A Novel Dual Fuel Reaction Mechanism for Ignition in Natural Gas–Diesel Combustion




Abstract
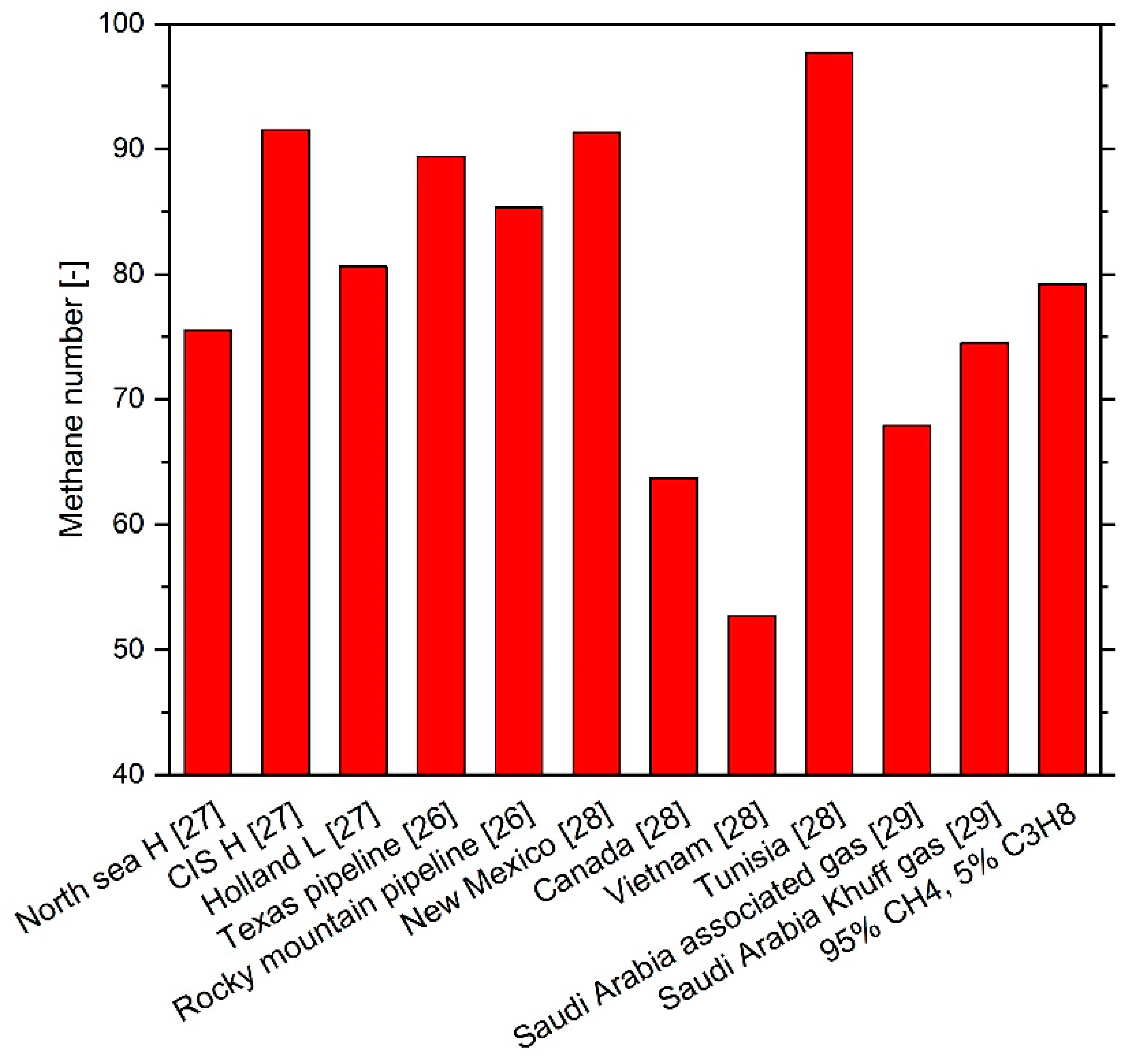
1. Introduction
