Abstract
In gas/solid conditions, different chemicals, such as diethylether, ethylene, butadiene, higher hydrocarbons, acetaldehyde, acetone and hydrogen, can be produced from ethanol with heterogeneous catalytic processes. The focus of this paper is the interplay of different reaction paths, which depend on thermodynamic factors as well as on kinetic factors, thus mainly from catalyst functionalities and reaction temperatures. Strategies for selectivity improvements in heterogeneously catalyzed processes converting (bio)ethanol into renewable chemicals and biofuels are also considered.
1. Introduction
In the near future, bioethanol produced by the fermentation of lignocellulosics or from microalgae feedstock [1] is expected to become a primary intermediate in the industrial organic chemistry based on renewables [2,3]. Somehow, this can be envisaged as a return to the past because bioethanol was largely used in the first half of twentieth century for the production of chemicals. As an example, ethylene has been produced from bioethanol in several countries since 1913 [4], while the production of butadiene from bioethanol was developed and applied industrially after 1920 [5]. In the past, the production of acetaldehyde from ethanol has also been realized industrially both through the dehydrogenation and oxidative dehydrogenation routes, although it has been successively substituted by the Wacker ethylene oxidation process [6].
A large number of different chemicals and fuels can be produced by converting ethanol [7]. Heterogeneous catalysis plays a fundamental role in developing and improving processes for the production of renewables from bioethanol [8].
In our laboratory, we undertook a number of investigations to efficiently convert ethanol into useful products at atmospheric pressure and in gas/solid catalytic processes. Working with different catalysts and in a range of conditions, we identified a series of different catalytic functionalities resulting in the production of several chemical intermediates. Similar reactions occur with different catalysts and sometimes compete with each other, thus reducing the selectivity of the desired product. In this paper, we will summarize data on the competition found for several of these reaction paths and we will address the selectivity limits of several processes.
2. Methods
Reaction enthalpy data were taken from [9], or when lacking, calculated using the same method referring to a gas phase with a temperature of 298.15 K and 1 bar using values reported by the Webbook of National Institute of Standards and Technology (NIST) and Poling et al. [10]. Reaction thermodynamics were evaluated through the calculation of ΔG0(T), as previously reported for a part of the considered reactions by Velu and Song [11,12].
Additionally, thermodynamic equilibria were also calculated in the diluted conditions used in our experiments using the Redlich–Kwong–Soave equation of state and a Gibbs reactor. Furnace temperature data were used, thus allowing a comparison of the experimental results. We did not use a reaction set but we only evaluated the thermodynamic equilibria of the given mixtures.
Specific and relevant data on the tested materials will be reported in each paragraph. The typical conditions applied for the laboratory catalytic tests are summarized in Table 1.

Table 1.
Experimental conditions applied in the catalytic tests (SS = steady state; TPSR = temperature-programmed surface reaction).
Most of the catalytic experiments were performed in quasi steady state (SS) conditions, as described in detail in Ref. [13]. Temperature-Programmed Surface Reaction (TPSR) experiments were also realized in the same line with continuous temperature increase and continuous online product analysis. Silica glass reactors (diameter = 6–7 mm) were used for testing the chosen reactions. Product analysis was realized by gas chromatography (Agilent 4890) by using both a flame ionization detector and thermo-conductivity detector and a Porabond Q/Molsieve 5A tandem column. In several cases, the product analyses were carried out by gas chromatography/mass spectrometry technique (FOCUS-ISQ instrument with a TG-SLQ column). For oxidative dehydrogenation and in many other cases to monitor eventual catalysts activation and deactivation, continuous online products analysis was performed using a Nicolet 6700 FT-IR instrument, equipped with a home-made gas cell. Definitions for the conversion of a reactant (R), XR, the selectivity and yield to a product (P), SP and YP, are the following:
3. Results
3.1. Evaluation of the Thermodynamic Equilibria in Ethanol Conversion to Chemicals
In Table 2 the enthalpy of the reactions considered in the discussion are summarized.
Table 2.
Chemical reactions considered and discussed in the text and their reaction enthalpies per mol of reactant.
In Figure 1, the data arising from the evaluation of ΔG0 as a function of temperature for several ethanol conversion reactions are included. In Figure 1 left, the dehydration reactions producing ethyl ether and ethylene, according to reactions (1) and (2), respectively, are considered. The two reactions have an opposite behavior, being the endothermic production of ethylene favored at a temperature higher than 373 K while the slightly exothermic production of diethyl ether (DEE) is only favored below 473 K.
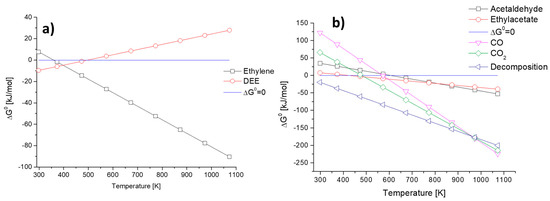
Figure 1.
The ΔG0 of selected ethanol conversion reactions as a function of temperature: (a) ethylene and DEE-forming reactions and (b) acetaldehyde, ethyl acetate, CO, CO2 and decomposition reactions.
In Figure 1 right, the reactions producing hydrogen are inserted. Reactions to acetaldehyde (3), ethyl acetate (4), CO (5) CO2 (6) and ethanol decomposition to methane CO and hydrogen (7) were considered.
It is possible to state that ethanol decomposition is far the most favored reaction as temperature increases and only above 973 K steam reforming reactions (reactions (5) and (6)) become even more favored. Ethyl acetate production through reaction (4) is favored for a temperature higher than 473 K but its less favored than all the other considered reactions. Ethanol dehydrogenation to acetaldehyde (3) starts to be favored (ΔG0 < 0) above 600 K, where also CO production through steam reforming starts to be favored. Instead, CO2 production through steam reforming displays a lower temperature where ΔG0 = 0, thus being favored at a temperature higher than 500 K. This clearly depicts that ethanol conversion into useful chemicals in high yield is possible by choosing the proper operation temperature and a selective catalyst toward the wanted reaction.
In order to have a suitable comparison with the experimental data obtained in diluted conditions, we additionally carried out a thermodynamic equilibrium evaluation for pure ethanol in the production of different chemicals and with different product mixtures in our experimental conditions.
In Figure 2a, the data concerning the equilibrium of the ethanol/ethylene/diethyl ether/water (DEE stands for diethyl ether) system are reported. It is evident that full conversion of ethanol into diethyl ether is possible only at very low temperature.
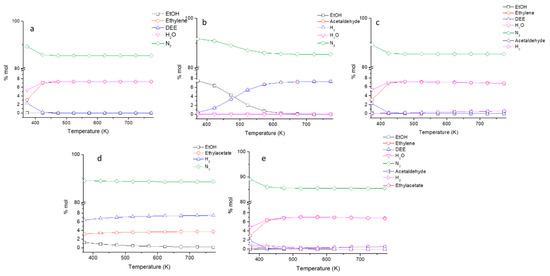
Figure 2.
Evaluated thermodynamic equilibrium in terms of molar fractions for the different investigated mixtures with the same reaction condition employed in the catalytic experiments; (a) N2/ethanol/water/DEE/ethylene, (b) N2/ethanol/acetaldehyde/H2, (c) N2/ethanol/water/DEE/ethylene/acetaldehyde/H2, (d) N2/ethanol/ethyl acetate/H2, and (e) N2/ethanol/water/DEE/ethylene/acetaldehyde/H2/ethyl acetate.
Starting from 423 K, the full conversion of ethanol into ethylene is possible. On the other hand, unitary ethanol conversion to acetaldehyde by dehydrogenation is possible above 600 K, while only partial conversion can be obtained at lower temperatures (Figure 2b). However, when dehydration and dehydrogenation reactions are considered together (Figure 2c), it becomes evident that dehydrogenation is far less favored than dehydration also at high temperature. Thus, to produce acetaldehyde from ethanol with high yields, the catalyst must be very active for dehydrogenation and inactive for dehydration. Finally, from the data in Figure 2d, it is evident that the ethyl acetate production through reaction (4) is favored for a temperature higher than 473 K but is less favored than all the other considered reactions, approaching a 100% yield to ethyl acetate only above 623 K. In all the cases and for several product mixtures, ethylene dehydration remains the most favored reaction (Figure 2e).
