Rational Design and Testing of Anti-Knock Additives



Abstract

1. Introduction
- Additive design using molecular thermodynamics from theory.
- Additive testing using small scale ignition measurements.
1.1. Knock in Spark Ignition (SI) Engines
1.1.1. Quantifying Knock—Research Octane Number (RON)
- Determination of the RON requires a relatively large volume of fuel. This can be restrictive to the testing of new fuel additives (see below).
- The compression ratio of a given RON test is dependent on the propensity of the test fuel to resist knock. Fuels more resistant to knock will require higher compression ratios, and thus experience higher end-gas temperatures and pressures. The temperature and pressure trajectories define the reaction rate of the flame and the end gas. This makes it difficult to objectively interpret the underlying chemistry contributing to any observed change in the RON.
1.1.2. The Chemical Causes of Knock
1.2. Anti-Knock Additives
1.2.1. Metallic Anti-Knock Additives
1.2.2. Non-Metallic Anti-Knock Additives
2. Methodology
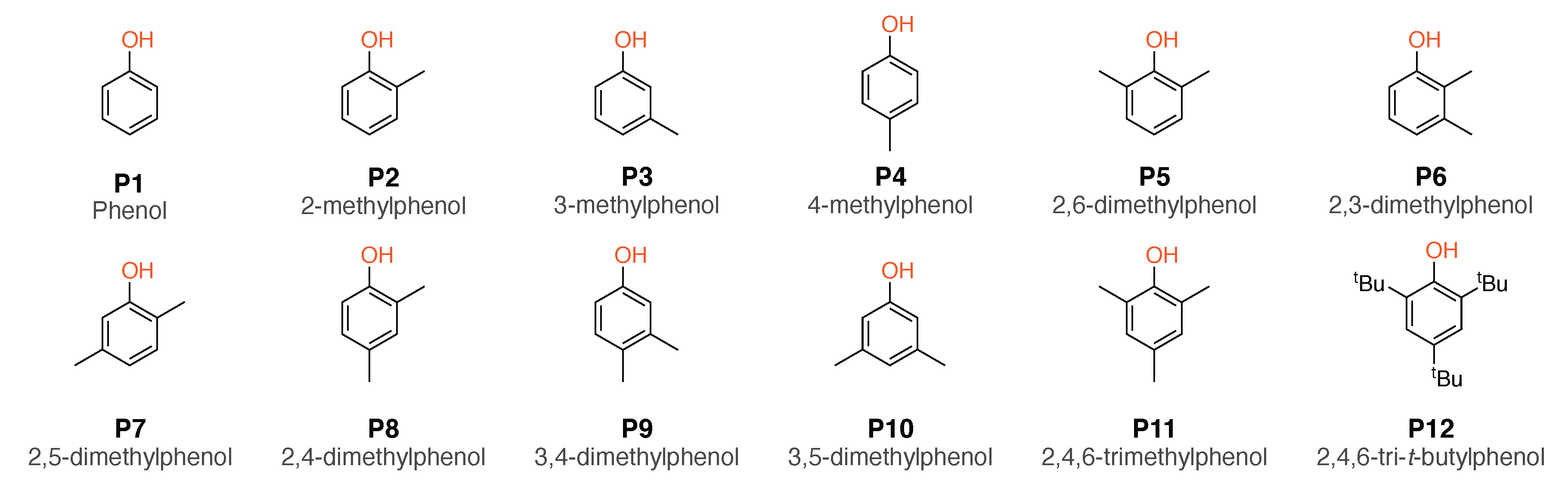
2.1. Selection of the Model Compounds
2.2. Additive Design by Molecular Thermodynamics from Theory
- For Reactions 7 and 8 to be competitive with hydrogen abstraction from the fuel, the additive must possess hydrogen atoms that are readily abstracted, i.e., the X-H bond dissociation energy must be as low as possible.
- In order to be a sink for HO2● and HO●, the formed radical (X●) must not decompose to produce active radicals at a timescale comparable to its rate of formation, i.e., the rate of Reaction 9 must be low compared to the rate of Reactions 7 and 8.