2. Materials and Methods
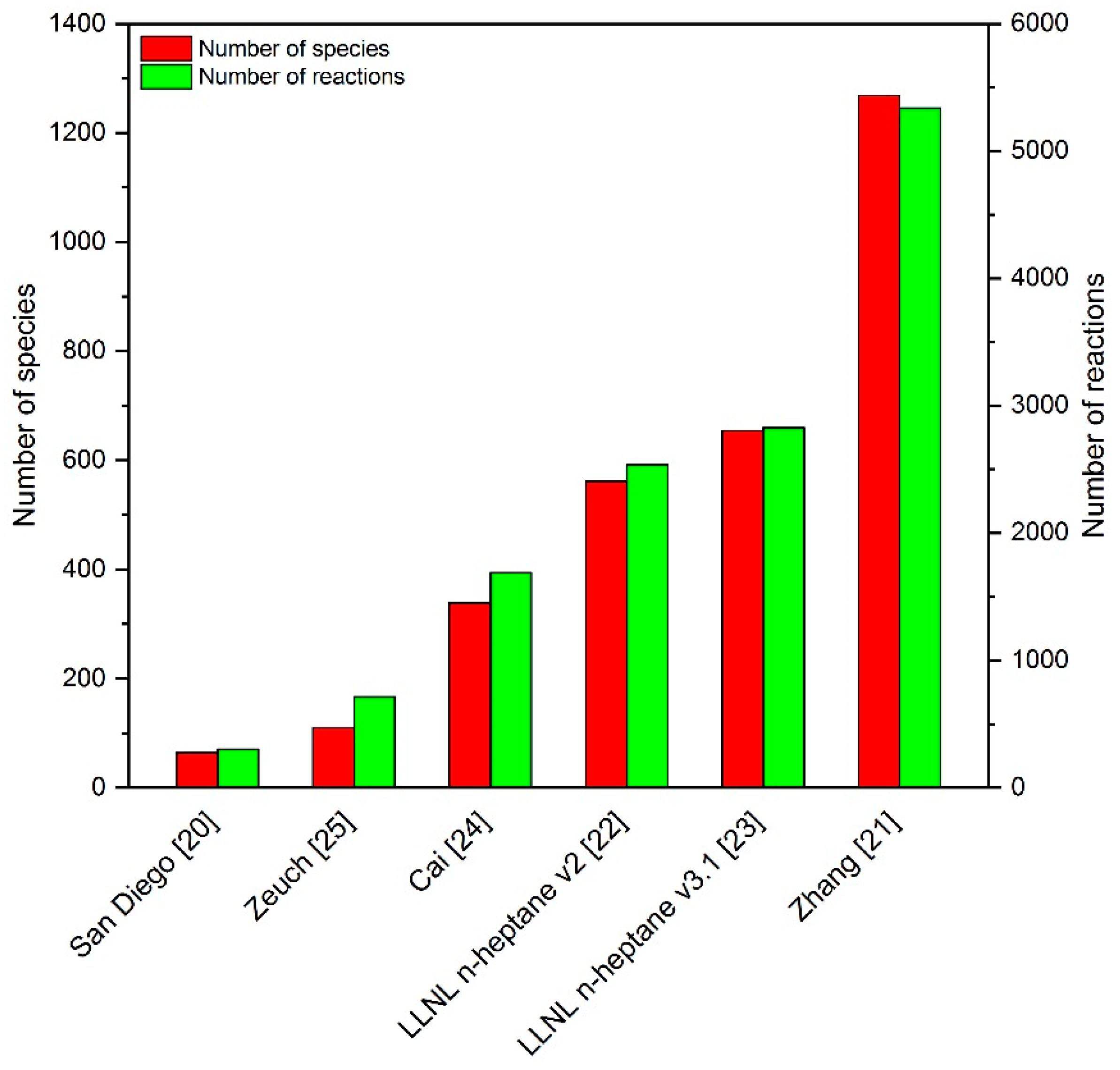
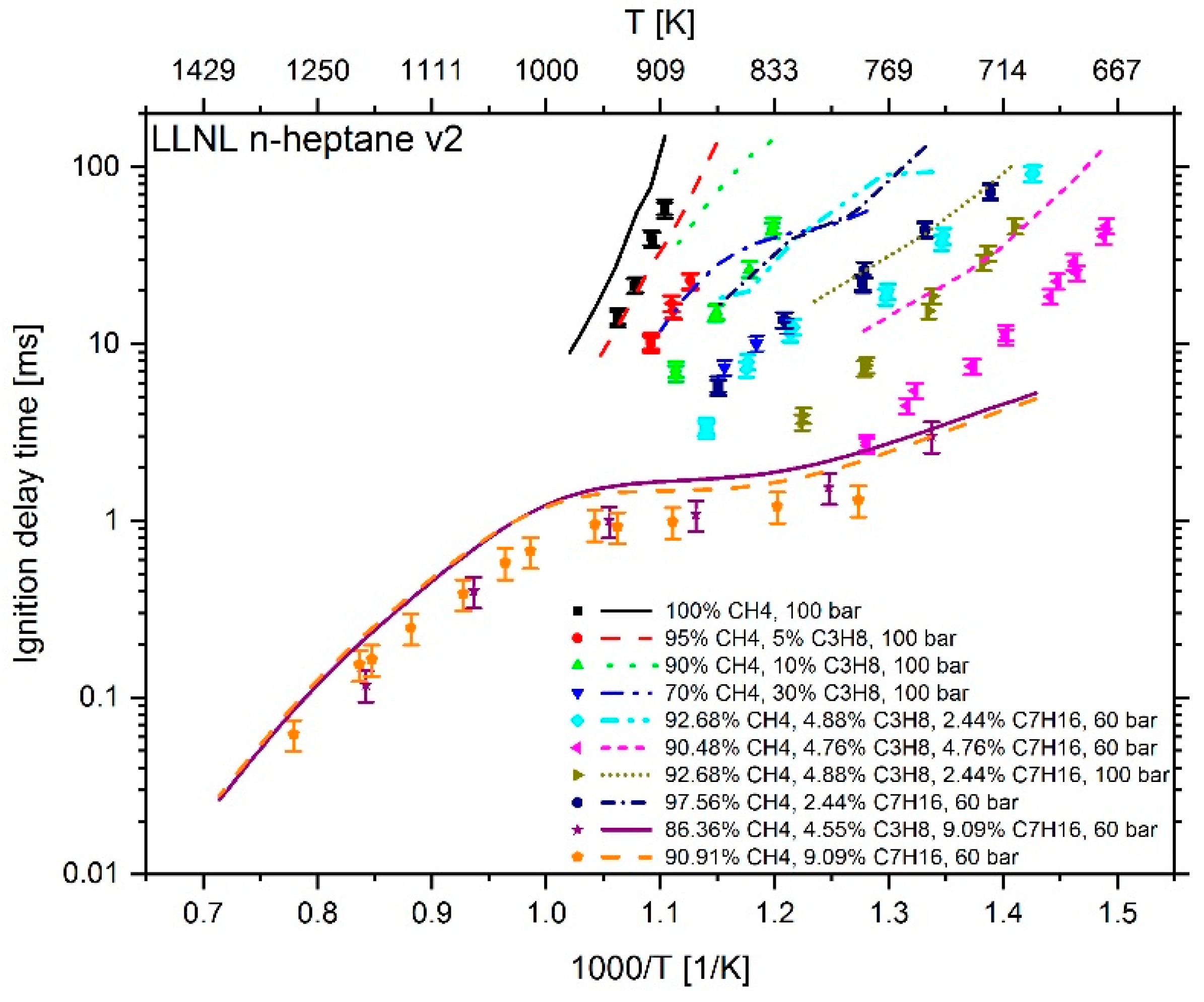
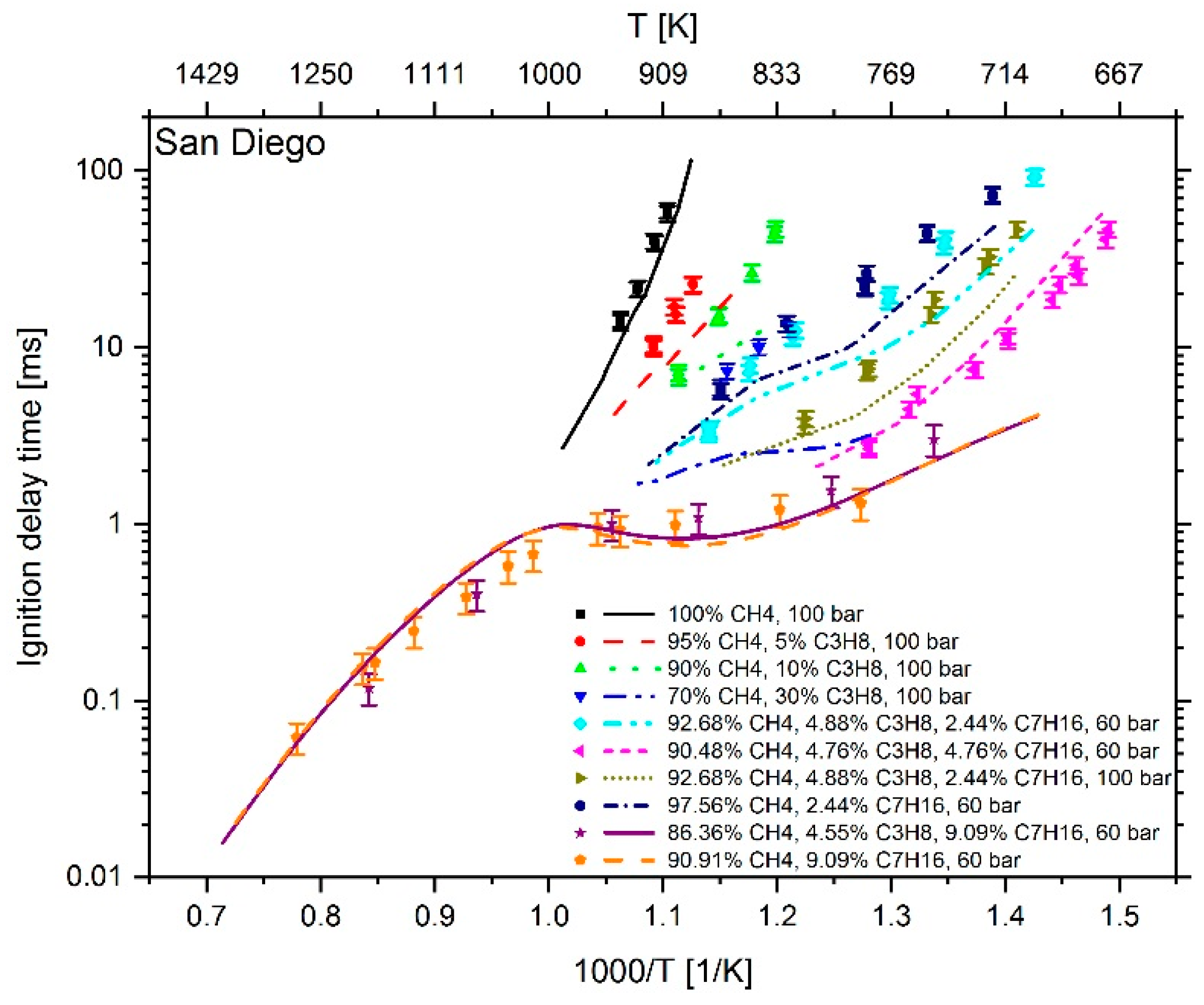