3.2. Ethanol to Ethyl Ether
Ethanol can be dehydrated to diethyl ether (DEE, or ethyl ether) according to reaction (1). This reaction is weakly exothermic with a limited change in entropy (ΔS0298 = −0.025 Jmol−1K−1), thus being an equilibrium reaction, and it can be completed only with selective water removal. This reaction also formally competes with the dehydration to ethylene, which is an endothermic process. For these reasons, the possibility to achieve a very high yield of DEE is practically impossible in a conventional flow reactor.
Over most acidic catalysts, ethanol is converted into DEE at moderate temperature and partial ethanol conversion. In Figure 3, the behavior of a typical acid catalyst (H-mordenite zeolite, H-MOR with a Si/Al2 = 20) for this reaction is shown.
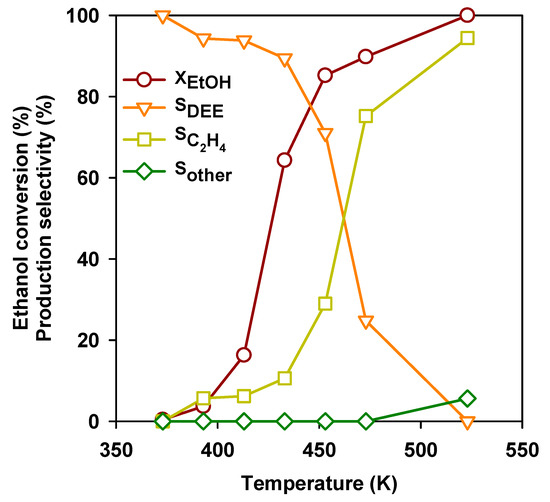
Figure 3.
Ethanol conversion and selectivities to the products as a function of temperature over the H-mordenite zeolite. GHSV = 10,000 h−1 and H-MOR with Si/Al2 = 20.
Over this catalyst, ethanol conversion starts to be significant above 390 K and achieves completion at 513 K. At low temperature, the most abundant product at low temperature is DEE, but its selectivity progressively declines. Above 473 K, ethylene becomes the predominant product, while starting from 493 K, other products, remarkably butenes, start to be observed. Over H-MOR zeolite and in the laboratory scale experiments, the maximum DEE yield can be achieved at 453 K. In Table 3, the behavior of a number of catalytic materials in DEE production are compared in the same experimental conditions.
Table 3.
Maximum DEE yields obtained on the different acid catalysts, and the byproduct yields.
Although alumina is reported to be industrially used for this reaction [14], we obtained higher DEE yields, and at a lower temperature, with different protonic zeolites [15] and over WO3/TiO2 catalysts [16]. Indeed, both protonic zeolites and WO3/TiO2 catalysts display strong Brønsted acidity together with Lewis acidity, while the alumina only possesses strong Lewis acid sites. Apparently, the best DEE yield can be obtained with protonic zeolite beta (H-BEA) and WO3/TiO2, while the best selectivity to DEE is obtained over protonic zeolite ferrierite (H-FER), although with lower conversion. With most of the tested zeolites, ethylene is the only detected (and useful) by-product in the conditions allowing maximum DEE yields. Over the other tested catalysts, several other by-products are also formed in very low amounts (acetaldehyde, butenes) already at moderate ethanol conversion, when the maximum yield to DEE is obtained.
Thus, the DEE yield is unavoidably affected by the equilibrium nature of reaction (1), and also by the concurrency of ethylene production occurring with two alternative formal reactions: the cracking (dehydration) of ethanol, reaction (2), and/or the cracking of DEE, reaction (8).
These reactions are endothermic and more favored at higher temperature than reaction (1). However, they are already favored in our reaction conditions at low temperature, i.e., when we find high DEE yields (e.g., at 400 K): thus, the control of selectivity to DEE is associated to kinetics and the catalysis of reactions (1), (2) and (8), not to thermodynamics. In other words, reaction (1) producing DEE is faster than reaction (2) at low temperature. As further proof of this, the DEE/ethylene selectivity ratio strongly depends on contact time [17]: the lower the contact time, the higher the DEE selectivity but the lower the ethanol conversion, in agreement with the nature of reaction (8) as a successive reaction with respect to reaction (1).
Spectroscopic data indicate a key role of alkoxide species [15,18], whose decomposition produces ethylene. The same intermediates may react with the gas-phase and/or weakly adsorbed undissociated ethanol, producing DEE. Exactly the opposite occurs when the DEE reacts with the clean surface producing ethoxy groups and ethanol [19].
A further crucial point is likely to be associated to catalyst stability. It seems likely that zeolites, due to their very strong Brønsted acidity, tend to produce also in mild conditions some carbon residues that finally may cause significant deactivation. Thus, this would require expensive steaming or oxidation steps for catalysts regeneration.
3.3. Ethanol to Ethylene
As aforementioned, the catalysts allowing high DEE selectivity at 450–550 K, also give rise to the highest yields to ethylene usually in the range of 500–600 K (Table 4). Although ethylene can be produced by reaction (8) at low ethanol conversion, it is quite clear that reaction (2), which is thermodynamically favored in our diluted conditions starting from near 400 K (refer to Figure 2), predominates in particular at both high temperatures and ethanol conversion. Yield to ethylene approaches 100% in our conditions on several catalysts, including protonic zeolites, sulphated and tungstated zirconia and amorphous silica–alumina (ASA). The best yields are obtained over catalysts that do not show maximum activity, while the most active ones tend to produce higher hydrocarbons that obviously contribute to reduce ethylene yields. Interestingly, it was also found that on several of these most active catalysts, a pronounced carbon deposition occurs.
Table 4.
Maximum ethylene yields obtained on different acid catalysts, and by-products.
Ethylene yields are clearly affected mainly by the over-conversion of ethylene into hydrocarbons, while coke deposition on the catalyst surface is also a clear drawback of the system. Both these phenomena are associated to a too high activity and are more relevant for Brønsted acid catalysts. i.e., zeolites [15], than for Lewis acid ones such as alumina. For these reasons, we attempted to modify alumina with the intention of increasing ethylene selectivity at almost total conversion. We approached different catalytic aluminas, from the same source and possibly with comparable surface areas; we also tested aluminas with different porosity, arising from different sources, extremely pure aluminas and aluminas with different dopants such as silica, lanthanum, potassium and calcium.
Alumina porosity does not seem to significantly influence catalyst performances. The use of ultrapure alumina may in fact further improve ethylene selectivity up to 99.8% yield, likely due to the lower extent of contamination by transition metals (Ti, Fe, Cr) that may be involved in ethylene over-conversion [20]. The addition of small amounts of silica to alumina result in a decrease in catalytic activity, due to the reduced amount of Lewis acid sites, without any addition of Brønsted acidity [21]. There is no evidence of improving ethylene selectivity as well. Moreover, the presence of small amounts of alkali and calcium decrease the activity without increasing, apparently, selectivity to ethylene at a high conversion [18,22]. The addition of lanthanum can increase selectivity at full conversion [19,23], depending on the samples and synthetic route, while a lower activity is observed at a low temperature.
The data confirm that both ethanol and some zeolites can give rise to very high yields in ethylene, while zeolite may be applied at lower temperatures. The main point for industrial application is probably related to the rate of deactivation, the procedure for regeneration and the stability of the catalyst over multiple regeneration cycles.