2.2.1. Bond Dissociation Energy (BDE)
2.2.2. Rate of Hydrogen Atom Abstraction
2.2.3. Free Energy of Formation
2.3. Additive Testing by Small Scale Ignition Measurements
3. Experimental
3.1. Computational Methodology
3.2. Combustion Measurements
3.3. Calculation of HO● and HO2● Concentrations
4. Results and Discussion
4.1. Effect of Phenols P1–P12 on DCN and RON
4.2. Calculation of Molecular Thermodynamic Properties Relative to Knock
4.2.1. Determination of the Rate of Hydrogen Atom Abstraction by HO● and HO2●
4.2.2. Gibbs Free Energy Terms
4.2.3. Number of Alkyl Substituents (#CH3)
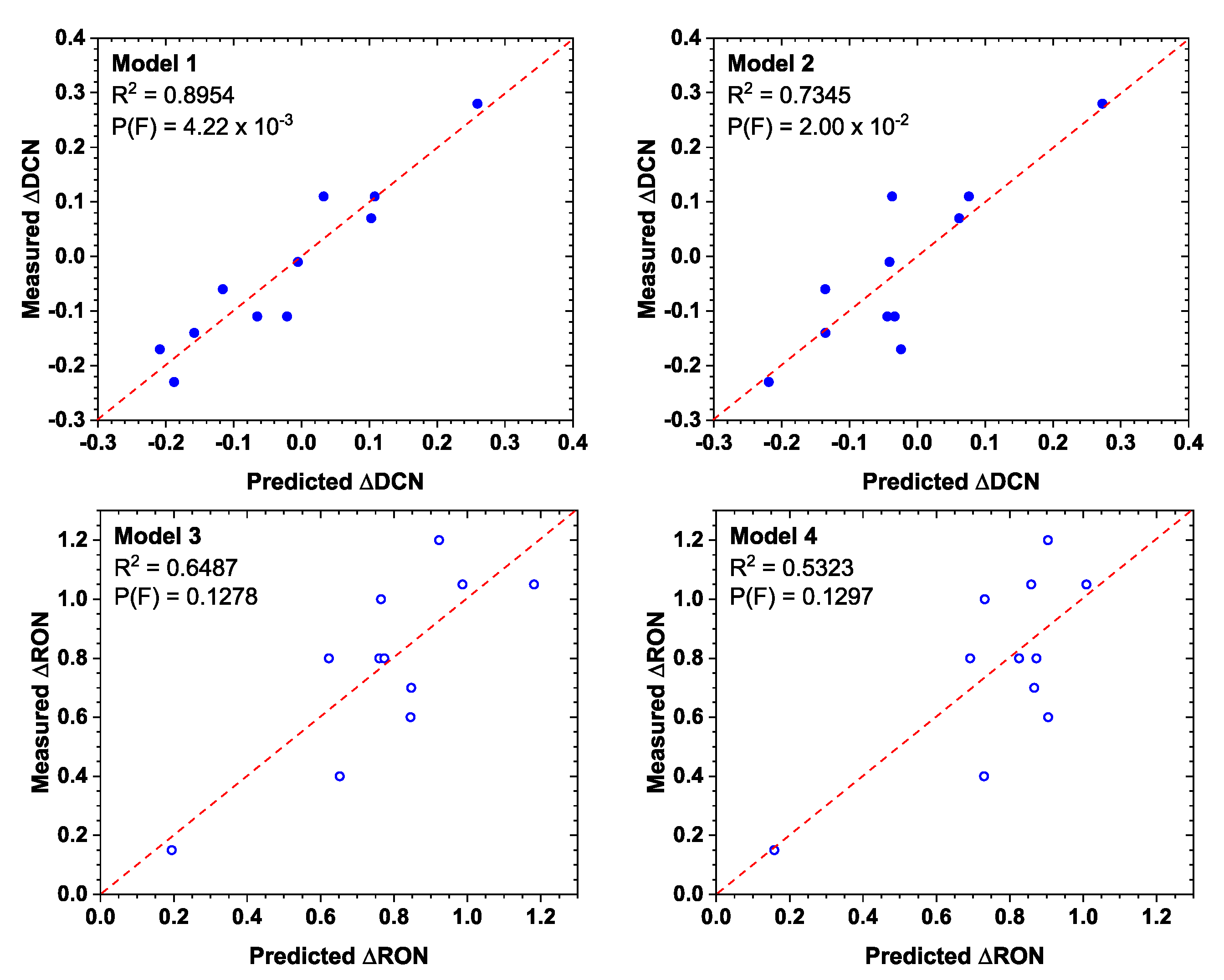
5. Numerical Models
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Splitter, D.; Pawlowski, A.; Wagner, R. A Historical Analysis of the Co-evolution of Gasoline Octane Number and Spark-Ignition Engines. Front. Mech. Eng. 2016, 1, 16. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EU) No 459/2012 of 29 May 2012 Amending Regulation (EC) No 715/2007 of the European Parliament and of the Council and Commission Regulation (EC) No 692/2008 as Regards Emissions from Light Passenger and Commercial Vehicles (Euro 6); European Commission: Brussels, Belgium, 2012. [Google Scholar]
- Directive (EU) 2018/2001 of the European Parliament and of the Council of 11 December 2018 on the Promotion of the Use of Energy from Renewable Sources. Available online: http://eur-lex.europa.eu/ (accessed on 5 June 2020).
- Ribeiro, N.M.; Pinto, A.C.; Quintella, C.M.; da Rocha, G.O.; Teixeira, L.S.G.; Guarieiro, L.L.N.; do Carmo Rangel, M.; Veloso, M.C.C.; Rezende, M.J.C.; Serpa da Cruz, R.; et al. The Role of Additives for Diesel and Diesel Blended (Ethanol or Biodiesel) Fuels: A Review. Energy Fuels 2007, 21, 2433–2445. [Google Scholar] [CrossRef]
- Speth, R.L.; Chow, E.W.; Malina, R.; Barrett, S.R.H.; Heywood, J.B.; Green, W.H. Economic and Environmental Benefits of Higher-Octane Gasoline. Environ. Sci. Technol. 2014, 48, 6561–6568. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.S.; Issayev, G.; Naser, N.; Sarathy, S.M.; Farooq, A.; Dooley, S. Ethanolic gasoline, a lignocellulosic advanced biofuel. Sustain. Energy Fuels 2019, 3, 409–421. [Google Scholar] [CrossRef]
- Seyferth, D. The Rise and Fall of Tetraethyllead. 2. Organometallics 2003, 22, 5154–5178. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, H.; Reitz, R.D. Knocking combustion in spark-ignition engines. Prog. Energy Combust. Sci. 2017, 61, 78–112. [Google Scholar] [CrossRef]
- Westbrook, C.K. Chemical kinetics of hydrocarbon ignition in practical combustion systems. Proc. Combust. Inst. 2000, 28, 1563–1577. [Google Scholar] [CrossRef]
