- Combustion of primary tar
- Combustion of secondary tar
2.2. Laboratory Experiments
3. Results
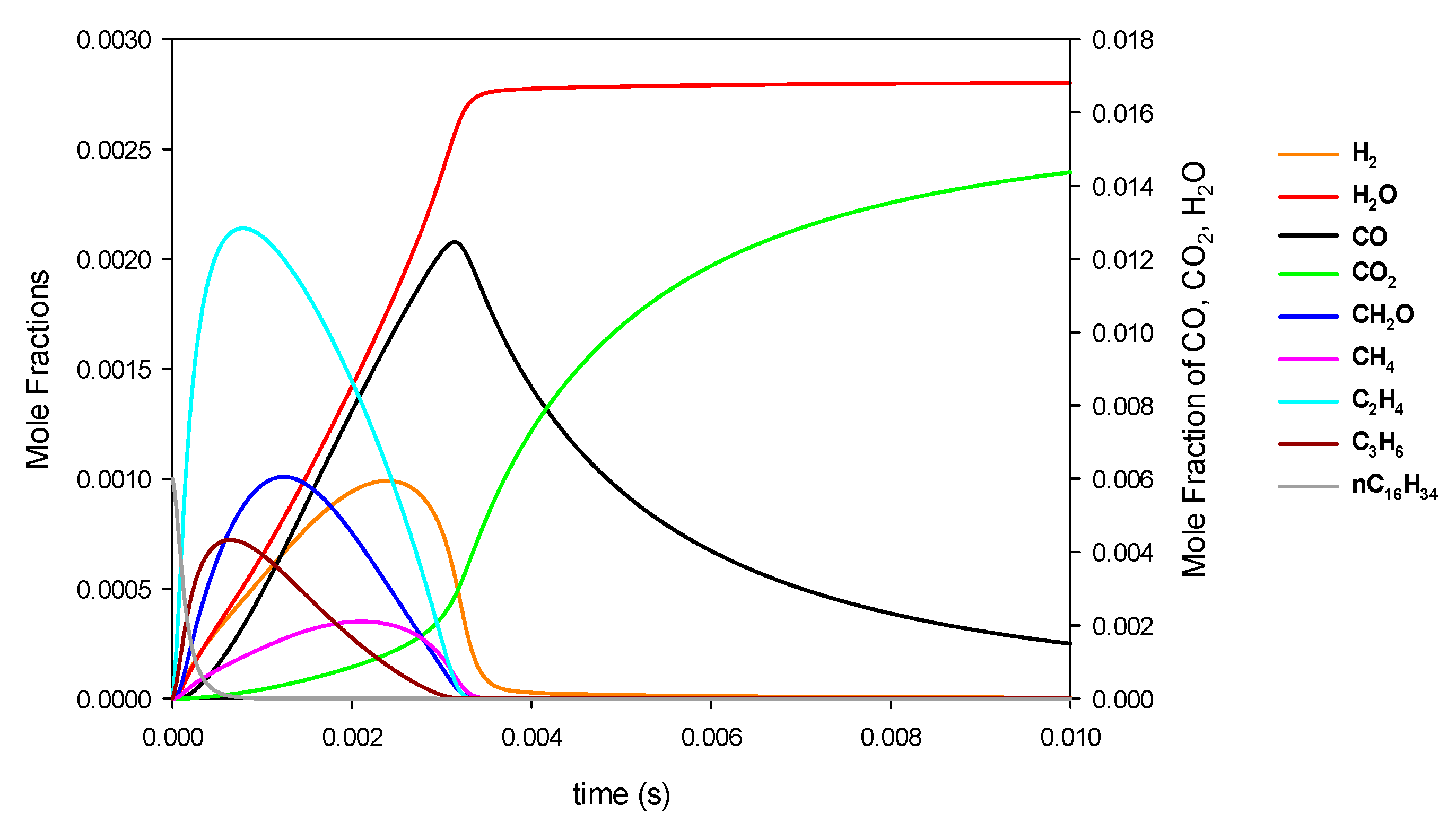
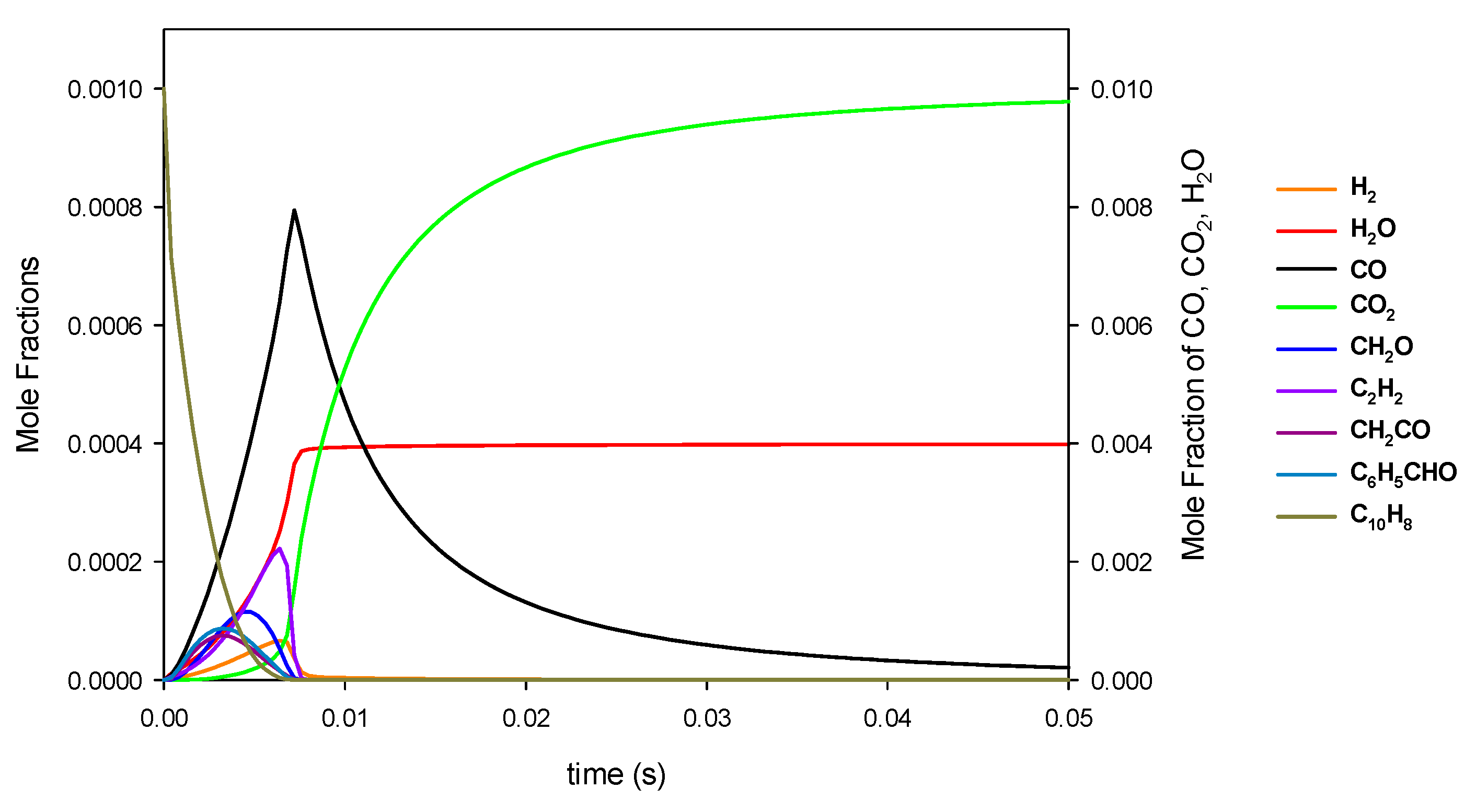
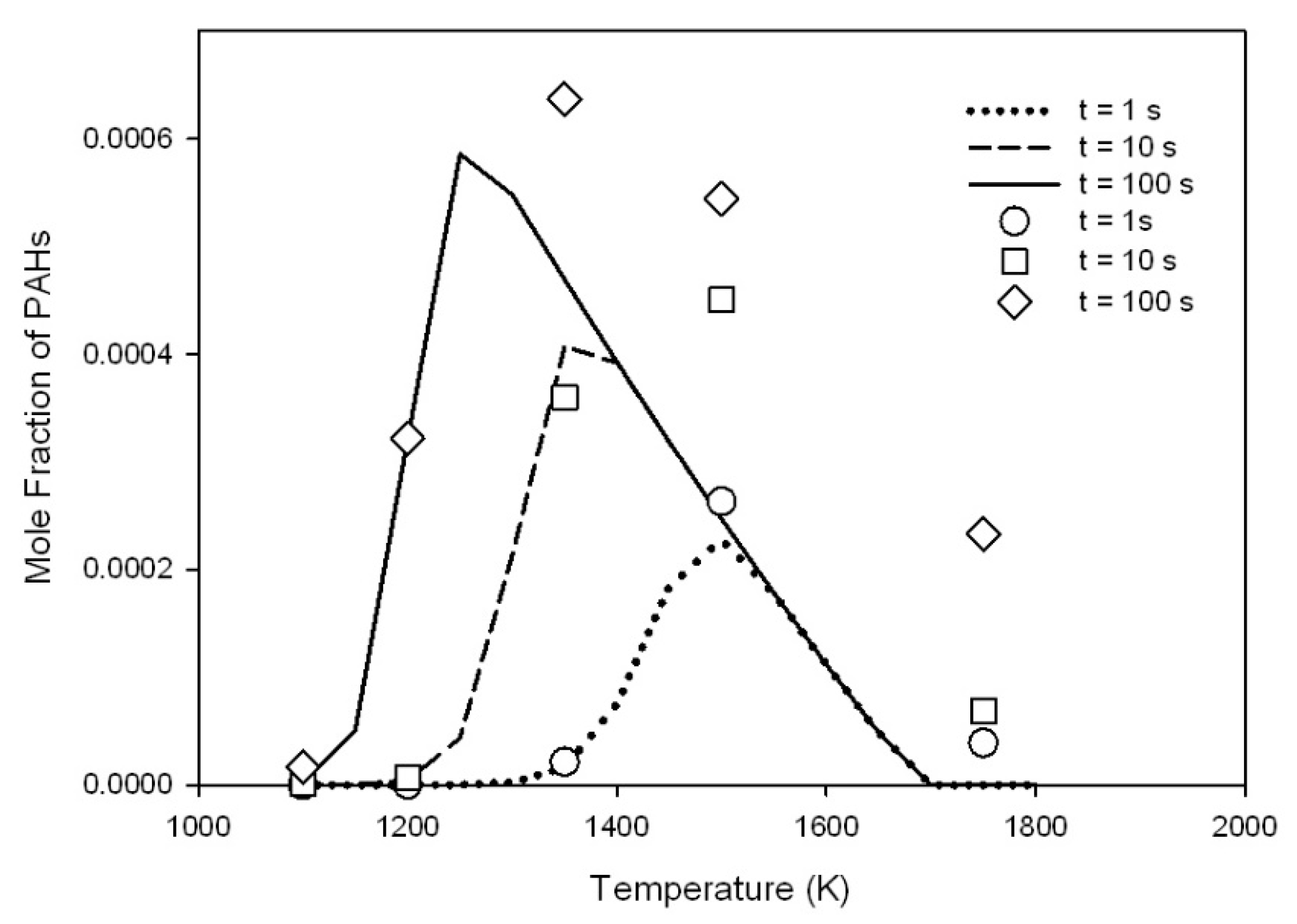
3.1. Reaction of Primary Tar Under Inert Conditions
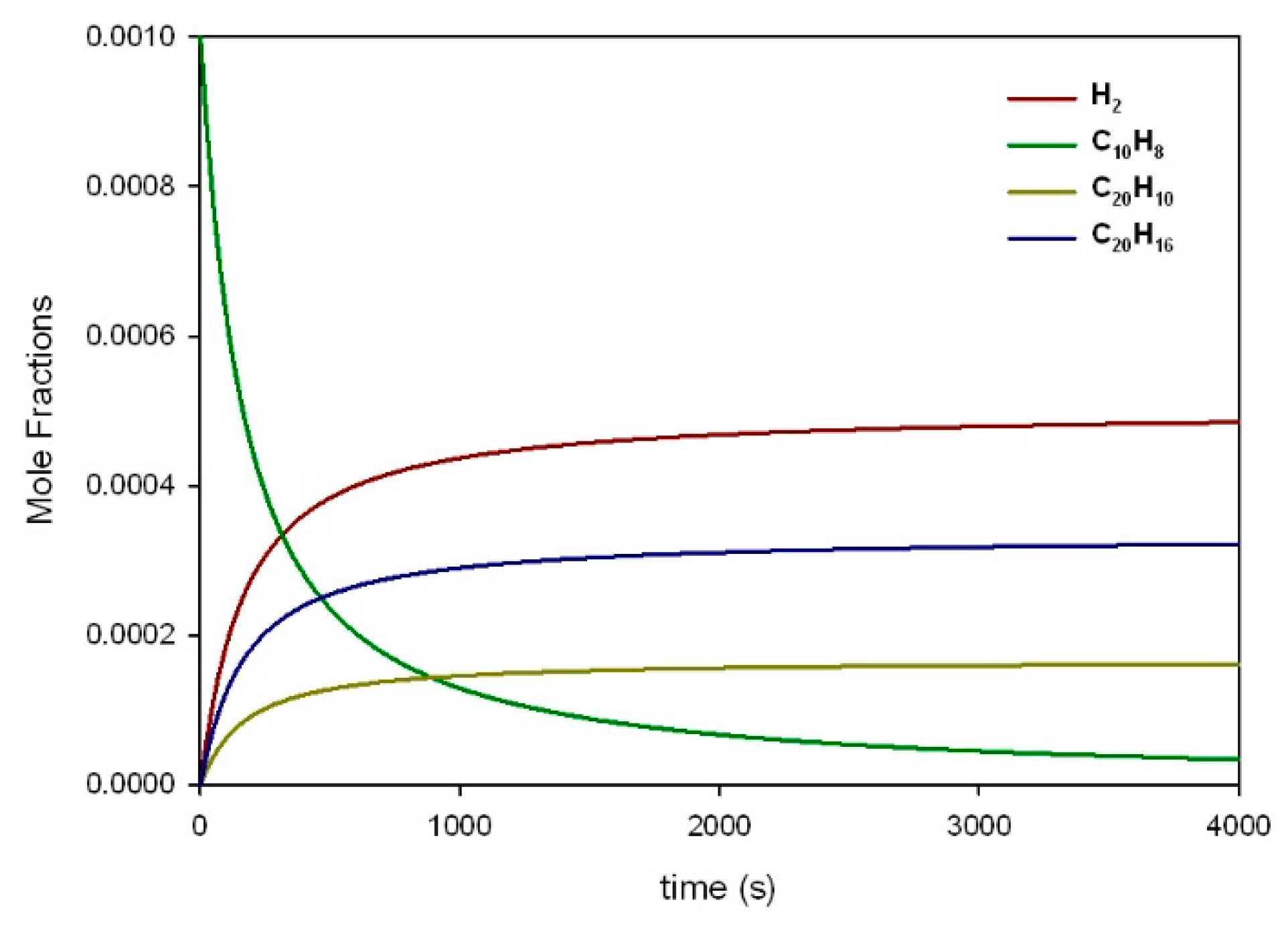
3.2. Reaction of Secondary Tar Under Inert Conditions
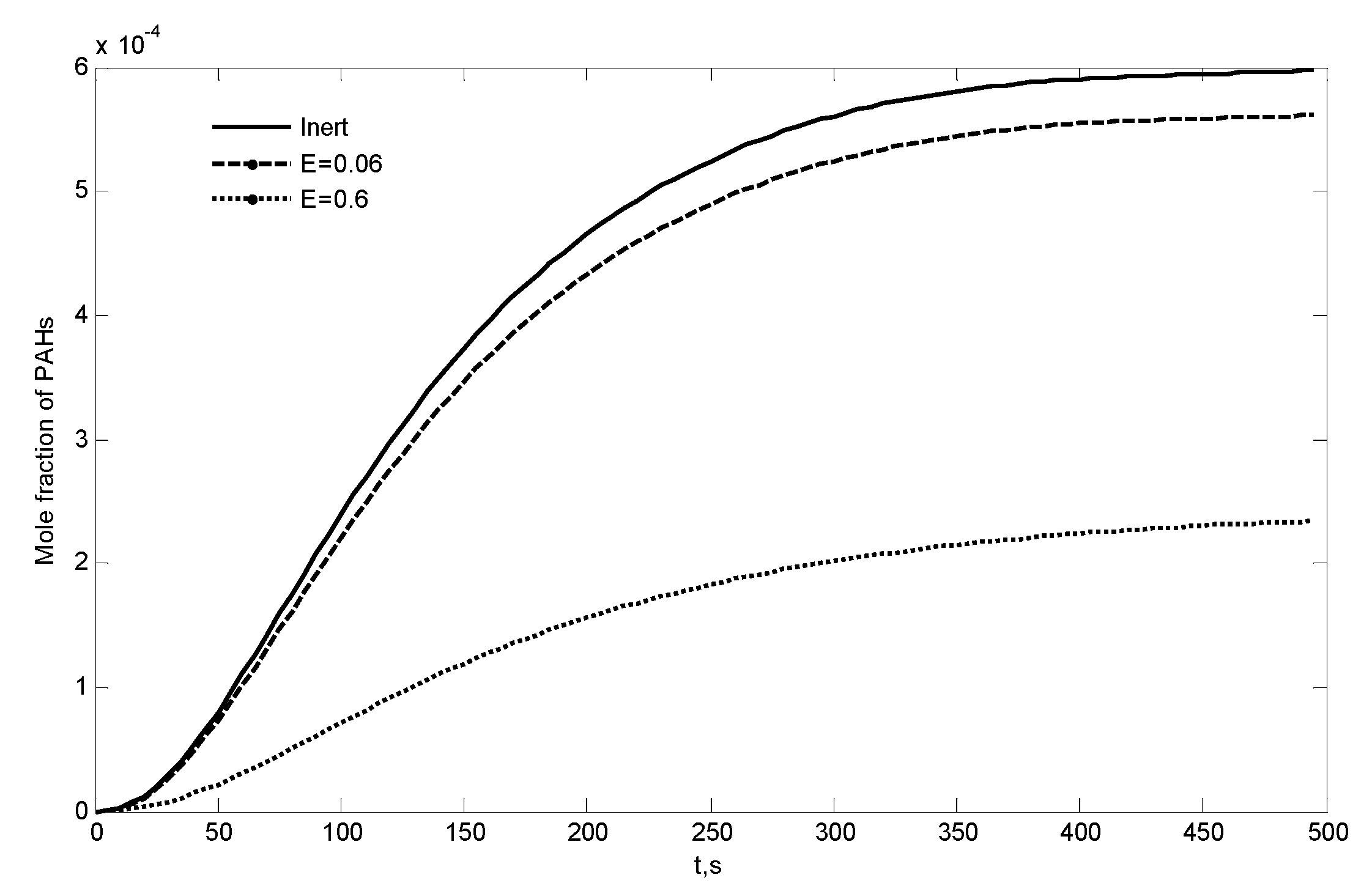
3.3. Combustion of Primary Tar
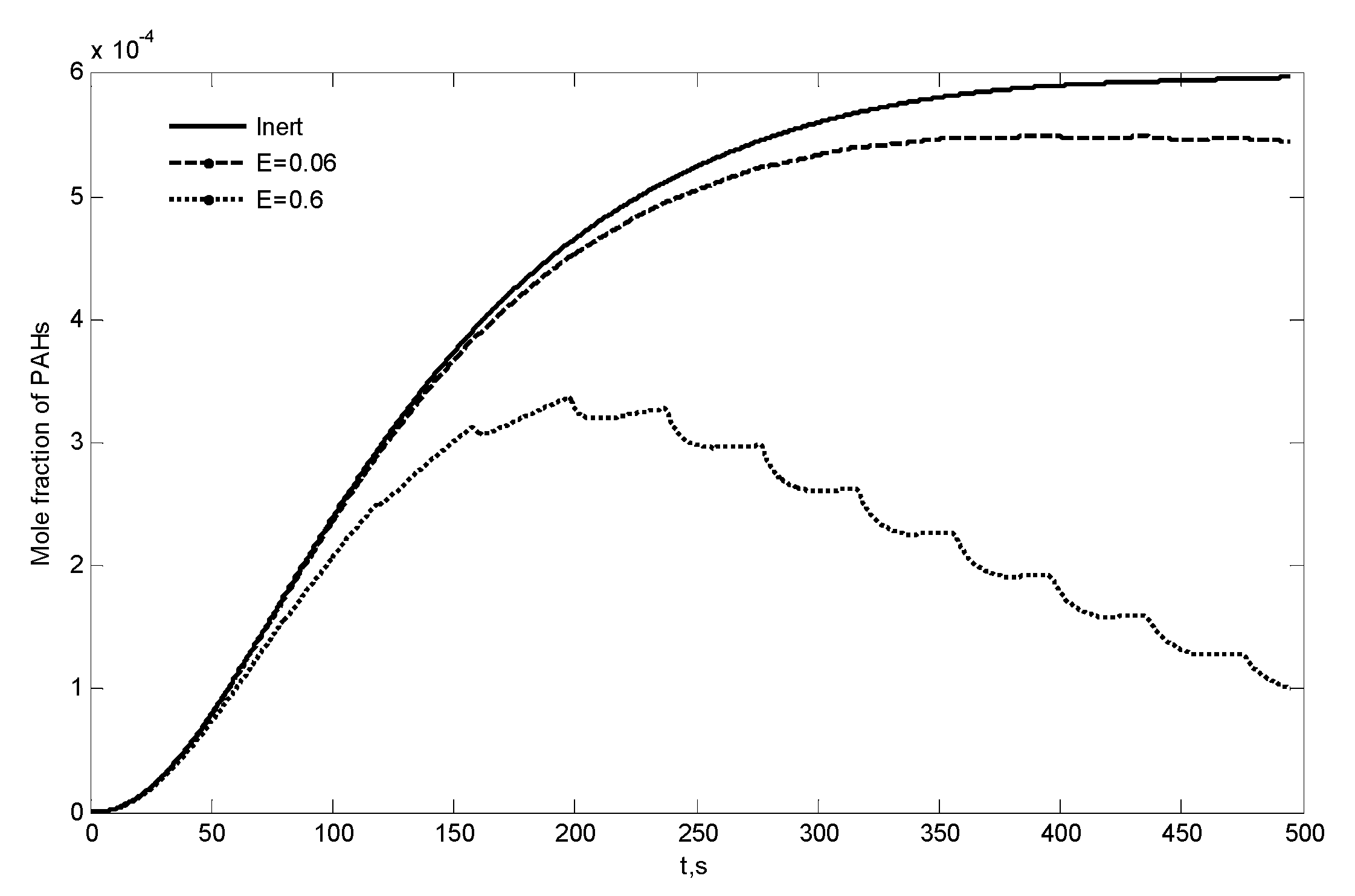
3.4. Combustion of Secondary Tar
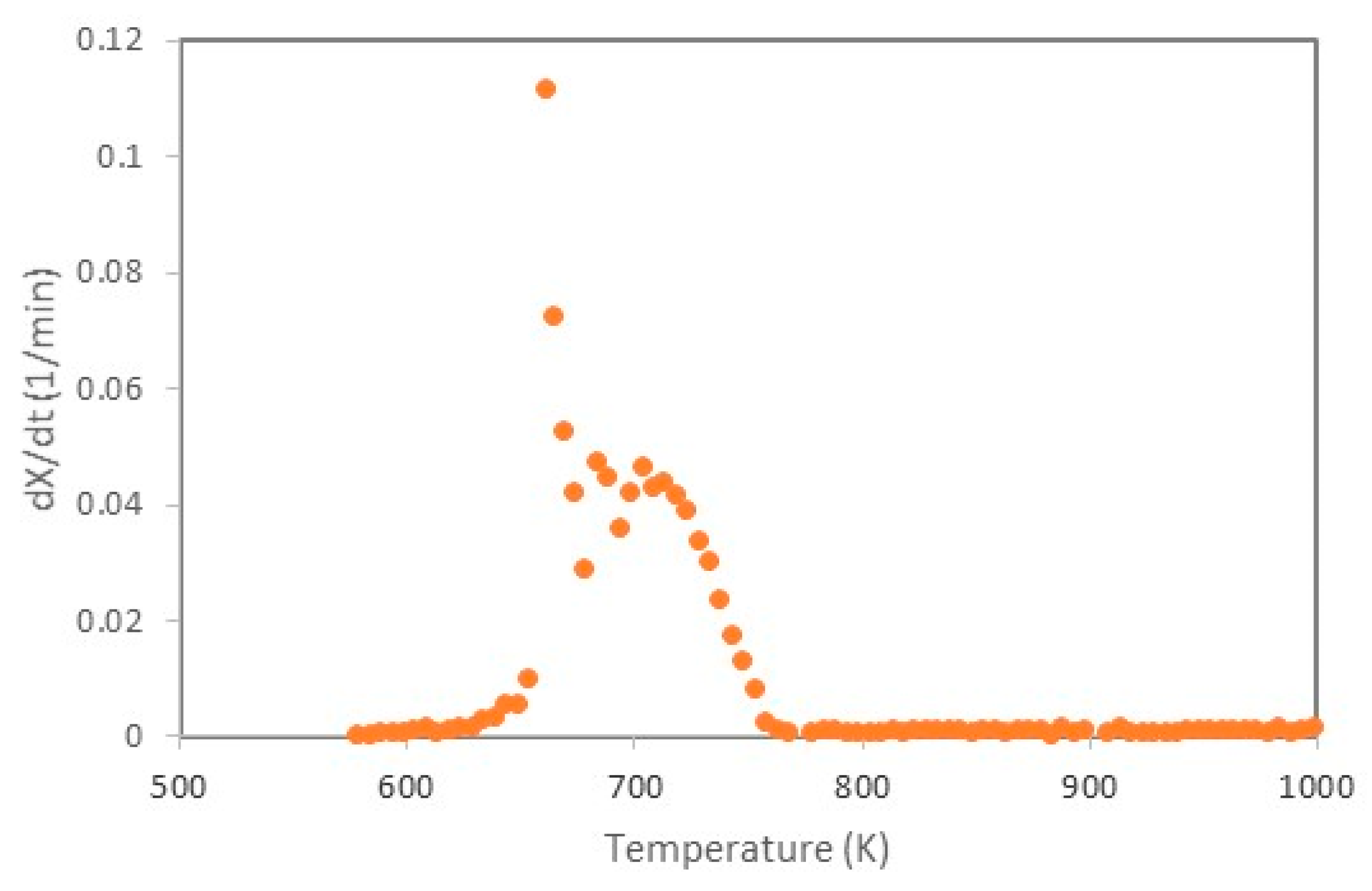
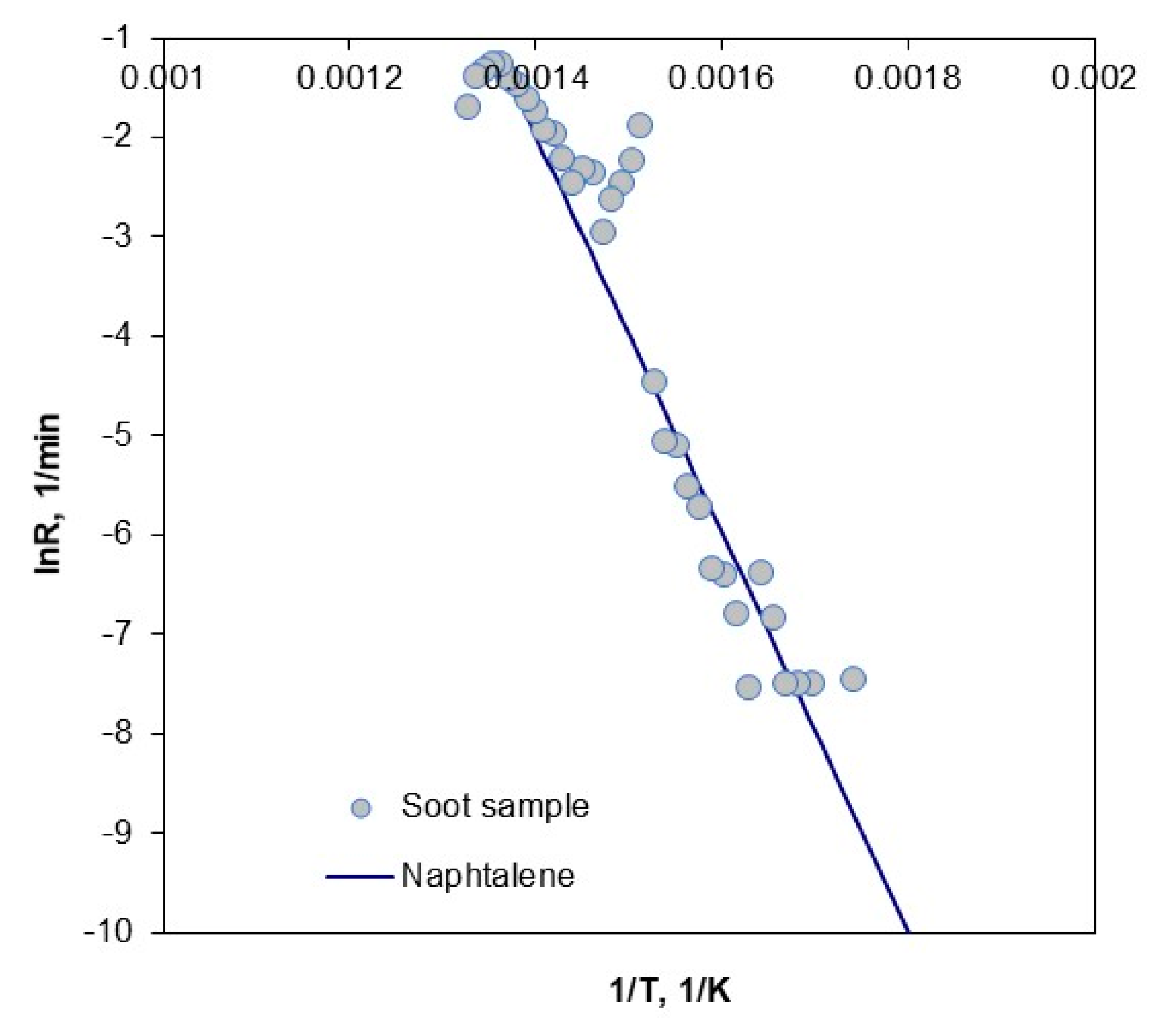
3.5. Combustion of Soot
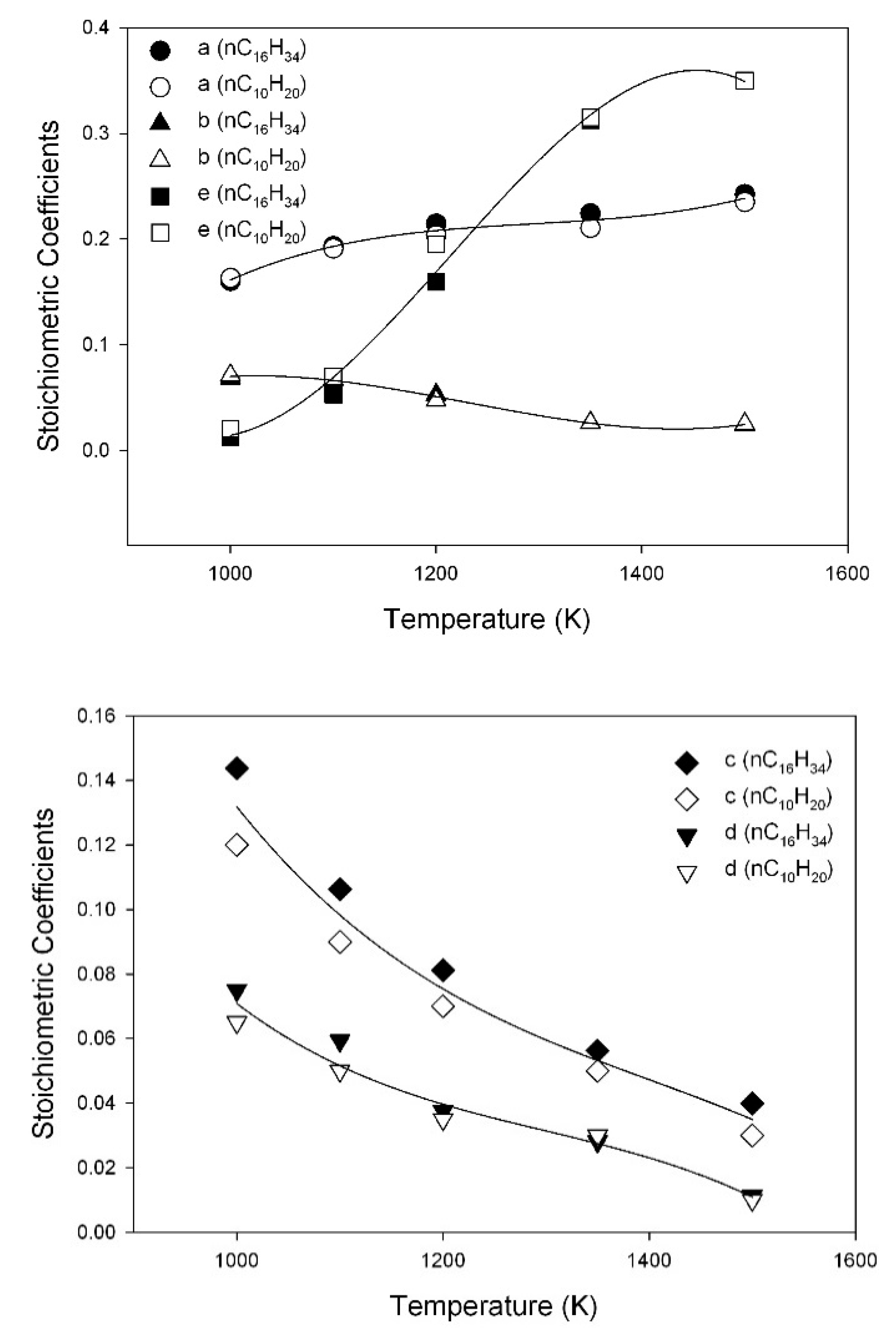
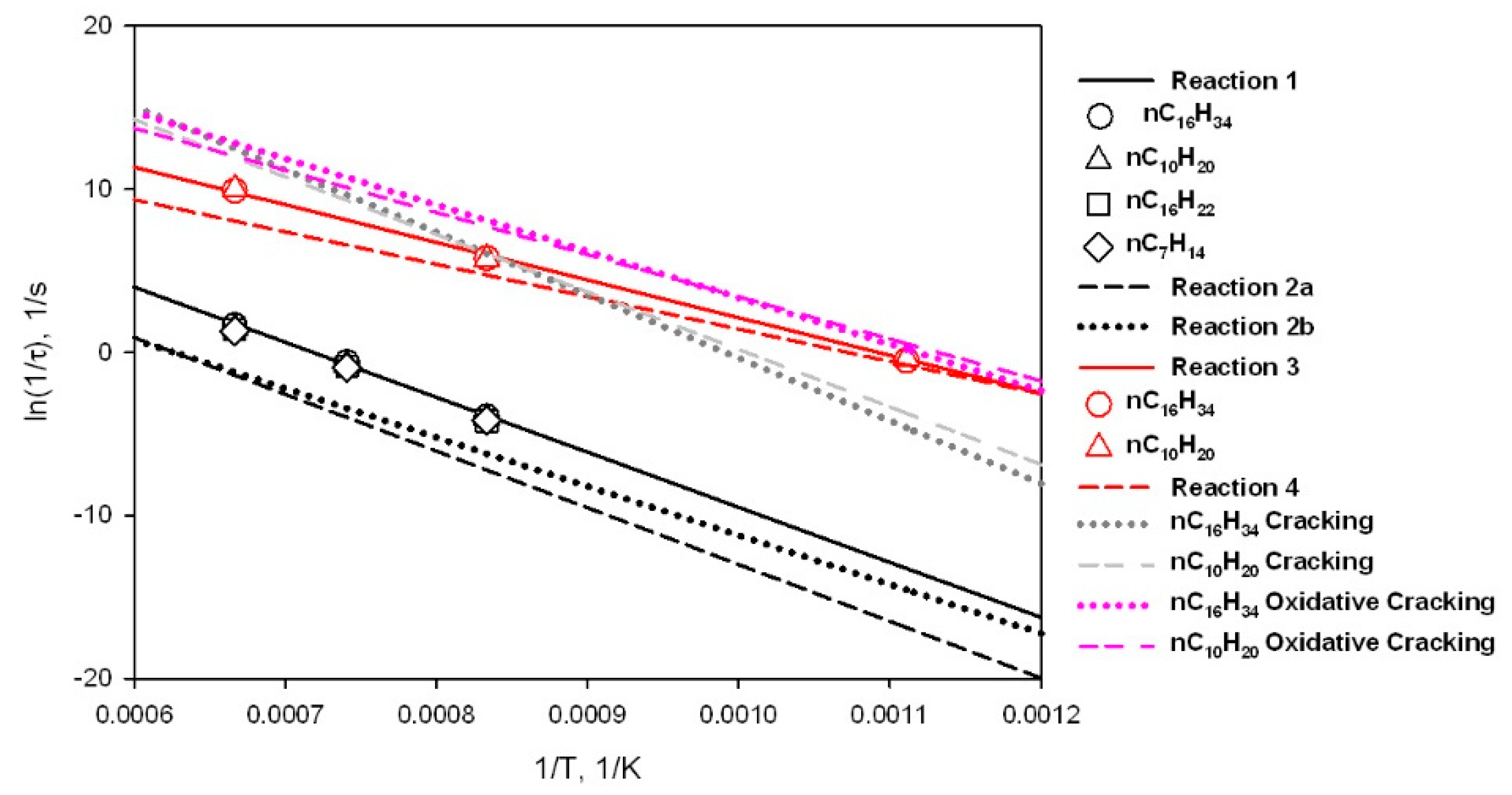
3.6. Assessment of Kinetic Rate Expressions
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fivga, A.; Dimitriou, I. Pyrolysis of plastic waste for production of heavy fuel substitute: A techno-economic assessment. Energy 2018, 149, 865–874. [Google Scholar] [CrossRef]
- Sharuddin, S.D.A.; Abnisa, F.; Daud, W.M.A.W.; Aroua, M.K. Pyrolysis of plastic waste for liquid fuel production as prospective energy resource. IOP Conf. Ser. Mater. Sci. Eng. 2018, 334. [Google Scholar] [CrossRef]
- Antelava, A.; Pallari, E.; Manos, G.; Constantinou, A. Design and limitations in polymer cracking fluidized beds for energy recovery. Plast. Energy Fuel Chem. Sustain. Implic. 2018, 221–231. [Google Scholar] [CrossRef]
- Al-Salem, S.M. Feedstock and optimal operation for plastics to fuel conversion in pyrolysis. Plast. Energy Fuel Chem. Sustain. Implic. 2018, 117–146. [Google Scholar] [CrossRef]
- Czajczyńska, D.; Anguilano, L.; Ghazal, H.; Krzyżyńska, R.; Reynolds, A.J.; Spencer, N.; Jouhara, H. Potential of pyrolysis processes in the waste management sector. Therm. Sci. Eng. Prog. 2017, 3, 171–197. [Google Scholar] [CrossRef]