3.4. Ethanol to Hydrocarbons
The conversion of ethanol into gasoline-range hydrocarbons over the H-ZSM5 zeolite was already reported many years ago [24] and is considered today as a potential way for the production of renewable fuels [25]. In Figure 4, the ethanol conversion and selectivities to main products over the H-ZSM5 zeolite (Si/Al2 = 50) are reported as a function of reaction temperature. As for most other acid catalysts, DEE is produced starting from 373 K, and later DEE yield decreases while ethylene selectivity grows up to above 90 % at 523 K. However, at even higher temperatures, ethylene selectivity also drops, with the growth of butenes and a number of other compounds including BTX aromatics.
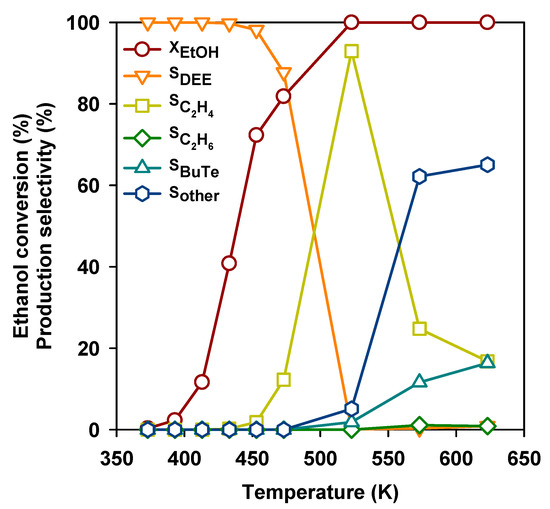
Figure 4.
Ethanol conversion and selectivities to the products as a function of temperature over the H-ZSM-5 (Si/Al2 = 50) zeolite.
The reported data suggest that ethylene is indeed a primary product of ethanol, over-converting into other hydrocarbons [26]. This is evident from the W/F data (weight/volumetric flow rate) reported in Figure 5, where the effect of contact time on the product distribution is reported at a total conversion (673 K) over the H-ZSM5 sample previously calcined at 700 K. It is evident that, while ethylene selectivity and yield progressively decrease, the formation of liquid hydrocarbons (C5+ aliphatics and BTX aromatics) but also of other hydrocarbon gases, increases. Additionally, DEE and also acetaldehyde are produced: CH3CHO detection might reveal the catalytic activity of carbon deposits as the dehydrogenation catalyst. Indeed, a significant amount of carbon deposits are formed during the reaction.
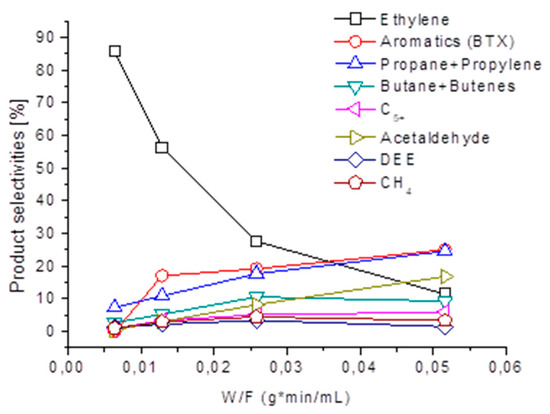
Figure 5.
Effect of W/F (weight/volumetric flow rate) on the selectivities to the gaseous products over H-ZSM5, at full ethanol conversion at 673 K.
This reaction likely consists of a first step where a proton-catalyzed oligomerization of ethylene occurs, i.e., the over-conversion of the product of the endothermic reaction (2), producing higher olefins. These two steps are followed by cyclization/dehydrogenation to aromatics or by heavy products cracking to light gases.
The proposed reaction network can be exemplified by the reactions (9) and (10), producing 1-hexene and benzene, respectively, although ethylene is very likely the primary intermediate in the formation of higher hydrocarbons.
Indeed, in the investigated temperature range, ethylene oligomerization is still possible, while aromatization and the cracking of longer chains is already favored. Interestingly, selectivities and yields are almost constant with increasing time on stream, suggesting that a steady state was attained in our timescale (few hours) with the co-presence of zeolite protonic sites and active carbon. The addition of the dehydrogenating activity of Ga2O3 to the protonic Mobil five (H-MFI) zeolite is reported to improve aromatics production from bioethanol [27].
It can be mentioned that the production of olefins from ethanol are formally dehydration reactions (e.g., reactions (2) and (9)). Moreover, the production of higher alcohols from ethanol are dehydration reactions. Among these, an interesting additional process is the coupling of ethanol to produce n-butanol, a useful fuel additive and intermediate. To produce this reaction, catalysts with a tuned acid-base but also redox properties are needed [28].
3.5. Ethanol to Acetaldehyde by Dehydrogenation
As already cited, over several catalysts acetaldehyde production by dehydrogenation competes with dehydration reactions, at high temperature. This is found, e.g., in the case of transition metal containing oxides like ZnAl2O4 [29] and Zn–Zr mixed oxides, TiO2, ZrO2, Nb2O5, as well as on basic catalysts, such as calcined hydrotalcite [16] and “basic” Y zeolites [30].
The dehydrogenation to acetaldehyde, reaction (3), is an endothermic reaction, thermodynamically favored above 600 K (refer to Figure 1) and is, as typical for dehydrogenation reactions, most efficiently catalyzed by metal-based catalysts.
The ethanol dehydrogenation process was already developed and used at the industrial level in the fifties using either metal or zinc oxide catalysts [6]. Very high selectivities can be obtained at a moderate conversion over some metallic catalysts such as gold [31] and copper [32], while over some copper catalysts, high selectivities were obtained also at quite high conversion. In our experimental conditions [33,34], we obtained acetaldehyde yields of 90 % over Cu/ZnAl2O4 catalysts at 573–623 K, with a copper loading of 17 wt.%. At higher temperatures, full ethanol conversion was achieved but acetaldehyde yield declines due to the over-conversion to acetone, ethyl acetate and other products including propene. However, copper deactivation in the dehydrogenation upon time on stream was found. In Figure 6, the effect of the time on stream is reported for ethanol conversion over a home-made catalyst (CuO/ZnAl2O4, CuO 9.1 wt.%) at 623 K, where still the Tamman temperature is not reached and thus copper sintering, it is assumed to be not a prevailing effect. By looking at the concentration of ethanol and acetaldehyde, it appears clear that selectivity to acetaldehyde as well as ethanol conversion are reduced upon time on stream, while both the ethyl ether and ethylene selectivities increase. This behavior is attributed to the deactivation of copper metal particles for the increasing formation of carbon residues. In fact, two reactions proceed in a parallel way: together with the deactivation of the dehydrogenation route (reaction (3)), the dehydration routes to DEE and ethylene (reactions (1) and (2)) catalyzed by the uncovered support become faster for the concurrent increased reactant availability. Instead, the production of ethyl acetate (reaction (4)) is almost stable, probably being due to acetaldehyde condensation catalyzed by the support too but depending on the acetaldehyde availability.
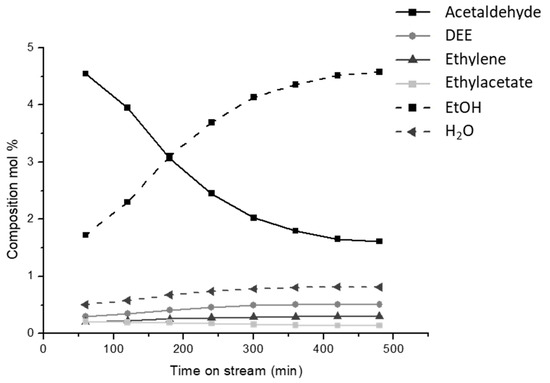
Figure 6.
Effect of the time on stream for the ethanol conversion 9.1% CuO/ZnAl2O4 catalyst at 623 K in terms of molar composition at a weight hourly space velocity (WHSV) = 12 h−1.