- Kalghatgi, G.T. Developments in internal combustion engines and implications for combustion science and future transport fuels. Proc. Combust. Inst. 2015, 35, 101–115. [Google Scholar] [CrossRef]
- ASTM International. ASTM D2699-18, Standard Test Method for Research Octane Number of Spark-Ignition Engine Fuel; ASTM International: West Conshohocken, PA, USA, 2018. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Warnatz, J.; Pitz, W.J. A detailed chemical kinetic reaction mechanism for the oxidation of iso-octane and n-heptane over an extended temperature range and its application to analysis of engine knock. Symp. (Int.) Combust. 1989, 22, 893–901. [Google Scholar] [CrossRef]
- Brown, J.E.; Lovell, W.G. A New Manganese Antiknock. Ind. Eng. Chem. 1958, 50, 1547–1550. [Google Scholar] [CrossRef]
- Fenard, Y.; Song, H.; Dauphin, R.; Vanhove, G. An engine-relevant kinetic investigation into the anti-knock effect of organometallics through the example of ferrocene. Proc. Combust. Inst. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Bradley, D.; Morley, C. Chapter 7 Autoignition in spark-ignition engines. In Comprehensive Chemical Kinetics; Pilling, M.J., Ed.; Elsevier: Amsterdam, The Netherlands, 1997; Volume 35, pp. 661–760. [Google Scholar] [CrossRef]
- Ballinger, P.R.; Ryason, P.R. Isolated stable cool flames of hydrocarbons. Symp. (Int.) Combust. 1971, 13, 271–277. [Google Scholar] [CrossRef]
- Erhard, K.H.L.; Norrish, R.G.W. Studies of knock and antiknock by kinetic spectroscopy. Proc. R. Soc. Lond. Ser. Math. Phys. Sci. 1956, 234, 178–191. [Google Scholar] [CrossRef]
- Erhard, K.H.L.; Norrish, R.G.W. Studies of knock and antiknock by kinetic spectroscopy. II. Proc. R. Soc. Lond. Ser. Math. Phys. Sci. 1960, 259, 297–303. [Google Scholar] [CrossRef]
- Benson, S.W. The mechanism of inhibition of knock by lead additives, a chain debranching reaction. J. Phys. Chem. 1988, 92, 1531–1533. [Google Scholar] [CrossRef]
- Chamberlain, G.H.N.; Walsh, A.D.; Egerton, A.C. The inhibiting effect of lead tetraethyl I. The effect of lead compounds on the vapour phase slow oxidation of diisopropyl ether and on the ignition of diethyl ether. Proc. R. Soc. Lond. Ser. Math. Phys. Sci. 1952, 215, 175–186. [Google Scholar] [CrossRef]
- Hoare, D.E.; Walsh, A.D.; Egerton, A.C. The inhibiting effect of lead tetraethyl II. The effect of lead monoxide on the slow oxidation of methane. Proc. R. Soc. Lond. Ser. Math. Phys. Sci. 1952, 215, 454–466. [Google Scholar] [CrossRef]
- Salooja, K.C. Studies relating to the mechanism of antiknock action of tetraethyl lead. Combust. Flame 1965, 9, 211–217. [Google Scholar] [CrossRef]