- Lopez, G.; Artetxe, M.; Amutio, M.; Alvarez, J.; Bilbao, J.; Olazar, M. Recent advances in the gasification of waste plastics. A critical overview. Renew. Sustain. Energy Rev. 2018, 82, 576–596. [Google Scholar] [CrossRef]
- Gopinath, K.P.; Nagarajan, V.M.; Krishnan, A.; Malolan, R. A critical review on the influence of energy, environmental and economic factors on various processes used to handle and recycle plastic wastes: Development of a comprehensive index. J. Clean. Prod. 2020, 274. [Google Scholar] [CrossRef]
- Lopez, G.; Artetxe, M.; Amutio, M.; Bilbao, J.; Olazar, M. Thermochemical routes for the valorization of waste polyolefinic plastics to produce fuels and chemicals. A review. Renew. Sustain. Energy Rev. 2018, 73, 346–368. [Google Scholar] [CrossRef]
- Miandad, R.; Barakat, M.A.; Aburiazaiza, A.S.; Rehan, M.; Nizami, A.S. Catalytic pyrolysis of plastic waste: A review. Process. Saf. Environ. Prot. 2016, 102, 822–838. [Google Scholar] [CrossRef]
- Al-Salem, S.M.; Antelava, A.; Constantinou, A.; Manos, G.; Dutta, A. A review on thermal and catalytic pyrolysis of plastic solid waste (PSW). J. Environ. Manag. 2017, 197, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Anuar Sharuddin, S.D.; Abnisa, F.; Wan Daud, W.M.A.; Aroua, M.K. A review on pyrolysis of plastic wastes. Energy Convers. Manag. 2016, 2016 115, 308–326. [Google Scholar] [CrossRef]
- Dwivedi, P.; Mishra, P.K.; Mondal, M.K.; Srivastava, N. Non-biodegradable polymeric waste pyrolysis for energy recovery. Heliyon 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Thunman, H.; Berdugo Vilches, T.; Seeman, M.; Nguyen, H.N.T. Circular use of plastics-transformation of existing petrochemical clusters into thermochemical recycling plants with 100% plastics recovery. Sustain. Mater. Technol. 2019, 22. [Google Scholar] [CrossRef]