Although over metal catalysts selectivity can be very high at a low conversion, the loss of selectivity at low temperature may come from the activity of the supports in dehydration reactions. Moreover, the condensation of acetaldehyde to ethyl acetate can contribute. Indeed, high selectivity in the production of ethyl acetate is achievable over copper catalysts working at higher pressures and contact times [35]. At high temperatures, a number of acetaldehyde over-conversion reactions start to be important.
These data confirm that copper-based catalysts can give rise to high yields in acetaldehyde also at high conversion, in contrast to what is mainly observed with noble metal catalysts that give rise to even 100% selectivities but only at a moderate to low conversion, while deactivation is a common problem [36].
3.6. Acetaldehyde Over-Conversion: Ethanol to Butadiene and Acetone
A number of other products can be obtained with a high selectivity from ethanol, probably through acetaldehyde as the main intermediate [7]. In particular, these reactions can be efficiently catalyzed by basic materials or transition metal oxides displaying some basicity. As an example, we found that over TiO2 (anatase) C4 hydrocarbons can be produced with a 40% yield [16], among which significant amounts of 1,3-butadiene. The production of butadiene from ethanol, the so-called Lebedev process [37], was carried out years ago industrially over basic catalysts (MgO–SiO2) and is an object of renewed interest today.
Among the interesting green chemicals, we obtained significant yields in acetone over a zinc oxide catalyst containing sulphate impurities. As shown in Figure 7, over this catalyst acetaldehyde selectivity is quite high also at a low conversion, together with ethylene selectivity. At 723 K, where ethanol conversion is almost complete, acetone becomes the most abundant product, with a 50 % yield. Thus, ethanol conversion to acetone might represent an alternative way for producing renewable acetone, an important chemical product as well as a useful intermediate.
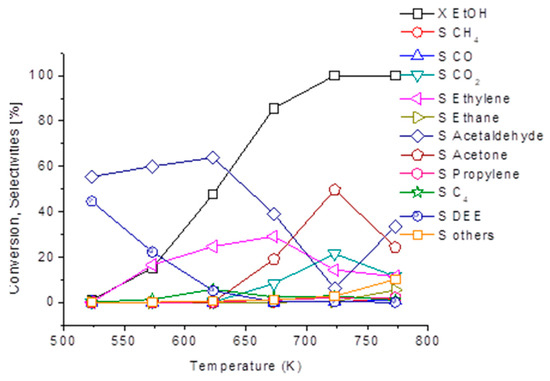
Figure 7.
Ethanol conversion and selectivity to products over the ZnO catalyst. Experimental conditions: 7% ethanol, 0.9% water, N2 balance. Total flow rate 40 NmL/min, catalysts weight 0.25 g.
It seems likely that acetone is mostly produced by the aldol condensation of acetaldehyde with the following cracking of the aldol intermediate (reaction 11), thus resulting in the overall stoichiometry reported in reaction (12).
As shown above, the reaction producing acetone is endothermic and thermodynamically favored at temperatures higher than 400 K. This reaction, together with dehydrations, may limit the production of acetaldehyde by ethanol dehydrogenation also over metal catalysts. Over ZnAl2O4 the effect of contact time on ethanol conversion was also investigated. Acetone yield increases with contact time while acetaldehyde yield decreases, up to reaching the maximum for acetone, followed by a drop. This supports the above mechanism, showing that also acetone was over-converted at higher temperatures and contact times.
3.7. Ethanol to Acetaldehyde by Oxidative Dehydrogenation
As cited above, the production of acetaldehyde from ethanol pure dehydrogenation is affected by equilibrium limitations, moderate selectivity and catalyst deactivation due to carbon deposition. To overcome these drawbacks, oxidative dehydrogenation might be considered as a suitable way for acetaldehyde production. The reaction (13) is exothermic and favored at any temperature, as any selective oxidation reaction. As reported by Ref. [38] for this reaction ΔG0298 = −190 kJ/molEtOH, nearly constant with temperature. This reaction has been carried out industrially using silver-based catalysts in air at 720–770 K, with a low conversion (30–50%) and quite high selectivity (up to 95%) [6]. Nowadays, a renewed interest in ethanol oxidative dehydrogenation has grown. This reaction is investigated both in liquid or gaseous phases mainly over metallic catalysts such as those based on gold [39,40] or silver. On the other hand, interesting reports were published concerning ethanol oxidation to acetaldehyde in gas phase with vanadia- [41] and molybdena-based catalysts [42]. In Figure 8, the data are reported concerning the oxidative dehydrogenation of ethanol over a wet impregnated 12% MoO3/Al2O3(5% SiO2) catalyst prepared home-made in our laboratories.
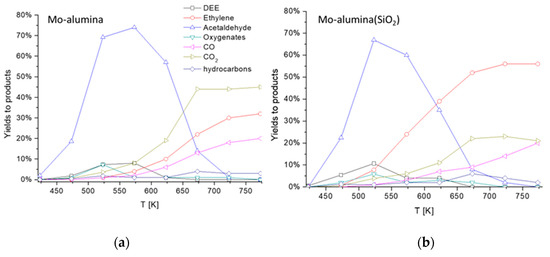
Figure 8.
Results in the oxidative ethanol dehydrogenation over a (a) 12% MoO3/Al2O3 and (b) 12% MoO3/Al2O3 (5% SiO2) catalyst. Yields to acetaldehyde, ethylene, ethyl ether and CO2. Experimental conditions: 2.1% C2H6O, 14.8% O2, 83.1% N2 total flow 80 NmL/min, catalyst weight 0.5000 g.
Over this yet-to-be optimized system, 75% yield to acetaldehyde is obtained at 523 K for alumina-supported catalysts, while 70% yield can be obtained over SiO2 (5 wt.%) containing alumina. At higher temperatures, acetaldehyde selectivity is limited by the coproduction of ethylene and ethyl ether. At a higher temperature, when conversion becomes total, ethylene and CO2 become the most abundant products. Thus, acetaldehyde yields are limited by the concurrency of reactions (1) and (2) which could occur on the support.
To determine whatever preparation effect the support and molybdena deposition could have, we prepared a home-made alumina and deposed silica on it using tetraethoxysilane (TEOS) as a silicon source. Molybdenum was added by an incipient wetness technique using ammonium molybdate tetrahydrate aqueous solution. The catalysts were tested in TPSR mode in order to achieve information as well on possible reaction intermediates. TPSR result is reported in Figure 9. In this case, an 80% yield to acetaldehyde is obtained in the unsteady state experiment, at 590 K, with the coproduction of ethylene and some DEE. Our experiments confirm that small amounts of silica may have a beneficial effect on acetaldehyde yield, and that the selectivity to acetaldehyde is reduced by the parallel formation of the dehydration products DEE and ethylene. Thus, attempts to reduce the acidity of the catalyst will be undertaken to increase the acetaldehyde yields.
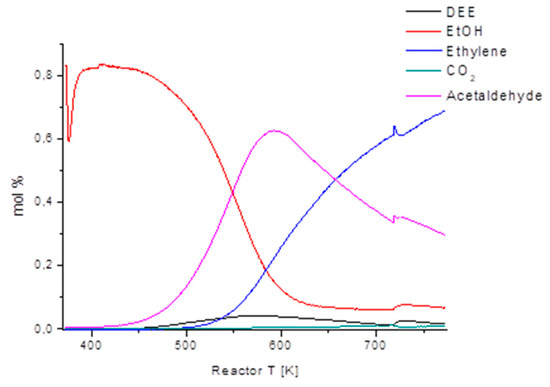
Figure 9.
Ethanol TPSR in the oxidative dehydrogenation experiment (0.8% ethanol/6% O2 and N2 balance, GHSV = 240,000 h−1) over 7%Mo/Al2O3 (2.5% SiO2) home-made catalyst.
Although oxidative dehydrogenation may have several advantages over the dehydrogenation route, the system we investigated to date does not seem to be still really competitive, due the insufficiently high acetaldehyde yields. Further investigation is needed either trying tomoderae its acidity, or using gold- and silver-based catalysts, or alternatively vanadia-based systems and other molybdate-based systems. Catalytic processes are also under study for the production of acetic acid [43] and acetonitrile [44] from ethanol by oxidation and ammoxidation, respectively.