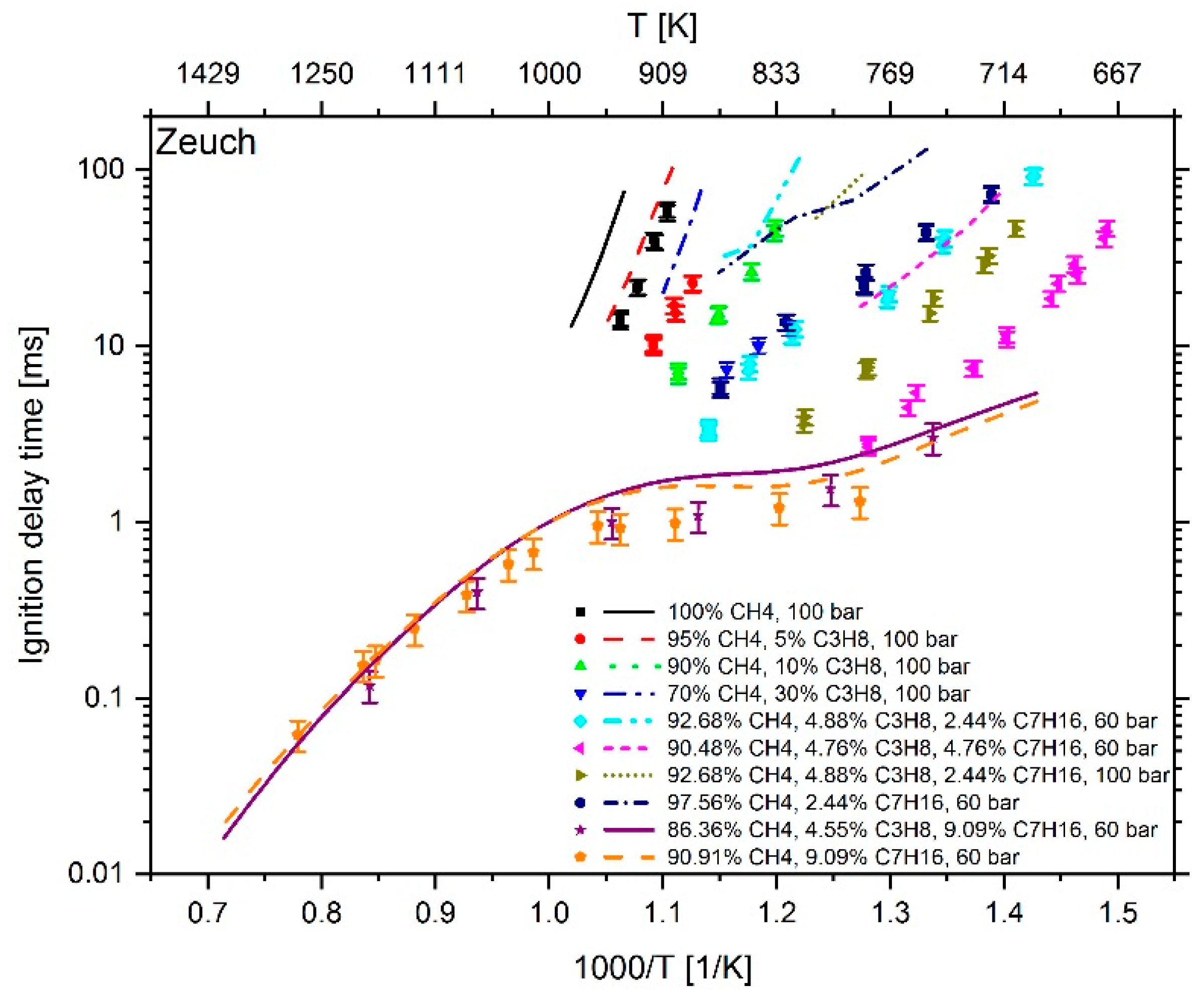
2.1. Reaction Mechanism Selection
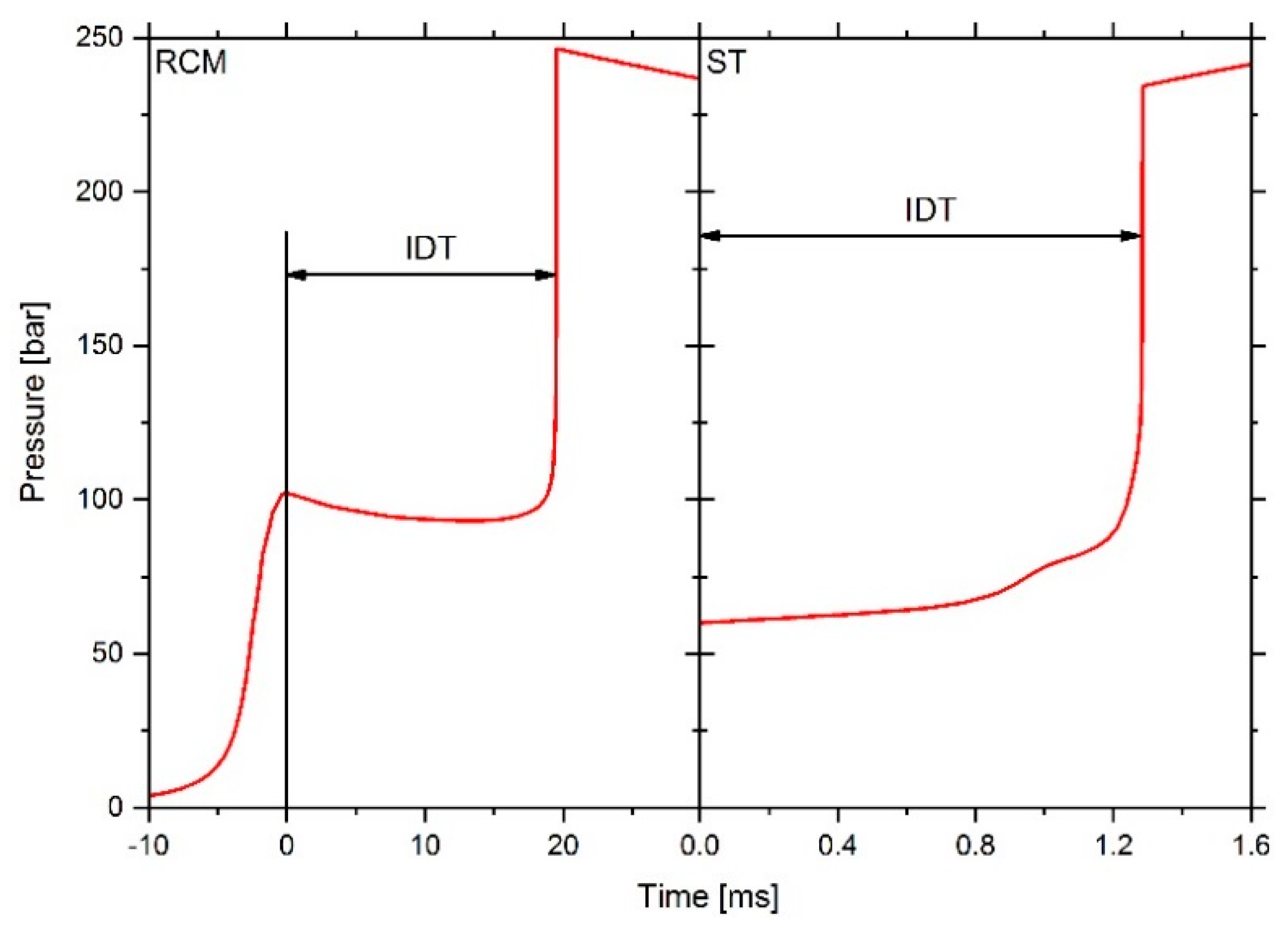
2.2. Investigation of