- Garrido, M.A.; Font, R.; Conesa, J.A. Pollutant emissions during the pyrolysis and combustion of flexible polyurethane foam. Waste Manag. 2016, 52, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Moltó, J.; López-Sánchez, B.; Domene-López, D.; Moreno, A.I.; Font, R.; Montalbán, M.G. Pollutant emissions during the pyrolysis and combustion of starch/poly(vinyl alcohol) biodegradable films. Chemosphere 2020, 256. [Google Scholar] [CrossRef]
- Nedjalkov, I.; Yoshiie, R.; Ueki, Y.; Naruse, I. Tar and soot generation behaviors from ABS, PC and PE pyrolysis. J. Mater. Cycles Waste Manag. 2017, 19, 682–693. [Google Scholar] [CrossRef]
- Russell, J.M.; Gracida-Alvarez, U.R.; Gracida-Alvarez, U.R.; Winjobi, O.; Winjobi, O.; Shonnard, D.R.; Shonnard, D.R. Update to Effect of Temperature and Vapor Residence Time on the Micropyrolysis Products of Waste High Density Polyethylene. Ind. Eng. Chem. Res. 2020, 59, 10716–10719. [Google Scholar] [CrossRef]
- Yan, G.X.; Jing, X.D.; Han, Q.Z.; Wen, H.; Xiang, S.G. Thermal cracking of HDPE/PP mixtures. Gao Xiao Hua Xue Gong Cheng Xue Bao J. Chem. Eng. Chin. Univ. 2015, 29, 571–577. [Google Scholar] [CrossRef]
- Abbas-Abadi, M.S. The effect of process and structural parameters on the stability, thermo-mechanical and thermal degradation of polymers with hydrocarbon skeleton containing PE, PP, PS, PVC, NR, PBR and SBR. J. Therm. Anal. Calorim. 2020. [Google Scholar] [CrossRef]