3.8. Ethanol Total Oxidation
As already said, the partial oxidation of ethanol to acetaldehyde competes with total oxidation, reaction (14), which is much more exothermic and thermodynamically favored. The catalytic combustion of VOCs (volatile organic compounds) like ethanol is of wide interest to remove organic pollutants and smelling compounds from industrial waste gases, as well from waste treatment plants, without producing NOx. Due to the application of bioethanol as a gasoline component, its catalytic combustion is also relevant in after-treatment systems for Otto engine cars. Noble metals, like mainly platinum and palladium, are mostly applied for such applications [45]. However, transition metal catalysts can also be used just for oxidizing oxygenated VOCs, which are more reactive than hydrocarbons. For example, as unsupported K-doped manganese oxides catalyze the complete oxidation of ethanol to CO2 at 430 K, alumina-supported catalysts with similar compositions allow full combustion at 473–500 K [46]. Poorly active catalysts indeed produce incomplete combustion with some acetaldehyde coproduction [47] by reaction (9), and due to the much higher toxicity of acetaldehyde with respect to ethanol, this needs to be completely avoided.
According to its high activity in undergoing oxidation, ethanol can be used as a reductant. One of the possible applications of ethanol as a reductant is the “cycle-reforming process” or “chemical loop reforming” [48], where ethanol is oxidized by iron oxide-based materials in a first step, producing metallic iron, CO2 and water. Metallic iron is re-oxidized in a second step by water-producing hydrogen and then regenerating iron oxide. Ethanol is mostly fully oxidized to CO2 and water, although some CO and methane are also formed, suggesting that some cracking also occurs upon this reaction [49]. Although this process essentially implies a total oxidation of ethanol, the stoichiometry of the overall cycle is that of steam reforming, see below.
3.9. Ethanol to Syngas by Partial Oxidation
As said, noble metals efficiently catalyze the total oxidation of ethanol usually realized in excess air. However, noble metals can also be applied to produce hydrogen from ethanol using partial oxidation with oxygen, reaction (15) [9,38]. This reaction can have advantages over the strongly endothermic ethanol steam-reforming process (see Section 3.10) because it is only slightly endothermic, because is the result of the strongly exothermic catalytic combustion (reaction (14)) together with the endothermic steam reforming. This reaction does not require an external heat source and can be spontaneously carried out in the short-contact-time mode [50].
3.10. Ethanol to Syngas by Steam Reforming
Ethanol steam reforming is a way to produce syngas and hydrogen from a renewable source [38,51,52]. The main reaction (5) is endothermic and reversible and thus favored at relatively high temperatures and low pressures. Due to the concurrence of the water gas shift equilibrium (16) which partially results in the “global” steam reforming process reaction (6).
Thus, by ethanol steam reforming, a syngas composed of CO, CO2 and H2 can be obtained. Indeed, hydrogen production is usually limited by the presence of variable amounts of methane, which can arise from two main paths: the methanation of carbon oxides (17) and (18) and/or the cracking of ethanol (7) or of the intermediate, i.e., acetaldehyde (22).
Typical catalysts for these reactions are the supported metals over a wide range of supports. Noble metals are very active, but the scientific interest is also largely devoted to the development of cheaper and more robust catalysts based on transition metals, i.e., Ni and Co.
We investigated catalysts based on Ni- and/or Co- supported over aluminates [53], zirconia [54], ceria [55], silica [56], and alumina [57,58,59,60,61,62,63,64,65,66]. Over Ni/Al2O3 catalysts, steam reforming predominates at 873 K and above. At low temperature, diethyl ether and ethylene form, due to the activity of the uncovered support, as well as ethanol dehydrogenation to acetaldehyde, likely occurring over nickel.
To better distinguish the activity of the supports from that of the metal particles, we investigated the activity of unsupported metal particles. On unsupported Ni particles [63], at a low reaction temperature (523 K), already the reaction occurs producing mainly CO and CH4 in a near 1:1 ratio, with small amounts of acetaldehyde, suggesting that dehydrogenation first occurs (reaction (6)) followed by the very fast acetaldehyde cracking (reaction (22)), globally giving ethanol cracking (reaction (21). Steam reforming, associated to water conversion, hydrogen production and reduced CH4 formation, becomes predominant at 673 K, and this can be considered as the required temperature for water activation. The higher activity of Ni nanoparticles at a lower temperature than the one observed on Ni/Al2O3 is likely due to the competition, in the latter case, with the dehydration reactions occurring on an alumina uncovered surface. Over Co nanoparticles [59,67,68], the catalytic activity per mass of catalyst is lower than on Ni, the selectivity to acetaldehyde is higher than on Ni, while the production of methane is lower, suggesting that acetaldehyde cracking (reaction (22)) is definitely faster on Nickel than on Cobalt. Moreover, considering the formulation of bimetallic nanoparticles, the presence of Co in Nickel decreases the selectivity to methane [67], thus increasing the hydrogen yield. This result is confirmed both for unsupported catalysts and for catalysts supported on zinc aluminates [53] as well as on zirconias [54].
Thus, while it seems likely that on cobalt catalysts ethanol steam reforming implies first a dehydrogenation to acetaldehyde, and later the steam reforming of acetaldehyde, on nickel it is still not known what is the origin of residual methane and what is the real molecule being steam reformed. To unravel this point, we investigated three reactions over a commercial catalyst: methane steam reforming (MSR), CO2 methanation [13,69] and ethanol steam reforming (ESR).
In similar conditions, ethanol steam reforming is faster than methane steam reforming, as indeed expected (Table 5).
Table 5.
Reactants conversion and the product yields in ethanol steam reforming (ESR) and methane steam reforming (MSR) over the commercial Ni/Calcium aluminate catalyst.
Over this catalyst, the methane coproduction selectivity upon ESR is 14% at 773 K, when the CO2/CH4 product molar ratio is 5.9, with small amounts of CO. At 893 K, selectivity to methane is essentially zero. At 773 K, the conversion of methane in MSR is 60% when the CO2/CH4 molar ratio is near 1. On the other hand, methane is fully converted at a temperature of 933 K. The CO2/CH4 ratio obtained in ESR is far higher than that expected for the sequence of ethanol (acetaldehyde) cracking + MSR, suggesting that the main molecule undergoing steam reforming is acetaldehyde, not methane, over this catalyst. On the other hand, at 773 K the CO2 conversion in methanation in excess hydrogen is 70% with a methane yield of 58%, thus with a CO2/CH4 molar ratio of 30 / 58 = 0.52. This datum, compared with the above, does not allow to exclude that CO2 methanation has a role in the origin of the CH4 produced during ESR over this catalyst, although over Ni/Al2O3 it seems clear that CH4 at room temperature originates at least in part from ethanol or acetaldehyde cracking. These data confirm that supported Ni catalysts are useful for renewable hydrogen production from bioethanol steam reforming and that commercial catalysts well proven for methane steam reforming can also be applied to ESR.
4. Conclusions
The data reported above clearly show that ethanol is a versatile platform chemical to produce a large number of chemicals and fuels. It is reactive over a wide range of different catalysts, such as Lewis and Brønsted acid catalysts, basic catalysts, oxidation catalysts and metallic catalysts, in reducing and oxidizing environments, producing well defined reaction paths. In Scheme 1, the reaction paths discussed here are schematically summarized by pointing out the fundamental role of heterogeneous catalysis in green industrial chemistry.
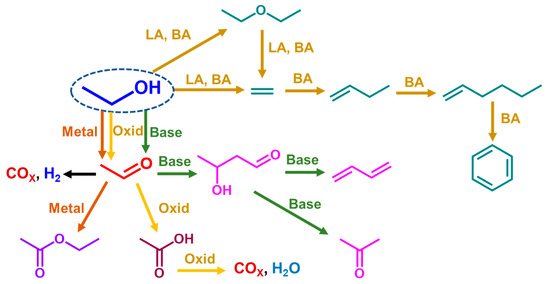
Scheme 1.