Inhomogeneous Fuel Mixtures
2.3. Mechanism Adjustment
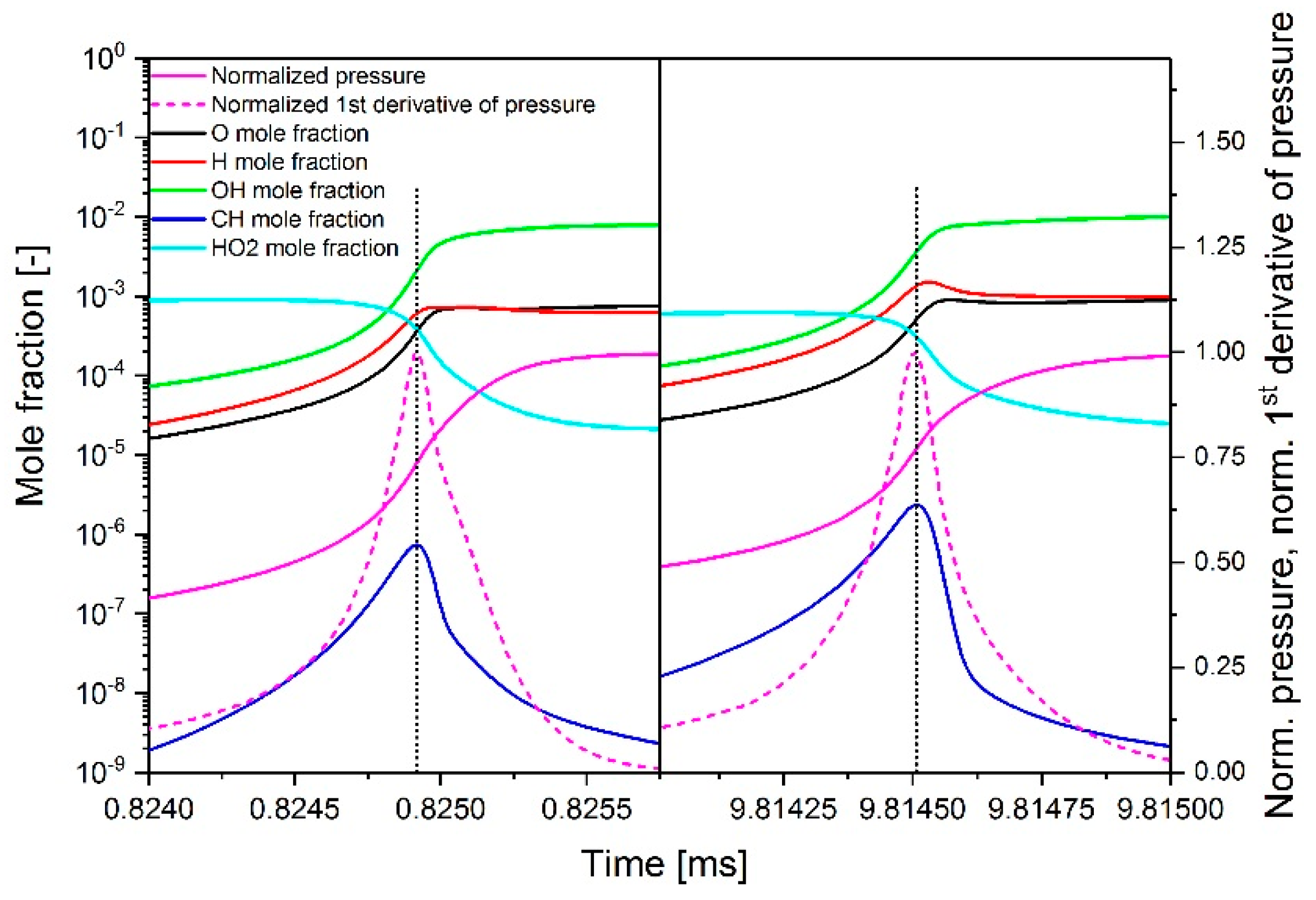
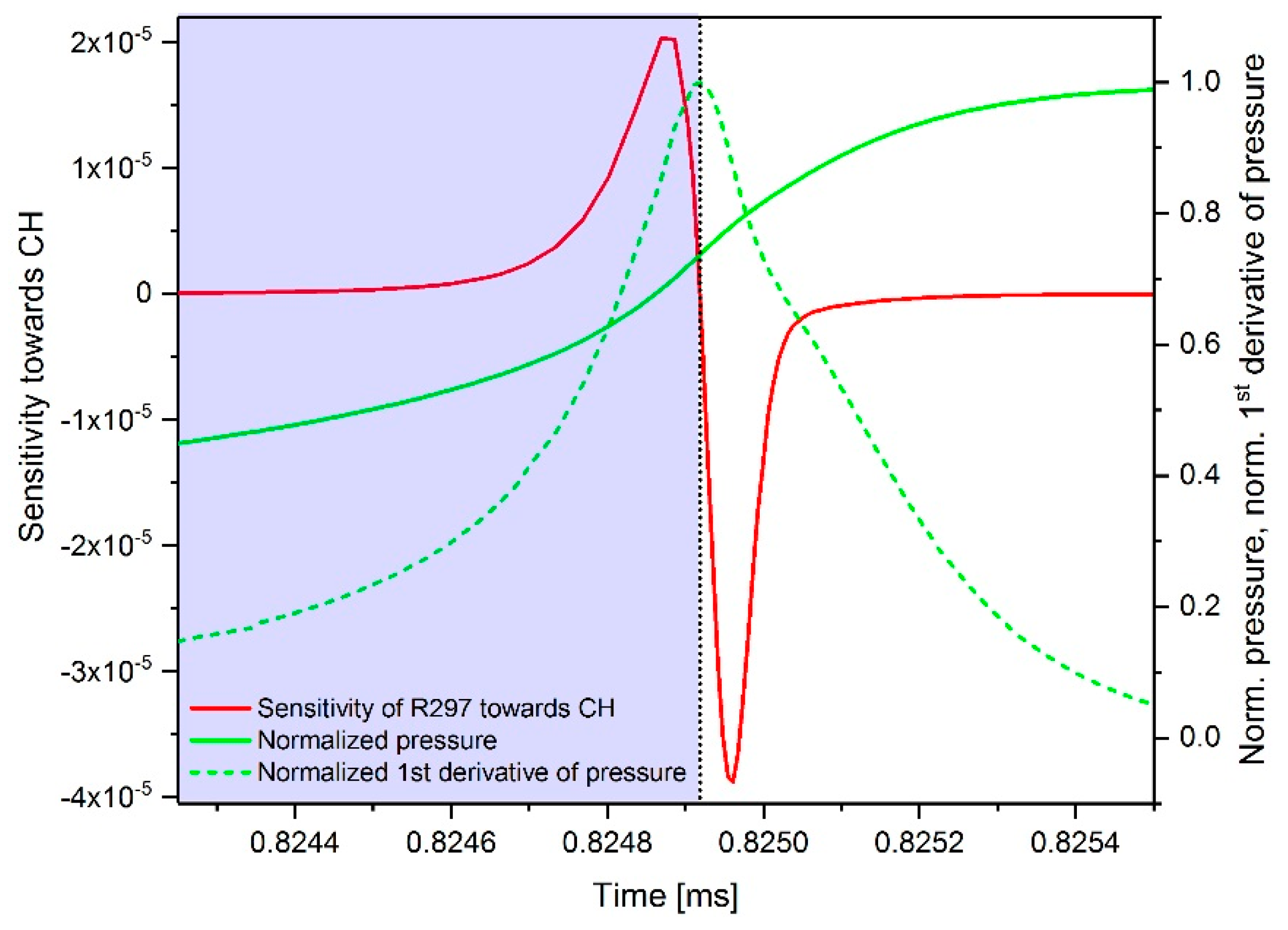