- Quesada, L.; Calero, M.; Martín-Lara, M.A.; Pérez, A.; Blázquez, G. Characterization of fuel produced by pyrolysis of plastic film obtained of municipal solid waste. Energy 2020, 186. [Google Scholar] [CrossRef]
- Ayodele, T.R.; Durodola, O.; Ogunjuyigbe, A.S.; Lange Munda, J. Effects of operating factors on the bio-oil produced from pyrolysis of plastic wastes using response surface methodology. In Proceedings of the IEEE PES/IAS PowerAfrica Conference: Power Economics and Energy Innovation in Africa, PowerAfrica, Abuja, Nigeria, 20–23 August 2019; pp. 527–532. [Google Scholar] [CrossRef]
- Hasan, N.U.; Rahman, M.M.; Rahat, R.I. Characteristics comparison of pyrolysed oils obtained from waste of plastic, tyres and biomass solid. In Proceedings of the (2017) 4th International Conference on Advances in Electrical Engineering (ICAEE) 2017, Dhaka, Bangladesh, 28–30 September 2017; pp. 450–454. [Google Scholar] [CrossRef]
- Vijayakumar, A.; Sebastian, J. Pyrolysis process to produce fuel from different types of plastic—A review. IOP Conf. Ser. Mater. Sci. Eng. 2018, 396. [Google Scholar] [CrossRef]
- Mastral, A.M.; Callea, M.S. A Review on Polycyclic Aromatic Hydrocarbon (PAH) Emissions from Energy Generation. Environ. Sci. Technol. 2000, 34, 3057. [Google Scholar] [CrossRef]
- Williams, P.T.; Besler, S. Polycyclic aromatic hydrocarbons in waste derived pyrolytic oils. J. Anal. Appl. Pyrolysis 1994, 30, 17–33. [Google Scholar] [CrossRef]
- Rausa, R.; Pollesel, P. Pyrolysis of automotive shredder residue (ASR) Influence of temperature on the distribution of products. J. Anal. Appl. Pyrolysis 1997, 40–41, 383–401. [Google Scholar] [CrossRef]