Main reaction paths concerned in the ethanol gas-phase catalytic conversions. LA = Lewis acid; BA = Brønsted acid; Oxid = oxide catalysts.
For most of the cited reactions, quite detailed surface chemical and mechanistic studies were realized, e.g., using infrared spectroscopy. In all cases, surface ethoxy groups are considered to be those involved in the ethanol adsorption step and consequently, represent the first surface molecular unit in the reactions. The ethoxy group can crack over acidic catalyst-producing ethylene by an elimination reaction or can react as a nucleophile, which attacks undissociated ethanol producing diethyl ether. Instead, when it is adsorbed over a reducible cation, a metallic center, or near strong basic sites, the ethoxy group can lose a hydride species producing adsorbed acetaldehyde. This introduces high reactivity at the C2 carbon atom, which is relevant over basic catalysts allowing aldol-like chemistry. This also allows steam reforming with the oxidation of both carbon atoms by water adsorbed on metal centers.
The relevant versatility of this molecule, however, is also a cause of poor selectivity in the conversion realized over many complex catalysts, in particular when still not really optimized. In fact, complex catalytic materials have, together with the desired functionality, also undesired functionalities. As for example, for the supported catalysts the inhibition of the support activity may be approached to obtain a high selectivity to the desired product. This is the case, e.g., of supported metal catalysts where dehydrogenation or steam reforming activity may compete with the acid-base activity of the support.
Author Contributions
Conceptualization, all authors; Formal analysis, G.G., P.R.; Investigation, G.G., T.K.P., G.P.; Methodology, G.G., P.R.; Supervision, G.B.; Writing—original draft, G.B.; Writing—review and editing, G.G., P.R., T.K.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors acknowledge Niccolò Ardoino, Andrea Crenna and Tullio Cavattoni for carrying out a part of the catalytic experiments here included.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ASA | Amorphous Silica–Alumina |
| BA | Brønsted Acid |
| BTX | Benzene-Toluene-Xylenes |
| C2 | Carbon Atom in Position 2 |
| DEE | Diethylether |
| ESR | Ethanol Steam Reforming |
| EtOH | Ethanol |
| FID | Flame Ionization Detector |
| GHSV | Gas Hourly Space Velocity |
| H-BEA | Protonic Zeolite Beta |
| H-FER | Protonic Zeolite Ferrierite |
| H-MFI | Protonic Zeolite Mobil Five |
| H-MOR | Protonic Zeolite Mordenite |
| HY | Protonic zeolite Faujasite |
| LA | Lewis Acid |
| MSR | Methanol Steam Reforming |
| NIST | National Institute of Standards and Technology |
| Oxid | Oxide Catalyst |
| SS | Steady State |
| TCD | Thermo-Conductivity Detector |
| TPSR | Temperature Programmed Surface Reaction |
| VOC | Volatile Organic Compounds |
| WHSV | Weight Hourly Space Velocity |
| USY | Ultra Stable Y zeolite |
| Symbols used | |
| G0 | standard reaction free energy (kJ/mol) |
| ΔH0298 | standard reaction enthalpy at 298 K (kJ/mol) |
| ΔS0298 | standard reaction entropy at 298 K (J/mol K) |
| F | molar flow (mol/time) |
| F | volumetric flow (mL/min) |
| n | stoichiometric coefficients ratio (-) |
| S | selectivity (-) |
| T | temperature (K) |
| X | conversion (-) |
| Y | yield (-) |
| W | catalyst weight (g) |
| Subscripts | |
| in | reactor entrance |
| out | reactor exit |
| P | product |
| R | reactant |
References
- Jambo, S.A.; Abdulla, R.; Mohd Azhar, S.H.; Marbawi, H.; Gansau, J.A.; Ravindra, P. A review on third generation bioethanol feedstock. Renew. Sustain. Energy Rev. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- Soccol, C.R.; Faraco, V.; Karp, S.G.; Vandenberghe, L.P.S.; Thomaz-Soccol, V.; Woiciechowski, A.L.; Pandey, A. Lignocellulosic Bioethanol: Current Status and Future Perspectives, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; ISBN 9780128168561. [Google Scholar]
- Cavani, F.; Albonetti, S.; Basile, F.; Gandini, A. Chemicals and Fuels from Biobased Bulding Blocks, 1st ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; ISBN 9789811518034. [Google Scholar]
- Mohsenzadeh, A.; Zamani, A.; Taherzadeh, M.J. Bioethylene Production from Ethanol: A Review and Techno-economical Evaluation. ChemBiolEng Rev. 2017, 4, 75–91. [Google Scholar] [CrossRef]
- Makshina, E.V.; Dusselier, M.; Janssens, W.; Degrève, J.; Jacobs, P.A.; Sels, B.F. Review of old chemistry and new catalytic advances in the on-purpose synthesis of butadiene. Chem. Soc. Rev. 2014, 43, 7917–7953. [Google Scholar] [CrossRef]
- Eckert, M.; Fleischmann, G.; Jira, R.; Bolt, H.M.; Golka, K. Acetaldehyde. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Sun, J.; Wang, Y. Recent Advances in Catalytic Conversion of Ethanol to Chemicals. ACS Catal. 2014, 4, 1078–1090. [Google Scholar] [CrossRef]
- Phung, T.K.; Busca, G. Selective Bioethanol Conversion to Chemicals and Fuels via Advanced Catalytic Approaches. In Biorefinery of Alternative Resources: Targeting Green Fuels and Platform Chemicals; Springer: Singapore, 2020; pp. 75–103. [Google Scholar]
- Sawatmongkhon, B.; Theinnoi, K.; Wongchang, T.; Haoharn, C.; Wongkhorsub, C.; Tsolakis, A. Hydrogen production via the catalytic partial oxidation of ethanol on a platinum-rhodium catalyst: Effect of the oxygen-to-ethanol molar ratio and the addition of steam. Energy Fuels 2019, 33, 6742–6753. [Google Scholar] [CrossRef]
- Poling, B.E.; Prausnitz, J.M.; O’Connell, J.P. Properties of Gases and Liquids, Fifth Edition; McGraw-Hill Education: New York, NY, USA, 2001; ISBN 9780070116825. [Google Scholar]
- Subramani, V.; Song, C. Catalysis; Spivey, J.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2007; Volume 20, ISBN 978-0-85404-244-9. [Google Scholar]
- Subramani, V.; Sharma, P.; Zhang, L.; Liu, K. Catalytic Steam Reforming Technology for the Production of Hydrogen and Syngas. In Hydrogen and Syngas Production and Purification Technologies, 1st ed.; Liu, K., Song, C., Subramani, V., Eds.; John Wiley & Sons Ltd, Inc.: Hoboken, NJ, USA, 2009; pp. 14–126. ISBN 9780470561256. [Google Scholar]
- Garbarino, G.; Pugliese, F.; Cavattoni, T.; Busca, G.; Costamagna, P. A Study on CO2 Methanation and Steam Methane Reforming over Commercial Ni/Calcium Aluminate Catalysts. Energies 2020, 13, 2792. [Google Scholar] [CrossRef]
- Sakuth, M.; Mensing, T.; Schuler, J.; Heitmann, W.; Strehlke, G.; Mayer, D. Ethers, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010. [Google Scholar]