- Salooja, K.C. Studies of combustion processes leading to ignition in hydrocarbons. Combust. Flame 1960, 4, 117–136. [Google Scholar] [CrossRef]
- Pahnke, A.J.; Cohen, P.M.; Sturgis, B.M. Preflame Oxidation of Hydrocarbons in a Motored Engine. Ind. Eng. Chem. 1954, 46, 1024–1029. [Google Scholar] [CrossRef]
- Walsh, A.D. Knock In Internal Combustion Engines and the Anti-Knock Effect of Lead Tetra Ethyl. J. R. Inst. Chem. 1951, 75, 319–371. [Google Scholar] [CrossRef]
- Cheaney, D.E.; Davies, D.A.; Davis, A.; Hoare, D.E.; Protheroe, J.; Walsh, A.D. Effects of surfaces on combustion of methane and mode of action of anti-knocks containing metals. Symp. (Int.) Combust. 1958, 7, 183–187. [Google Scholar] [CrossRef]
- Graiff, L.B. The Mode of Action of Tetraethyllead and Supplemental Antiknock Agents; SAE Transactions: Warrendale, PA, USA, 1966. [Google Scholar] [CrossRef]
- Boot, M.D.; Tian, M.; Hensen, E.J.M.; Mani Sarathy, S. Impact of fuel molecular structure on auto-ignition behavior—Design rules for future high performance gasolines. Prog. Energy Combust. Sci. 2017, 60, 1–25. [Google Scholar] [CrossRef]
- Poulopoulos, S.; Philippopoulos, C. Influence of MTBE addition into gasoline on automotive exhaust emissions. Atmos. Environ. 2000, 34, 4781–4786. [Google Scholar] [CrossRef]
- Brocard, J.C.; Baronnet, F.; O’Neal, H.E. Chemical kinetics of the oxidation of methyl tert-butyl ether (MTBE). Combust. Flame 1983, 52, 25–35. [Google Scholar] [CrossRef]
- Le, Q.N.; Thomson, R.T. Process for the Production of Tertiary Alkyl Ether Rich FCC Gasoline. U.S. Patent 5,489,719, 6 February 1996. [Google Scholar]
- Tian, M.; McCormick, R.L.; Luecke, J.; de Jong, E.; van der Waal, J.C.; van Klink, G.P.M.; Boot, M.D. Anti-knock quality of sugar derived levulinic esters and cyclic ethers. Fuel 2017, 202, 414–425. [Google Scholar] [CrossRef]
- Christensen, E.; Williams, A.; Paul, S.; Burton, S.; McCormick, R.L. Properties and performance of levulinate esters as diesel blend components. Energy Fuels 2011, 25, 5422–5428. [Google Scholar] [CrossRef]
- Christensen, E.; Yanowitz, J.; Ratcliff, M.; McCormick, R.L. Renewable oxygenate blending effects on gasoline properties. Energy Fuels 2011, 25, 4723–4733. [Google Scholar] [CrossRef]
- Heyne, J.S.; Dryer, F.L. Uncertainty analysis in the use of chemical thermometry: A case study with cyclohexene. J. Phys. Chem. A 2013, 117, 5401–5406. [Google Scholar] [CrossRef]
- Heyne, J.S.; Dooley, S.; Dryer, F.L. Dehydration rate measurements for tertiary -butanol in a variable pressure flow reactor. J. Phys. Chem. A 2013, 117, 8997–9004. [Google Scholar] [CrossRef]