- Elliott, D.C. Pyrolysis Oils from Biomass, Producing, Analysing and Upgrading; Soltes, J., Milne, T.A., Eds.; ACS Symp. Ser. 376; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Mastral, J.F.; Esperanza, E.; García, P.; Juste, M. Pyrolysis of high-density polyethylene in a fluidised bed reactor. Influence of the temperature and residence time. J. Anal. Appl. Pyrolysis 2002, 63, 1–15. [Google Scholar] [CrossRef]
- Niksa, S.; Kerstein, A.R. Flashchain Theory for Rapid Coal Devolatilization Kinetics. 1. Formulation. Energy Fuels 1991, 5, 647–664. [Google Scholar] [CrossRef]
- Solomon, P.R.; Hamblen, D.G.; Carangelo, R.; Serio, M.; Deshpande, G.V. General Model of Coal Devolatilization. Energy Fuels 1988, 2, 405. [Google Scholar] [CrossRef]
- Fletcher, T.H.; Kerstein, A.R.; Pugmire, R.J.; Grant, D.M. Chemical Percolation Model for Devolatilization. 2. Temperature and Heating Rate Effects on Product Yields. Energy Fuels 1990, 4, 54–60. [Google Scholar] [CrossRef]
- Grant, D.M.; Pugmire, R.J.; Fletcher, T.H.; Kerstein, A.R. Chemical Model of Coal Devolatilization Using Percolation Lattice Statistics. Energy Fuels 1989, 3, 175–186. [Google Scholar] [CrossRef]
- Ranzi, E.; Frassoldati, A.; Grana, R.; Cuoci, A.; Faravelli, T.; Kelley, A.P.; Law, C.K. Hierarchical and comparative kinetic modeling of laminar flame speeds of hydrocarbon and oxygenated fuels. PECS 2012, 38, 468–501. [Google Scholar] [CrossRef]
- Ranzi, E.; Dente, M.; Goldaniga, A.; Bozzano, G.; Faravelli, T. Lumping procedures in detailed kinetic modeling of gasification, pyrolysis, partial oxidation and combustion of hydrocarbon mixtures. Prog. Energy Combust. Sci. 2001, 27, 99–139. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, J.; Zhao, L.; Yang, J.; Jin, H.; Cheng, Z.; Li, Y.; Zhang, L.; Qi, F. An experimental and kinetic modeling study of three butene isomers pyrolysis at low pressure. Combust. Flame 2012, 159, 905–917. [Google Scholar] [CrossRef]