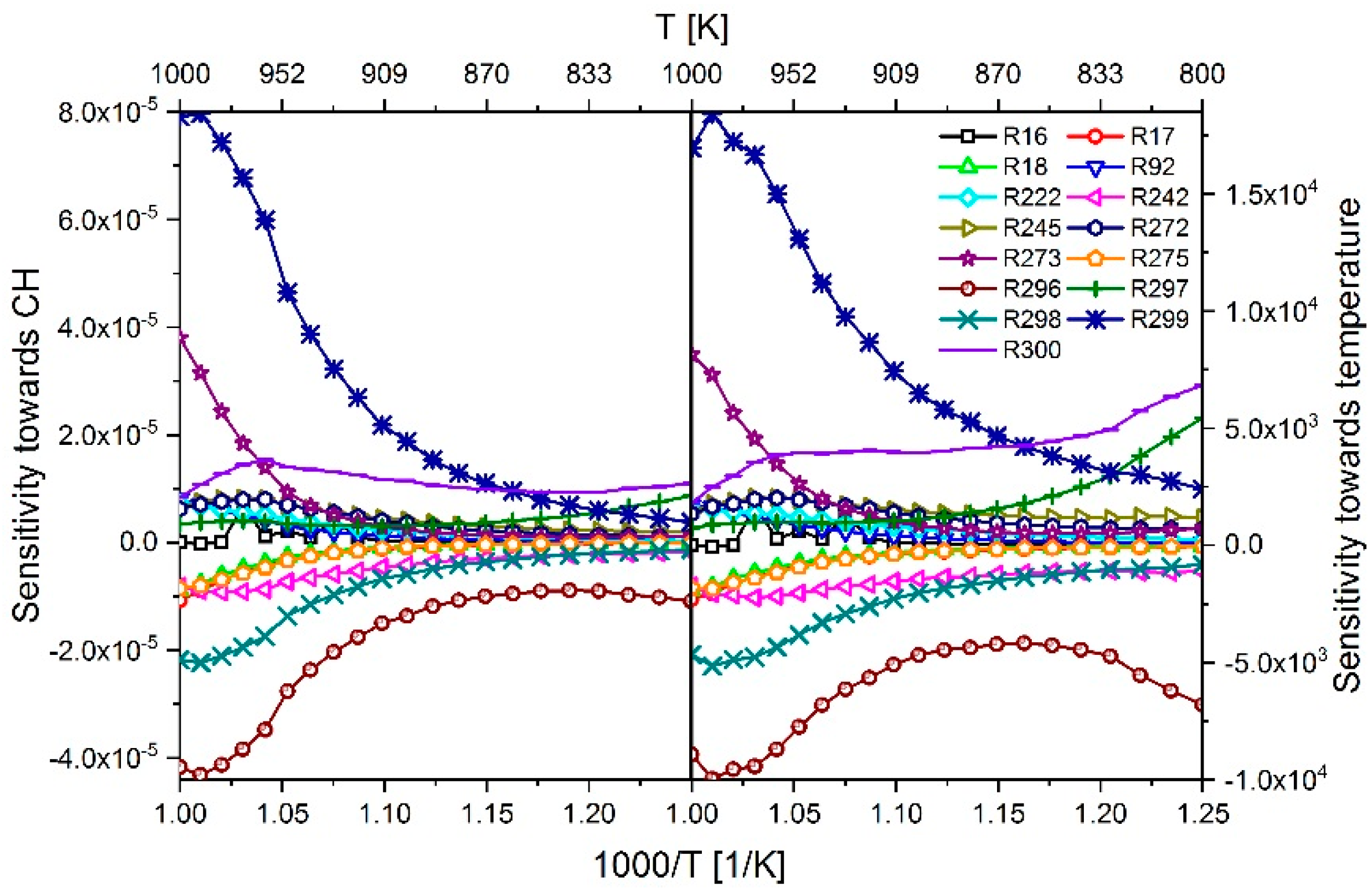
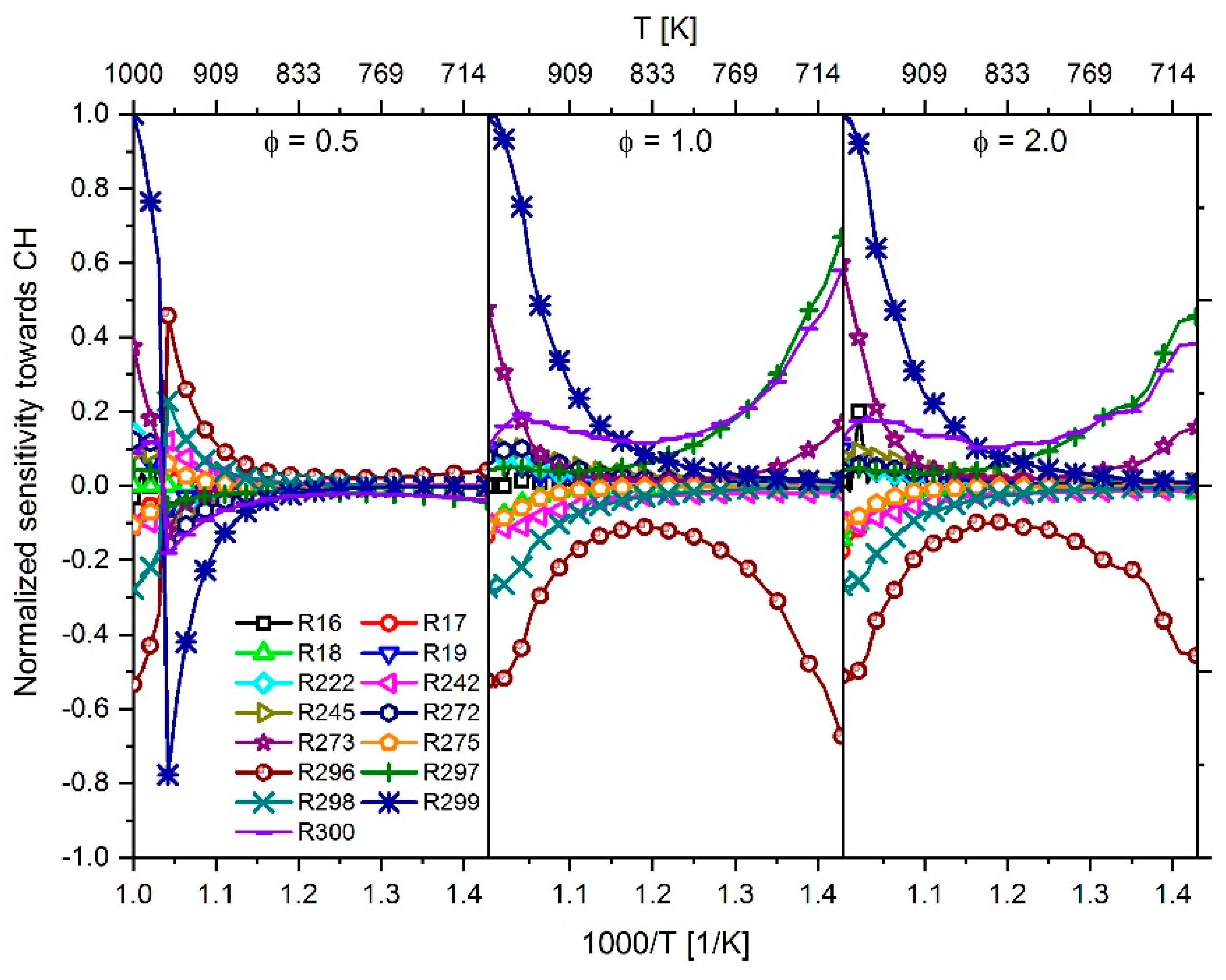
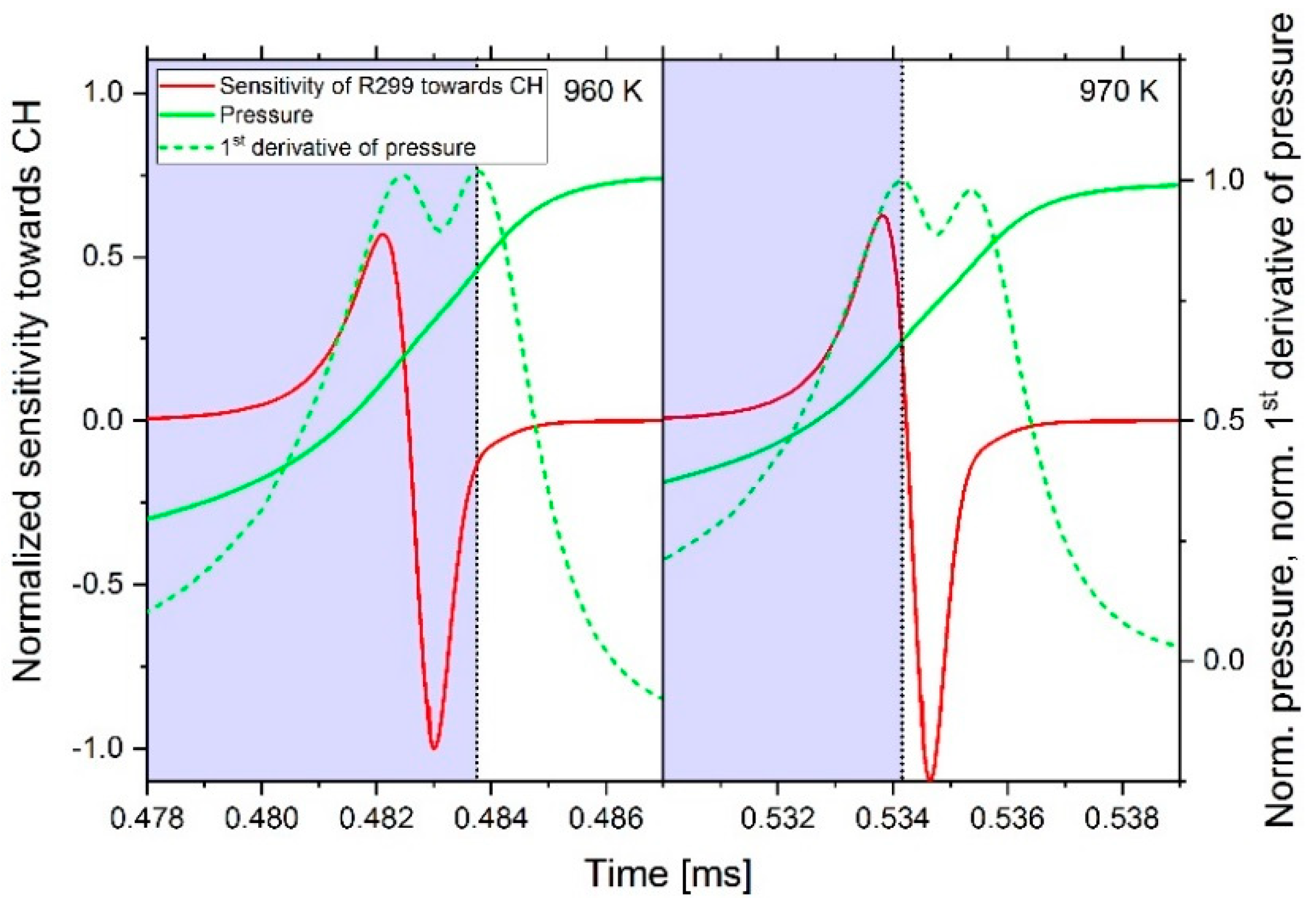
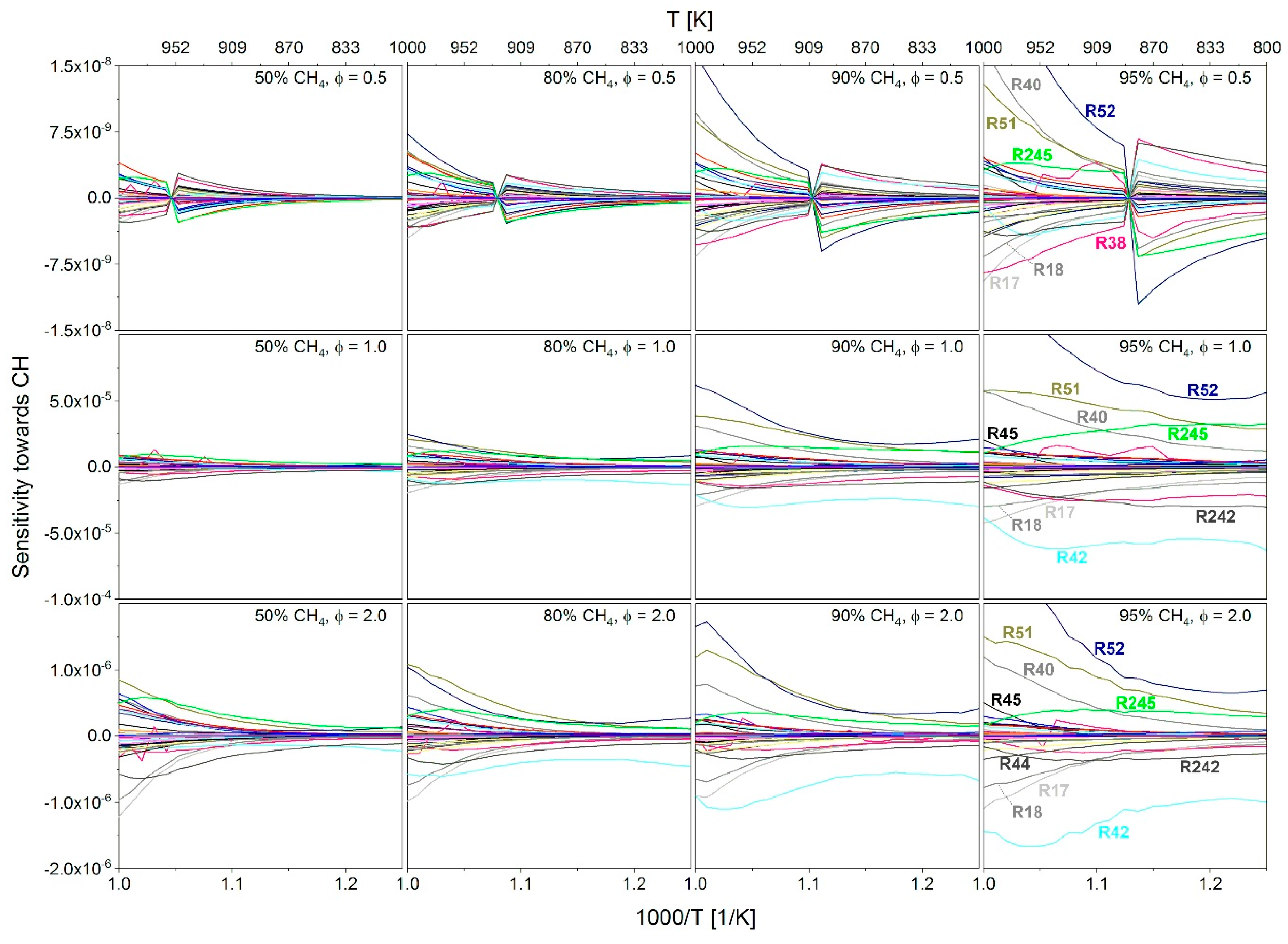