- Walsh, A.D. The effect of aromatic compounds on the vapour-phase oxidation of fuels. Part II.— The anti-knock effect of aromatic compounds in engines. Trans. Faraday Soc. 1949, 45, 1043–1048. [Google Scholar] [CrossRef]
- Dussan, K.; Won, S.H.; Ure, A.D.; Dryer, F.L.; Dooley, S. Chemical functional group descriptor for ignition propensity of large hydrocarbon liquid fuels. Proc. Combust. Inst. 2019, 37, 5083–5093. [Google Scholar] [CrossRef]
- Brown, J.E.; Markley, F.X.; Shapiro, H. Mechanism of Aromatic Amine Antiknock Action. Ind. Eng. Chem. 1955, 47, 2141–2146. [Google Scholar] [CrossRef]
- Mohamed, C. Suppression of reaction during rapid compression and its effect on ignition delay. Combust. Flame 1998, 112, 438–444. [Google Scholar] [CrossRef]
- Halstead, M.P.; Quinn, C.P. Inhibition of autoignition at high pressure. Combust. Flame 1973, 20, 223–230. [Google Scholar] [CrossRef]
- Zhang, P.; Yee, N.W.; Filip, S.V.; Hetrick, C.E.; Yang, B.; Green, W.H. Modeling study of the anti-knock tendency of substituted phenols as additives: An application of the reaction mechanism generator (RMG). Phys. Chem. Chem. Phys. 2018, 20, 10637–10649. [Google Scholar] [CrossRef]
- Dooley, S.; Won, S.H.; Chaos, M.; Heyne, J.; Ju, Y.; Dryer, F.L.; Kumar, K.; Sung, C.J.; Wang, H.; Oehlschlaeger, M.A.; et al. A jet fuel surrogate formulated by real fuel properties. Combust. Flame 2010, 157, 2333–2339. [Google Scholar] [CrossRef]
- Mueller, C.J.; Cannella, W.J.; Bays, J.T.; Bruno, T.J.; DeFabio, K.; Dettman, H.D.; Gieleciak, R.M.; Huber, M.L.; Kweon, C.B.; McConnell, S.S.; et al. Diesel Surrogate Fuels for Engine Testing and Chemical-Kinetic Modeling: Compositions and Properties. Energy Fuels 2016, 30, 1445–1461. [Google Scholar] [CrossRef]
- Naser, N.; Sarathy, S.M.; Chung, S.H. Ignition delay time sensitivity in ignition quality tester (IQT) and its relation to octane sensitivity. Fuel 2018, 233, 412–419. [Google Scholar] [CrossRef]
- McCormick, R.L.; Fioroni, G.; Fouts, L.; Christensen, E.; Yanowitz, J.; Polikarpov, E.; Albrecht, K.; Gaspar, D.J.; Gladden, J.; George, A. Selection Criteria and Screening of Potential Biomass-Derived Streams as Fuel Blendstocks for Advanced Spark-Ignition Engines. SAE Int. J. Fuels Lubr. 2017, 10, 442–460. [Google Scholar] [CrossRef]
- Naser, N.; Yang, S.Y.; Kalghatgi, G.; Chung, S.H. Relating the octane numbers of fuels to ignition delay times measured in an ignition quality tester (IQT). Fuel 2017, 187, 117–127. [Google Scholar] [CrossRef]