- Al-Salem, S.M. Thermal pyrolysis of high density polyethylene (HDPE) in a novel fixed bed reactor system for the production of high value gasoline range hydrocarbons (HC). Process. Saf. Environ. Prot. 2019, 127, 171–179. [Google Scholar] [CrossRef]
- Bradley, J.N. A General Mechanism for the High-Temperature Pyrolysis of Alkanes. The Pyrolysis of Isobutane. Proc. R. Soc. London A 1974, 337, 199–216. [Google Scholar]
- Held, T.J.; Marchese, A.J.; Dryer, F.L. A semi-empirical reaction mechanism for n-heptane oxidation and pyrolysis. Combust. Sci. Technol. 1997, 123, 107–146. [Google Scholar] [CrossRef]
- Zeppieri, S.P.; Klotz, S.D.; Dryer, F.L. Modeling concepts for larger carbon number alkanes: A partially reduced skeletal mechanism for n-decane oxidation and pyrolysis. Proc. Combust. Inst. 2000, 28, 1587. [Google Scholar] [CrossRef]
- Ranzi, E.; Frassoldati, A.; Granata, S.; Faravelli, T. Wide-range kinetic modeling study of the pyrolysis, partial oxidation, and combustion of heavy n-alkanes. Ind. Eng. Chem. Res. 2005, 44, 5170–5183. [Google Scholar] [CrossRef]
- Granata, S.; Faravelli, T.; Ranzi, E. A wide range kinetic modeling study of the pyrolysis and combustion of naphthenes. Combust. Flame 2003, 132, 533–544. [Google Scholar] [CrossRef]
- Maffei, T.; Frassoldati, A.; Cuoci, A.; Ranzi, E.; Faravelli, T. Predictive one step kinetic model of coal pyrolysis for CFD applications. Proc. Combust. Inst. 2013, 34, 2401–2410. [Google Scholar] [CrossRef]
- Ranzi, E.; Cuoci, A.; Faravelli, T.; Frassoldati, A.; Migliavacca, G.; Pierucci, S. Chemical kinetics of biomass pyrolysis. Energy Fuels 2008, 22, 4292–4300. [Google Scholar] [CrossRef]