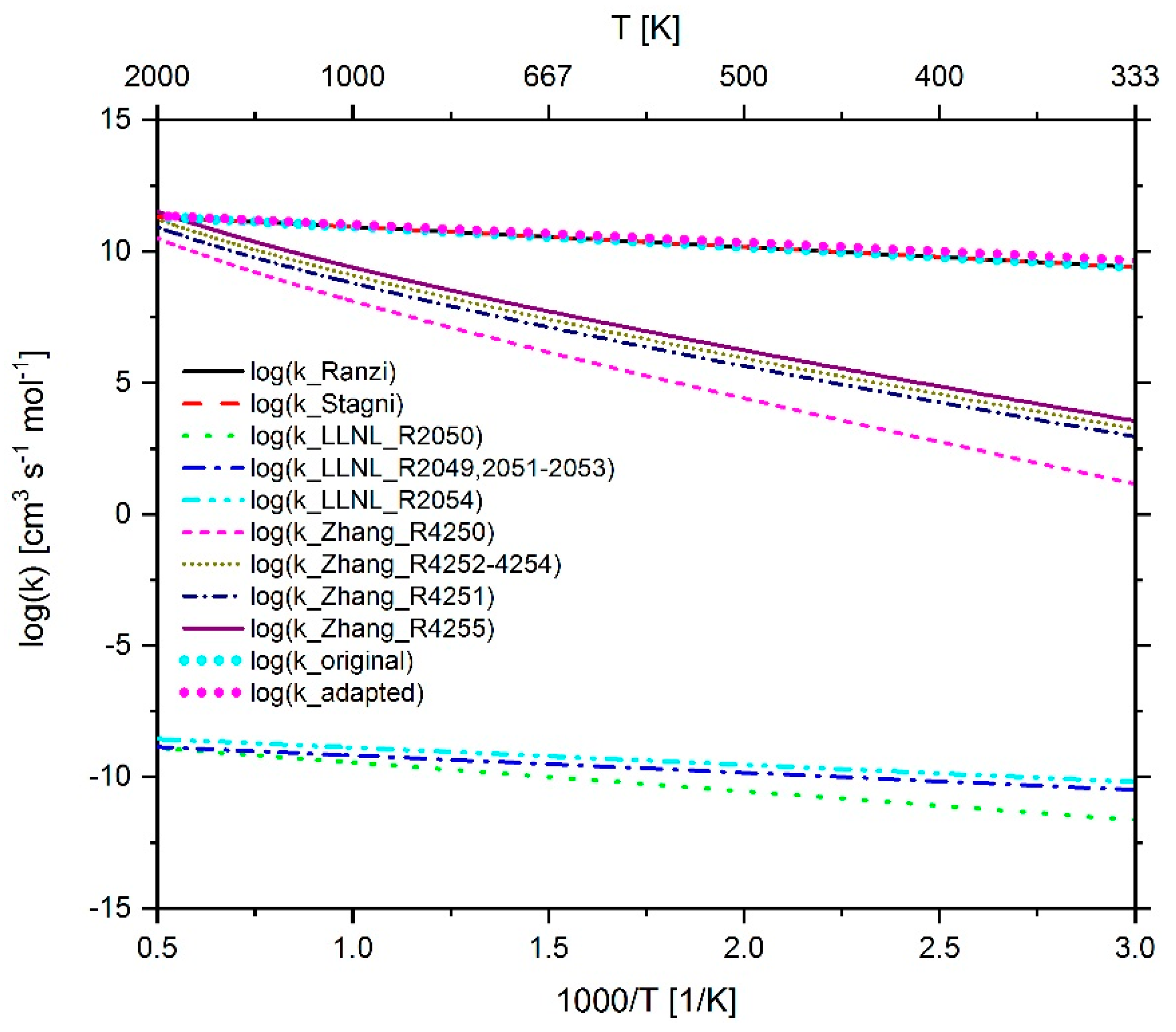
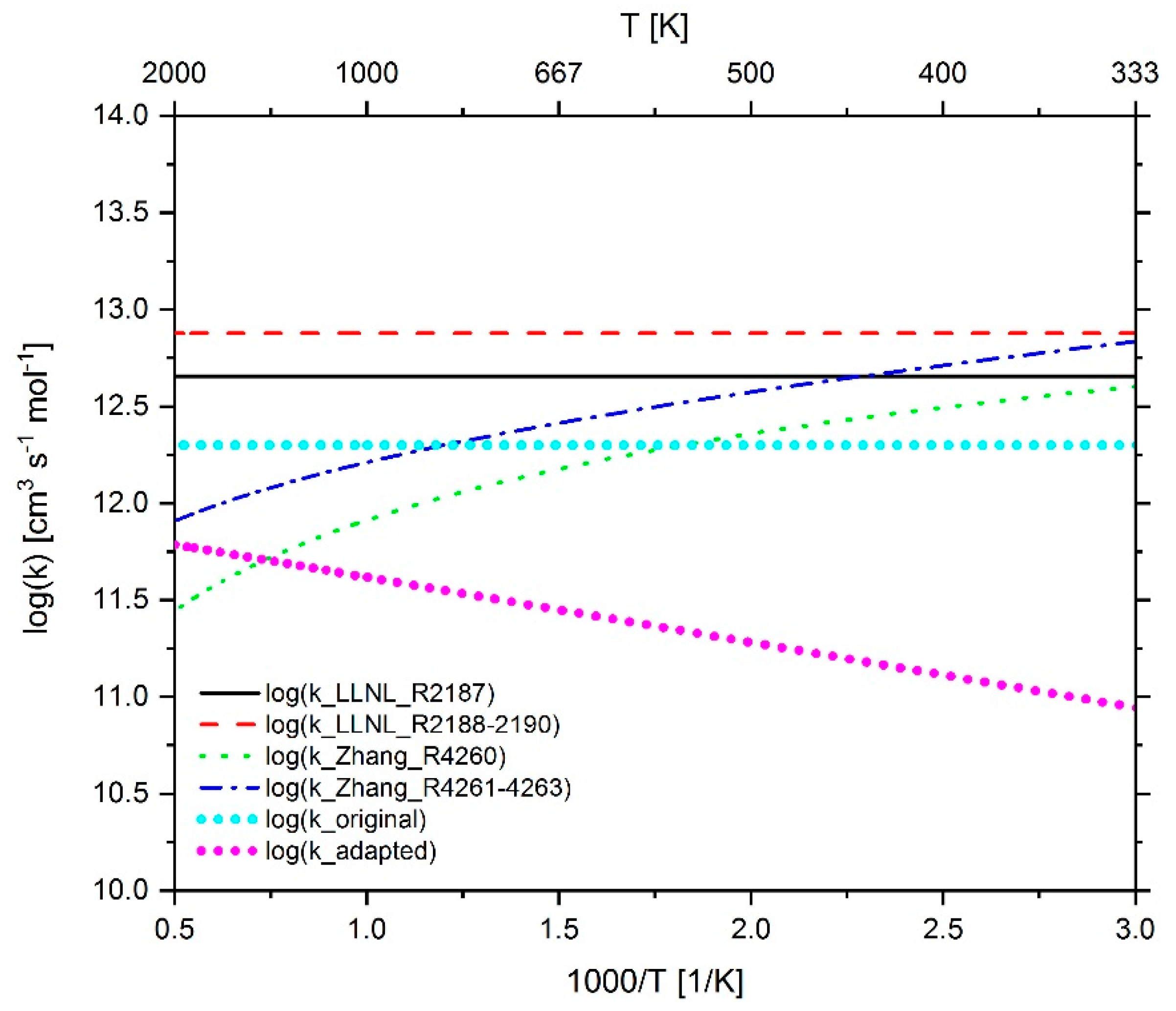
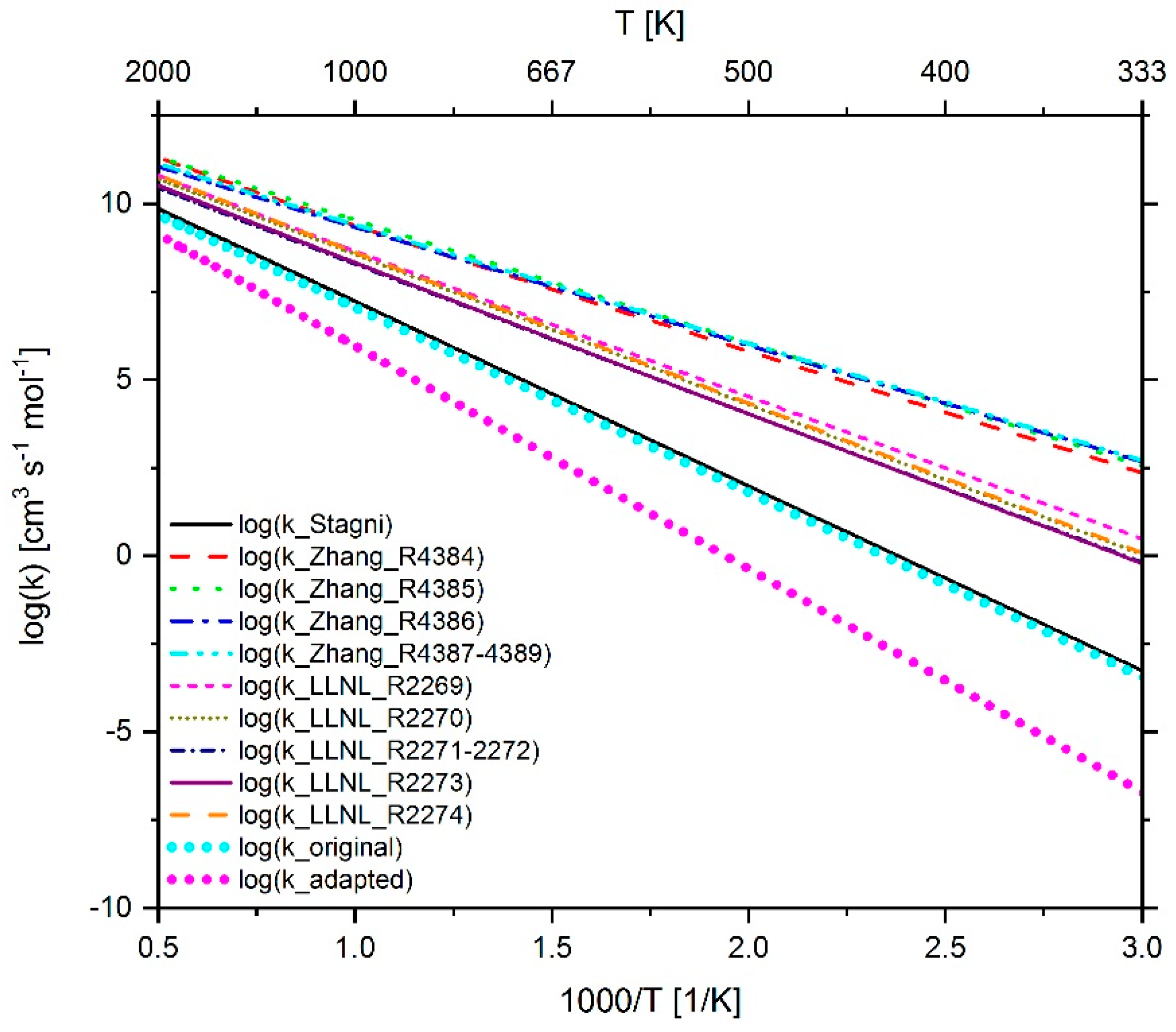
2.3.1. Determination of