- Phung, T.K.; Proietti Hernández, L.; Lagazzo, A.; Busca, G. Dehydration of ethanol over zeolites, silica alumina and alumina: Lewis acidity, Brønsted acidity and confinement effects. Appl. Catal. A Gen. 2015, 493, 77–89. [Google Scholar] [CrossRef]
- Phung, T.K.; Hernández, L.P.; Busca, G. Conversion of Ethanol over transition metal oxide catalysts: Effect of tungsta addition on catalytic behaviour of titania and zirconia. Appl. Catal. A Gen. 2015, 489, 180–187. [Google Scholar] [CrossRef]
- Phung, T.K.; Busca, G. Diethyl ether cracking and ethanol dehydration: Acid catalysis and reaction paths. Chem. Eng. J. 2015, 272, 92–101. [Google Scholar] [CrossRef]
- Phung, T.K.; Lagazzo, A.; Rivero Crespo, M.Á.; Sánchez Escribano, V.; Busca, G. A study of commercial transition aluminas and of their catalytic activity in the dehydration of ethanol. J. Catal. 2014, 311, 102–113. [Google Scholar] [CrossRef]
- Garbarino, G.; Prasath Parameswari Vijayakumar, R.; Riani, P.; Finocchio, E.; Busca, G. Ethanol and diethyl ether catalytic conversion over commercial alumina and lanthanum-doped alumina: Reaction paths, catalyst structure and coking. Appl. Catal. B Environ. 2018, 236, 490–500. [Google Scholar] [CrossRef]
- Garbarino, G.; Travi, I.; Pani, M.; Carnasciali, M.M.; Busca, G. Pure vs. ultra-pure γ-alumina: A spectroscopic study and catalysis of ethanol conversion. Catal. Commun. 2015, 70, 77–81. [Google Scholar] [CrossRef]
- Phung, T.K.; Busca, G. Ethanol dehydration on silica-aluminas: Active sites and ethylene/diethyl ether selectivities. Catal. Commun. 2015, 68, 110–115. [Google Scholar] [CrossRef]
- Phung, T.K.; Herrera, C.; Larrubia, M.Á.; García-Diéguez, M.; Finocchio, E.; Alemany, L.J.; Busca, G. Surface and catalytic properties of some γ-Al2O3 powders. Appl. Catal. A Gen. 2014, 483, 255–264. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Valsamakis, I.; Chitsazan, S.; Riani, P.; Finocchio, E.; Flytzani-Stephanopoulos, M.; Busca, G. Acido-basicity of lanthana/alumina catalysts and their activity in ethanol conversion. Appl. Catal. B Environ. 2017, 200, 458–468. [Google Scholar] [CrossRef]
- Costa, E.; Uguina, A.; Aguado, J.; Hernandez, P.J. Ethanol to gasoline process: Effect of variables, mechanism, and kinetics. Ind. Eng. Chem. Process Des. Dev. 1985, 24, 239–244. [Google Scholar] [CrossRef]
- Eagan, N.M.; Kumbhalkar, M.D.; Buchanan, J.S.; Dumesic, J.A.; Huber, G.W. Chemistries and processes for the conversion of ethanol into middle-distillate fuels. Nat. Rev. Chem. 2019, 3, 223–249. [Google Scholar] [CrossRef]
- Phung, T.K.; Radikapratama, R.; Garbarino, G.; Lagazzo, A.; Riani, P.; Busca, G. Tuning of product selectivity in the conversion of ethanol to hydrocarbons over H-ZSM-5 based zeolite catalysts. Fuel Process. Technol. 2015, 137, 290–297. [Google Scholar] [CrossRef]
- Li, Z.; Lepore, A.W.; Salazar, M.F.; Foo, G.S.; Davison, B.H.; Wu, Z.; Narula, C.K. Selective conversion of bio-derived ethanol to renewable BTX over Ga-ZSM-5. Green Chem. 2017, 19, 4344–4352. [Google Scholar] [CrossRef]
- Cimino, S.; Lisi, L.; Romanucci, S. Catalysts for conversion of ethanol to butanol: Effect of acid-base and redox properties. Catal. Today 2018, 304, 58–63. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Villa García, M.; Finocchio, E.; Sánchez Escribano, V.; Busca, G. A study of ethanol conversion over zinc aluminate catalyst. React. Kinet. Mech. Catal. 2018, 124, 503–522. [Google Scholar] [CrossRef]
- Lari, G.M.; Desai, K.; Mondelli, C.; Pérez-Ramírez, J. Selective dehydrogenation of bioethanol to acetaldehyde over basic USY zeolites. Catal. Sci. Technol. 2016, 6, 2706–2714. [Google Scholar] [CrossRef]
- Wang, C.; Garbarino, G.; Allard, L.F.; Wilson, F.; Busca, G.; Flytzani-Stephanopoulos, M. Low-Temperature Dehydrogenation of Ethanol on Atomically Dispersed Gold Supported on ZnZrOX. ACS Catal. 2016, 6, 210–218. [Google Scholar] [CrossRef]
- Finger, P.H.; Osmari, T.A.; Costa, M.S.; Bueno, J.M.C.; Gallo, J.M.R. The role of the interface between Cu and metal oxides in the ethanol dehydrogenation. Appl. Catal. A Gen. 2020, 589, 117236. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Villa García, M.; Finocchio, E.; Sanchez Escribano, V.; Busca, G. A study of ethanol dehydrogenation to acetaldehyde over copper/zinc aluminate catalysts. Catal. Today 2020, 354, 167–175. [Google Scholar] [CrossRef]
- Pampararo, G.; Garbarino, G.; Riani, P.; Garcia, M.V.; Escribano, V.S.; Busca, G. A study of ethanol dehydrogenation to acetaldehyde over supported copper catalysts: Catalytic activity, deactivation and regeneration. Appl. Catal. A Gen. 2020, 117710. [Google Scholar] [CrossRef]
- Carotenuto, G.; Tesser, R.; Di Serio, M.; Santacesaria, E. Kinetic study of ethanol dehydrogenation to ethyl acetate promoted by a copper/copper-chromite based catalyst. Catal. Today 2013, 203, 202–210. [Google Scholar] [CrossRef]
- Morales, M.V.; Asedegbega-Nieto, E.; Castillejos-López, E.; Bachiller-Baeza, B.; Guerrero-Ruiz, A. Difference in the deactivation of Au catalysts during ethanol transformation when supported on ZnO and on TiO2. RSC Adv. 2018, 8, 7473–7485. [Google Scholar] [CrossRef]
- Ochoa, J.V.; Bandinelli, C.; Vozniuk, O.; Chieregato, A.; Malmusi, A.; Recchi, C.; Cavani, F. An analysis of the chemical, physical and reactivity features of MgO–SiO2 catalysts for butadiene synthesis with the Lebedev process. Green Chem. 2016, 18, 1653–1663. [Google Scholar] [CrossRef]
- Subramani, V.; Song, C. Advances in Catalysis and Processes for Hydrogen Production from Ethanol Reforming. In Catalysis; Royal Society of Chemistry: Cambridge, UK, 2007; pp. 65–106. [Google Scholar]
- Liu, P.; Hensen, E.J.M. Highly Efficient and Robust Au/MgCuCr2O4 Catalyst for Gas-Phase Oxidation of Ethanol to Acetaldehyde. J. Am. Chem. Soc. 2013, 135, 14032–14035. [Google Scholar] [CrossRef]
- Mostrou, S.; Nagl, A.; Ranocchiari, M.; Föttinger, K.; van Bokhoven, J.A. The catalytic and radical mechanism for ethanol oxidation to acetic acid. Chem. Commun. 2019, 55, 11833–11836. [Google Scholar] [CrossRef] [PubMed]
- Čičmanec, P.; Ganjkhanlou, Y.; Kotera, J.; Hidalgo, J.M.; Tišler, Z.; Bulánek, R. The effect of vanadium content and speciation on the activity of VOx/ZrO2 catalysts in the conversion of ethanol to acetaldehyde. Appl. Catal. A Gen. 2018, 564, 208–217. [Google Scholar] [CrossRef]