- Jess, A. Mechanisms and kinetics of thermal reactions of aromatic hydrocarbons from pyrolysis of solid fuels. Fuel 1996, 75, 1441–1448. [Google Scholar] [CrossRef]
- Umeki, K.; Yamamoto, K.; Namioka, T.; Yoshikawa, K. High temperature steam-only gasification of woody biomass. Appl. Energy 2010, 87, 791–798. [Google Scholar] [CrossRef]
- Corella, J.; Sanz, A. Modeling circulating fluidized bed biomass gasifiers. A pseudo-rigorous model for stationary state. Fuel Process. Technol. 2005, 86, 1021–1053. [Google Scholar] [CrossRef]
- Maki, T.; Miura, K. A simulation model for the pyrolysis of oil emulsion. Energy Fuels 1996, 11, 819–824. [Google Scholar] [CrossRef]
- Zou, B.; Lou, Q.; Mo, S.; Feng, S. Study of a kinetic model of atmospheric gas oil pyrlolysis and coke deposition. Ind. Eng. Chem. Res. 1993, 32, 843–847. [Google Scholar] [CrossRef]
- Jackson, K.J.; Burnham, A.K.; Braun, R.L.; Knauss, K.G. Temperature and pressure dependence of n-hexadecane cracking. Org. Geochem. 1995, 23, 941–954. [Google Scholar] [CrossRef]
- Kumar, S.; Panda, A.K.; Singh, R.K. A review on tertiary recycling of high-density polyethylene to fuel. Resour. Conserv. Recycl. 2011, 55, 893–910. [Google Scholar] [CrossRef]
- Conesa, J.A.; Font, R.; Marcilla, A.; García, A.N. Pyrolysis of polyethylene in a fluidized bed reactor. Energy Fuels 1994, 8, 1238–1246. [Google Scholar] [CrossRef]
- Walendziewski, J. Engine fuel derived from waste plastics by thermal treatment. Fuel 2000, 81, 473–481. [Google Scholar] [CrossRef]
- Walendziewski, J. Continuous flow cracking of waste plastics. Fuel Process. Technol. 2005, 86, 1265–1278. [Google Scholar] [CrossRef]