- Olarte, M.V.; Albrecht, K.O.; Bays, J.T.; Polikarpov, E.; Maddi, B.; Linehan, J.C.; O’Hagan, M.J.; Gaspar, D.J. Autoignition and select properties of low sample volume thermochemical mixtures from renewable sources. Fuel 2019, 238, 493–506. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Somers, K.P.; Simmie, J.M. Benchmarking Compound Methods (CBS-QB3, CBS-APNO, G3, G4, W1BD) against the Active Thermochemical Tables: Formation Enthalpies of Radicals. J. Phys. Chem. A 2015, 119, 8922–8933. [Google Scholar] [CrossRef] [PubMed]
- Simmie, J.M.; Somers, K.P. Benchmarking Compound Methods (CBS-QB3, CBS-APNO, G3, G4, W1BD) against the Active Thermochemical Tables: A Litmus Test for Cost-Effective Molecular Formation Enthalpies. J. Phys. Chem. A 2015, 119, 7235–7246. [Google Scholar] [CrossRef]
- Ghosh, M.K.; Howard, M.S.; Dooley, S. Accurate and standard thermochemistry for oxygenated hydrocarbons: A case study of ethyl levulinate. Proc. Combust. Inst. 2019, 37, 337–346. [Google Scholar] [CrossRef]
- ASTM International. ASTM D6890-18, Standard Test Method for Determination of Ignition Delay and Derived Cetane Number (DCN) of Diesel Fuel Oils by Combustion in a Constant Volume Chamber; ASTM International: West Conshohocken, PA, USA, 2018; Available online: www.astm.org (accessed on 7 November 2019).
- Singh, E.; Badra, J.; Mehl, M.; Sarathy, S.M. Chemical Kinetic Insights into the Octane Number and Octane Sensitivity of Gasoline Surrogate Mixtures. Energy Fuels 2017, 31, 1945–1960. [Google Scholar] [CrossRef]
- Richard, L.S.; Bernardes, C.E.S.; Diogo, H.P.; Leal, J.P.; da Piedade, M.E. Energetics of Cresols and of Methylphenoxyl Radicals. J. Phys. Chem. A 2007, 111, 8741–8748. [Google Scholar] [CrossRef]
- Curran, H.J.; Gaffuri, P.; Pitz, W.J.; Westbrook, C.K. A Comprehensive Modeling Study of n-Heptane Oxidation. Combust. Flame 1998, 114, 149–177. [Google Scholar] [CrossRef]
- Pelucchi, M.; Cavallotti, C.; Faravelli, T.; Klippenstein, S.J. H-Abstraction reactions by OH, HO2, O, O2 and benzyl radical addition to O2 and their implications for kinetic modelling of toluene oxidation. Phys. Chem. Chem. Phys. 2018, 20, 10607–10627. [Google Scholar] [CrossRef]
- Yuan, W.; Li, Y.; Pengloan, G.; Togbé, C.; Dagaut, P.; Qi, F. A comprehensive experimental and kinetic modeling study of ethylbenzene combustion. Combust. Flame 2016, 166, 255–265. [Google Scholar] [CrossRef]
- O’Malley, P.J. Hybrid Density Functional Studies of Phenoxyl Free Radicals Modeling α-Tocopheroxyl. J. Phys. Chem. B 2002, 106, 12331–12335. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Compound | RON | DCN | BDEOH | BDECH3 | KOH | KCH3 | KB | GTot | #CH3 |
|---|---|---|---|---|---|---|---|---|---|