the Most Important Reactions
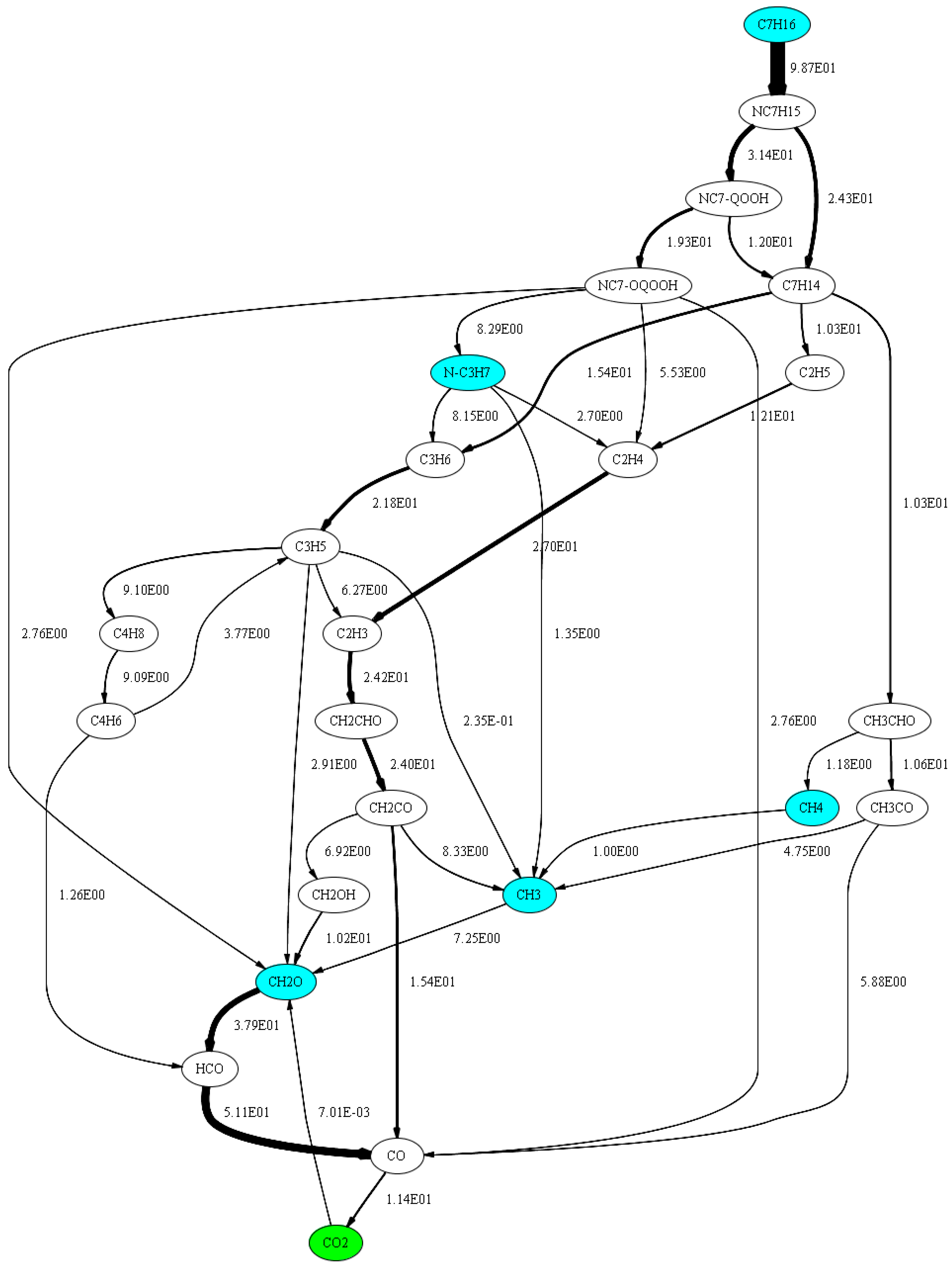
2.3.2. Investigation of n-Heptane Oxidation
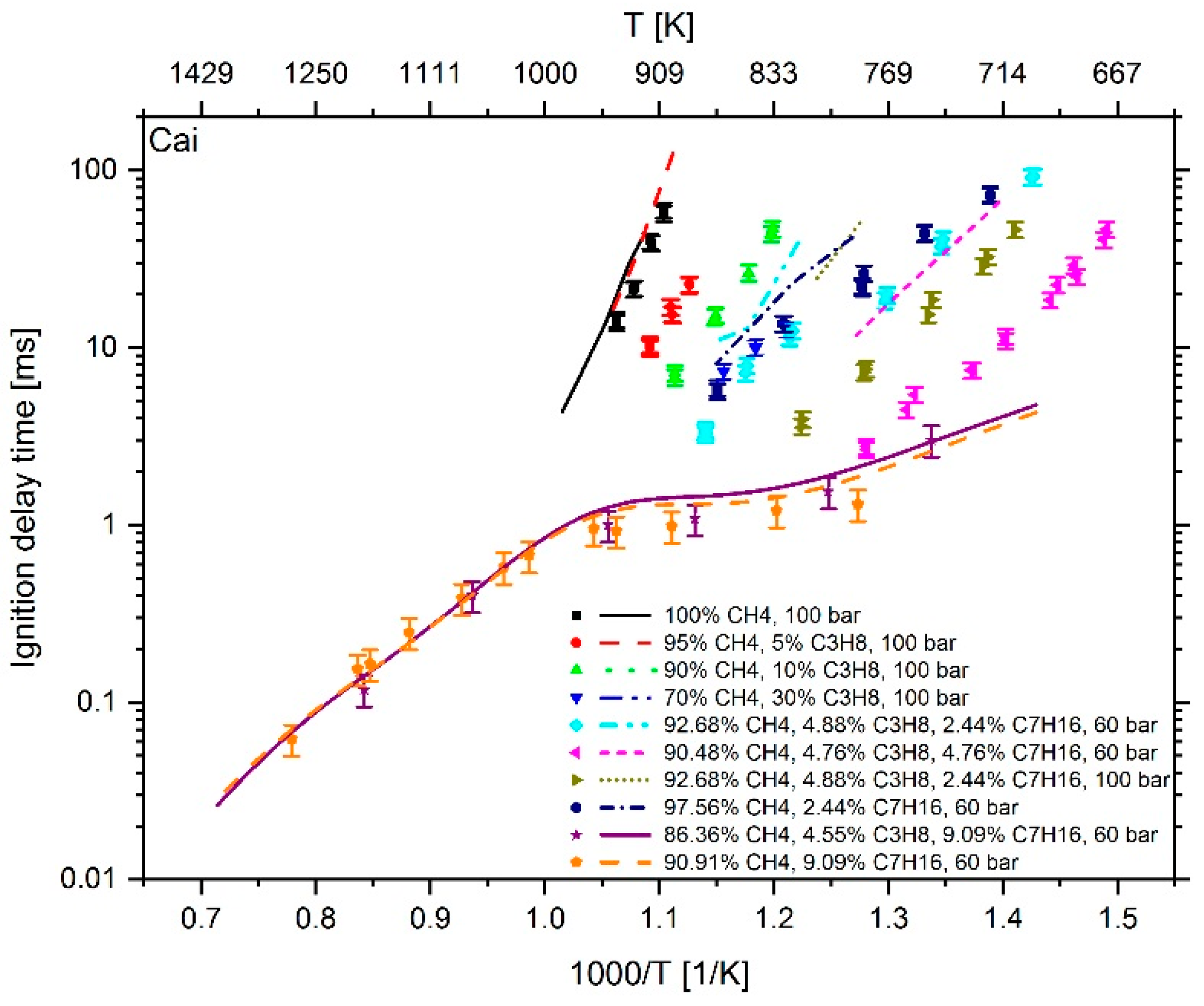