- Chumbhale, V.R.; Sonsale, A.Y.; Awasarkar, P.A. Oxidative Dehydrogenation of Ethanol over Binary Oxides of Sn and Mo. In Studies in Surface Science and Catalysis; Elsevier Inc.: Amsterdams, The Netherlands, 1998; Volume 113, pp. 479–485. [Google Scholar]
- Li, X.; Iglesia, E. Selective Catalytic Oxidation of Ethanol to Acetic Acid on Dispersed Mo-V-Nb Mixed Oxides. Chem. A Eur. J. 2007, 13, 9324–9330. [Google Scholar] [CrossRef] [PubMed]
- Folco, F.; Velasquez Ochoa, J.; Cavani, F.; Ott, L.; Janssen, M. Ethanol gas-phase ammoxidation to acetonitrile: The reactivity of supported vanadium oxide catalysts. Catal. Sci. Technol. 2017, 7, 200–212. [Google Scholar] [CrossRef]
- Liotta, L.F. Catalytic oxidation of volatile organic compounds on supported noble metals. Appl. Catal. B Environ. 2010, 100, 403–412. [Google Scholar] [CrossRef]
- Peluso, M.A.; Pronsato, E.; Sambeth, J.E.; Thomas, H.J.; Busca, G. Catalytic combustion of ethanol on pure and alumina supported K-Mn oxides: An IR and flow reactor study. Appl. Catal. B Environ. 2008, 78. [Google Scholar] [CrossRef]
- Gallegos, M.V.; Peluso, M.A.; Finocchio, E.; Thomas, H.J.; Busca, G.; Sambeth, J.E. Removal of VOCs by catalytic process. A study of MnZnO composites synthesized from waste alkaline and Zn/C batteries. Chem. Eng. J. 2017, 313. [Google Scholar] [CrossRef]
- Trevisanut, C.; Mari, M.; Millet, J.-M.M.; Cavani, F. Chemical-loop reforming of ethanol over metal ferrites: An analysis of structural features affecting reactivity. Int. J. Hydrogen Energy 2015, 40, 5264–5271. [Google Scholar] [CrossRef]
- Ochoa, J.V.; Trevisanut, C.; Millet, J.-M.M.; Busca, G.; Cavani, F. In situ DRIFTS-MS study of the anaerobic oxidation of ethanol over spinel mixed oxides. J. Phys. Chem. C 2013, 117, 23908–23918. [Google Scholar] [CrossRef]
- Livio, D.; Diehm, C.; Donazzi, A.; Beretta, A.; Deutschmann, O. Catalytic partial oxidation of ethanol over Rh/Al2O3: Spatially resolved temperature and concentration profiles. Appl. Catal. A Gen. 2013, 467, 530–541. [Google Scholar] [CrossRef]
- Nahar, G.; Dupont, V. Hydrogen via steam reforming of liquid biofeedstock. Biofuels 2012, 3, 167–191. [Google Scholar] [CrossRef]
- Bion, N.; Duprez, D.; Epron, F. Design of Nanocatalysts for Green Hydrogen Production from Bioethanol. ChemSusChem 2012, 5, 76–84. [Google Scholar] [CrossRef]
- Busca, G.; Costantino, U.; Montanari, T.; Ramis, G.; Resini, C.; Sisani, M. Nickel versus cobalt catalysts for hydrogen production by ethanol steam reforming: Ni-Co-Zn-Al catalysts from hydrotalcite-like precursors. Int. J. Hydrog. Energy 2010, 35, 5356–5366. [Google Scholar] [CrossRef]
- Resini, C.; Concepción Herrera Delgado, M.; Presto, S.; Alemany, L.J.; Riani, P.; Marazza, R.; Ramis, G.; Busca, G. Yttria-stabilized zirconia (YSZ) supported Ni-Co alloys (precursor of SOFC anodes) as catalysts for the steam reforming of ethanol. Int. J. Hydrog. Energy 2008, 33, 3728–3735. [Google Scholar] [CrossRef]
- Moretti, E.; Storaro, L.; Talon, A.; Chitsazan, S.; Garbarino, G.; Busca, G.; Finocchio, E. Ceria-zirconia based catalysts for ethanol steam reforming. Fuel 2015, 153, 166–175. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Cavattoni, T.; Busca, G. CO2 hydrogenation and ethanol steam reforming over Co/SiO2 catalysts: Deactivation and selectivity switches. Catal. Today 2020. [Google Scholar] [CrossRef]
- Garbarino, G.; Sanchez Escribano, V.; Finocchio, E.; Busca, G. Steam reforming of phenol-ethanol mixture over 5% Ni/Al2O3. Appl. Catal. B Environ. 2012, 147, 113–114. [Google Scholar] [CrossRef]
- Garbarino, G.; Romero Perez, A.; Finocchio, E.; Busca, G. A study of the deactivation of low loading Ni/Al2O3 steam reforming catalyst by tetrahydrothiophene. Catal. Commun. 2013, 38, 67–73. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Lucchini, M.A.; Canepa, F.; Kawale, S.; Busca, G. Cobalt-based nanoparticles as catalysts for low temperature hydrogen production by ethanol steam reforming. Int. J. Hydrogen Energy 2013, 38, 82–91. [Google Scholar] [CrossRef]
- Garbarino, G.; Lagazzo, A.; Riani, P.; Busca, G. Steam reforming of ethanol-phenol mixture on Ni/Al2O3: Effect of Ni loading and sulphur deactivation. Appl. Catal. B Environ. 2013, 129. [Google Scholar] [CrossRef]
- Garbarino, G.; Campodonico, S.; Perez, A.R.; Carnasciali, M.M.; Riani, P.; Finocchio, E.; Busca, G. Spectroscopic characterization of Ni/Al2O3 catalytic materials for the steam reforming of renewables. Appl. Catal. A Gen. 2013, 452, 163–173. [Google Scholar] [CrossRef]
- Chitsazan, S.; Sepehri, S.; Garbarino, G.; Carnasciali, M.M.; Busca, G. Steam reforming of biomass-derived organics: Interactions of different mixture components on Ni/Al2O3 based catalysts. Appl. Catal. B Eniron. 2016, 187, 386–398. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Lucchini, M.A.; Canepa, F.; Busca, G. Unsupported versus alumina-supported Ni nanoparticles as catalysts for steam/ethanol conversion and CO2 methanation. J. Mol. Catal. A Chem. 2014, 383, 10–16. [Google Scholar] [CrossRef]
- Garbarino, G.; Chitsazan, S.; Phung, T.K.; Riani, P.; Busca, G. Preparation of supported catalysts: A study of the effect of small amounts of silica on Ni/Al2O3 catalysts. Appl. Catal. A Gen. 2015, 505, 86–97. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Valsamakis, I.; Chitsazan, S.; Riani, P.; Finocchio, E.; Flytzani-Stephanopoulos, M.; Busca, G. A study of Ni/Al2O3 and Ni-La/Al2O3 catalysts for the steam reforming of ethanol and phenol. Appl. Catal. B Environ. 2015, 174, 21–34. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Canepa, F.; Busca, G. Cobalt nanoparticles mechanically deposited on α-Al2O3: A competitive catalyst for the production of hydrogen through ethanol steam reforming. J. Chem. Technol. Biotechnol. 2019, 94, 538–546. [Google Scholar] [CrossRef]
- Garbarino, G.; Cavattoni, T.; Riani, P.; Brescia, R.; Canepa, F.; Busca, G. On the Role of Support in Metallic Heterogeneous Catalysis: A Study of Unsupported Nickel–Cobalt Alloy Nanoparticles in Ethanol Steam Reforming. Catal. Lett. 2019, 149, 929–941. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Cavattoni, T.; Canepa, F.; Busca, G. Unsupported cobalt nanoparticles as catalysts: Effect of preparation method on catalytic activity in CO2 methanation and ethanol steam reforming. Int. J. Hydrogen Energy 2019, 44, 27319–27328. [Google Scholar] [CrossRef]
- Costamagna, P.; Pugliese, F.; Cavattoni, T.; Busca, G.; Garbarino, G. Modeling of Laboratory Steam Methane Reforming and CO2 Methanation Reactors. Energies 2020, 13, 2624. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).