| kcal mol−1 | s−1 | kcal mol−1 | |||||||
| P1 | 0.15 | 0.28 | 87.09 | n/a | 10.33 | n/a | 0.31 | −472.8 | 0 |
| P2 | 0.40 | −0.01 | 85.30 | 90.60 | 11.04 | 25.63 | 0.73 | −432.0 | 1 |
| P3 | 0.80 | 0.07 | 87.00 | 91.00 | 10.37 | 25.20 | 0.74 | −432.4 | 0 |
| P4 | 1.00 | −0.11 | 84.70 | 89.90 | 11.28 | 26.49 | 0.73 | −432.4 | 1 |
| P5 | 0.60 | −0.06 | 83.50 | 90.80 | 11.76 | 50.89 | 0.82 | −409.8 | 2 |
| P6 | 0.80 | 0.11 | 85.40 | 89.90 | 11.01 | 52.81 | 0.84 | −410.1 | 1 |
| P7 | 1.05 | −0.17 | 83.40 | 90.70 | 11.80 | 51.98 | 0.83 | −409.8 | 1 |
| P8 | 1.20 | −0.14 | 83.10 | 90.60 | 11.92 | 51.40 | 0.82 | −409.9 | 2 |
| P9 | 0.70 | −0.11 | 85.00 | 90.50 | 11.16 | 51.56 | 0.83 | −410.4 | 1 |
| P10 | 0.80 | 0.11 | 87.00 | 91.10 | 10.37 | 50.19 | 0.84 | −410.0 | 0 |
| P11 | 1.05 | −0.23 | 81.30 | 90.30 | 12.63 | 78.03 | 0.86 | −396.0 | 3 |
| P12 | 0.00 | 0.98 | n/a | n/a | n/a | n/a | n/a | n/a | n/a |
| Model 1 | Model 2 | |||||
|---|---|---|---|---|---|---|
| a0 + a1KCH3 + a2KOH + a3GTot + a4#CH3 | a0 + a1KB + a2GTot + a3#CH3 | |||||
| Estimate | Est | p-Value | Estimate | Est | p-Value | |
| a0 | −0.724 | 2.056 | 0.737 | 3.002 | 3.174 | 0.376 |
| a1 | 0.008 | 0.004 | 0.117 | −0.982 | 0.744 | 0.228 |
| a2 | −0.293 | 0.093 | 0.020 | 0.005 | 0.006 | 0.434 |
| a3 | −0.008 | 0.004 | 0.069 | −0.114 | 0.046 | 0.041 |
| a4 | 0.090 | 0.072 | 0.262 | |||
| Model 3 | Model 4 | |||||
|---|---|---|---|---|---|---|
| a0 + a1KCH3 + a2KOH + a3GTot + a4#CH3 | a0 + a1KB + a2GTot + a3#CH3 | |||||
| Estimate | Est | p-Value | Estimate | Est | p-Value | |
| a0 | 1.666 | 7.655 | 0.835 | −0.054 | 8.557 | 0.995 |
| a1 | −0.007 | 0.016 | 0.679 | 1.220 | 2.006 | 0.562 |
| a2 | 0.518 | 0.345 | 0.184 | 0.000 | 0.017 | 0.984 |
| a3 | 0.014 | 0.014 | 0.352 | 0.049 | 0.123 | 0.700 |
| a4 | −0.323 | 0.269 | 0.275 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ure, A.D.; Ghosh, M.K.; Rappo, M.; Dauphin, R.; Dooley, S. Rational Design and Testing of Anti-Knock Additives. Energies 2020, 13, 4923. https://doi.org/10.3390/en13184923
Ure AD, Ghosh MK, Rappo M, Dauphin R, Dooley S. Rational Design and Testing of Anti-Knock Additives. Energies. 2020; 13(18):4923. https://doi.org/10.3390/en13184923
Chicago/Turabian StyleUre, Andrew D., Manik K. Ghosh, Maria Rappo, Roland Dauphin, and Stephen Dooley. 2020. "Rational Design and Testing of Anti-Knock Additives" Energies 13, no. 18: 4923. https://doi.org/10.3390/en13184923
APA StyleUre, A. D., Ghosh, M. K., Rappo, M., Dauphin, R., & Dooley, S. (2020). Rational Design and Testing of Anti-Knock Additives. Energies, 13(18), 4923. https://doi.org/10.3390/en13184923