- Seo, Y.H.; Lee, K.H.; Shin, D.H. Investigation of catalytic degradation of high-density polyethylene by hydrocarbon group type analysis. J. Anal. Appl. Pyrolysis 2003, 70, 383–398. [Google Scholar] [CrossRef]
- Wu, Y.; Isarov, A.V.; Connell, C.O. Thermal analysis of high density polethylene-maple woodflour composites. Thermochim. Acta 1999, 340–341, 205–220. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | |
| b | |
| c | |
| d | |
| e | |
| k0 (1/s) | Ea (kcal/mol) | |
|---|---|---|
| Reaction 1 | ||
| 3.32 × 1010 | 67.0 | |
| Reaction 2a | ||
| 2.86 × 109 | 69.1 | |
| Reaction 2b | ||
| 1.90 × 1010 | 59.6 | |
| Reaction 3 | ||
| 8.6 × 1010 | 45.8 | 0 (xO2 > 0.2) |
| 8.4 × 1011 | 45.8 | 1.4 (xO2 < 0.2) |
| Reaction 4–5 | ||
| 1.7 × 109 | 39.4 | 0 (xO2 > 0.2) |
| 1.9 × 1010 | 39.4 | 1.5 (xO2 < 0.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senneca, O.; Tucciullo, T. Lumped Kinetics for Homogeneous Reactions of n-Hexadecane and n-Decene as Model Compounds for PE Pyrolysis Primary Tars. Energies 2020, 13, 5466. https://doi.org/10.3390/en13205466
Senneca O, Tucciullo T. Lumped Kinetics for Homogeneous Reactions of n-Hexadecane and n-Decene as Model Compounds for PE Pyrolysis Primary Tars. Energies. 2020; 13(20):5466. https://doi.org/10.3390/en13205466
Chicago/Turabian StyleSenneca, Osvalda, and Teresa Tucciullo. 2020. "Lumped Kinetics for Homogeneous Reactions of n-Hexadecane and n-Decene as Model Compounds for PE Pyrolysis Primary Tars" Energies 13, no. 20: 5466. https://doi.org/10.3390/en13205466
APA StyleSenneca, O., & Tucciullo, T. (2020). Lumped Kinetics for Homogeneous Reactions of n-Hexadecane and n-Decene as Model Compounds for PE Pyrolysis Primary Tars. Energies, 13(20), 5466. https://doi.org/10.3390/en13205466