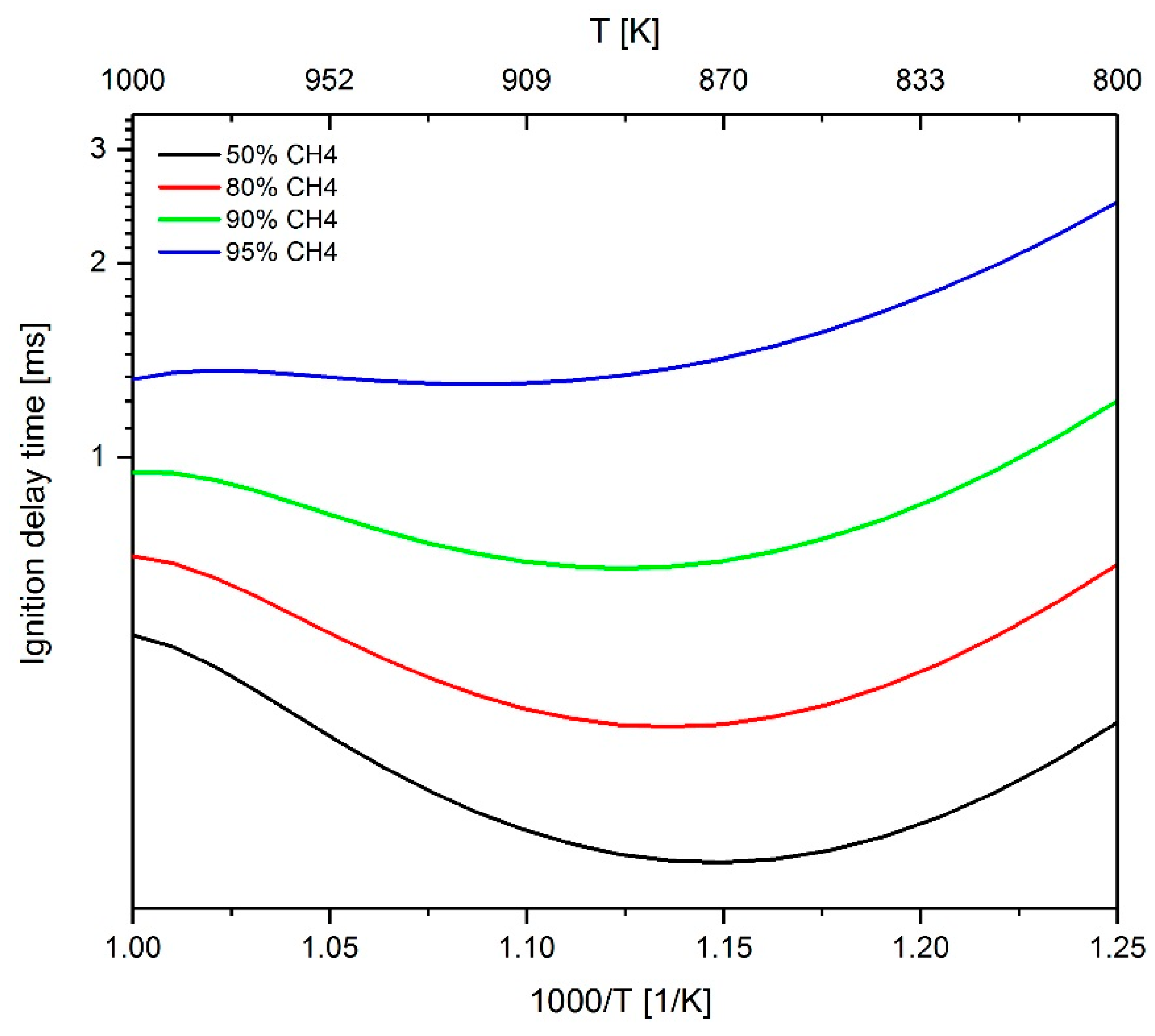
2.3.3. Investigation of the Influence of Methane on the IDT
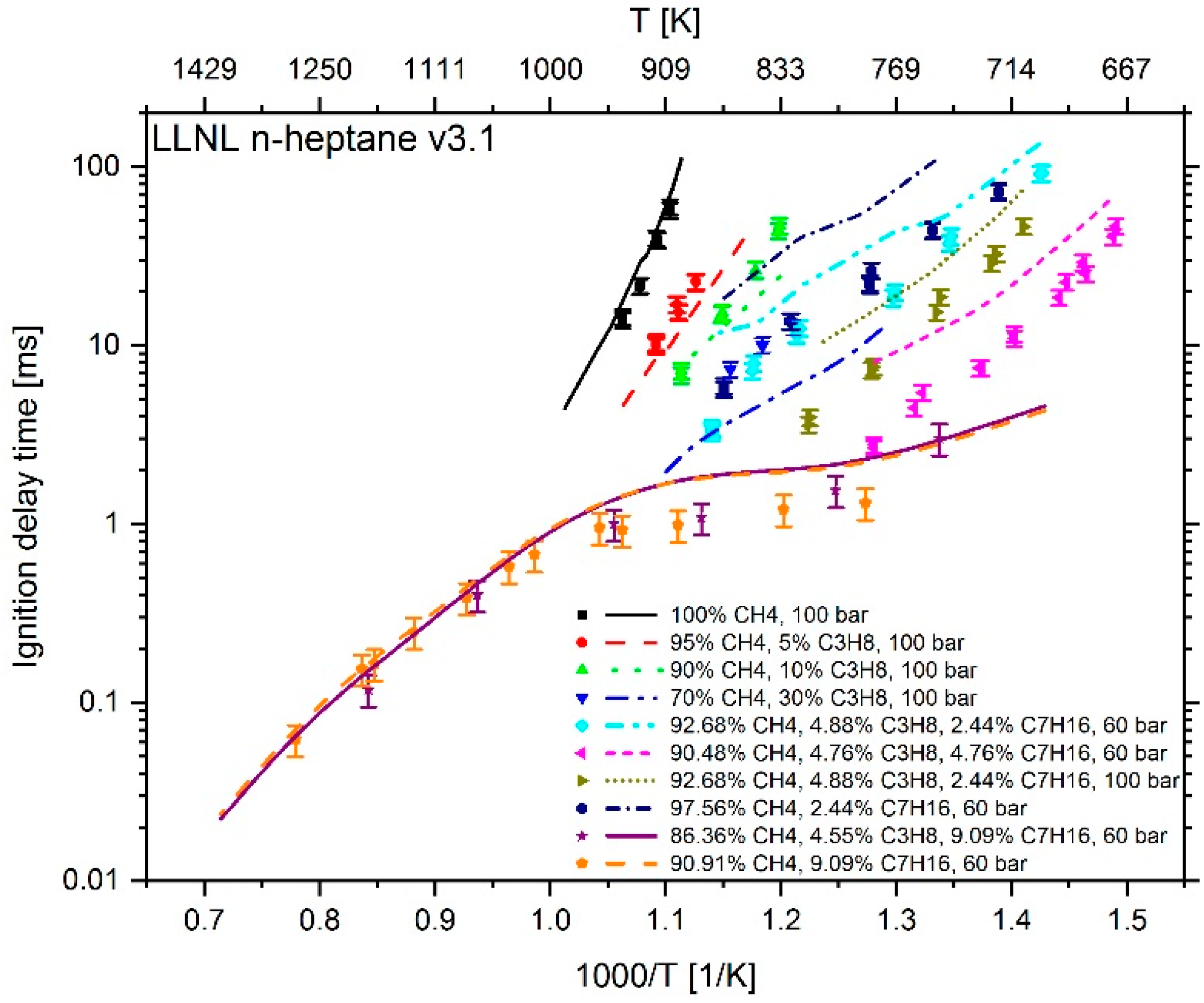
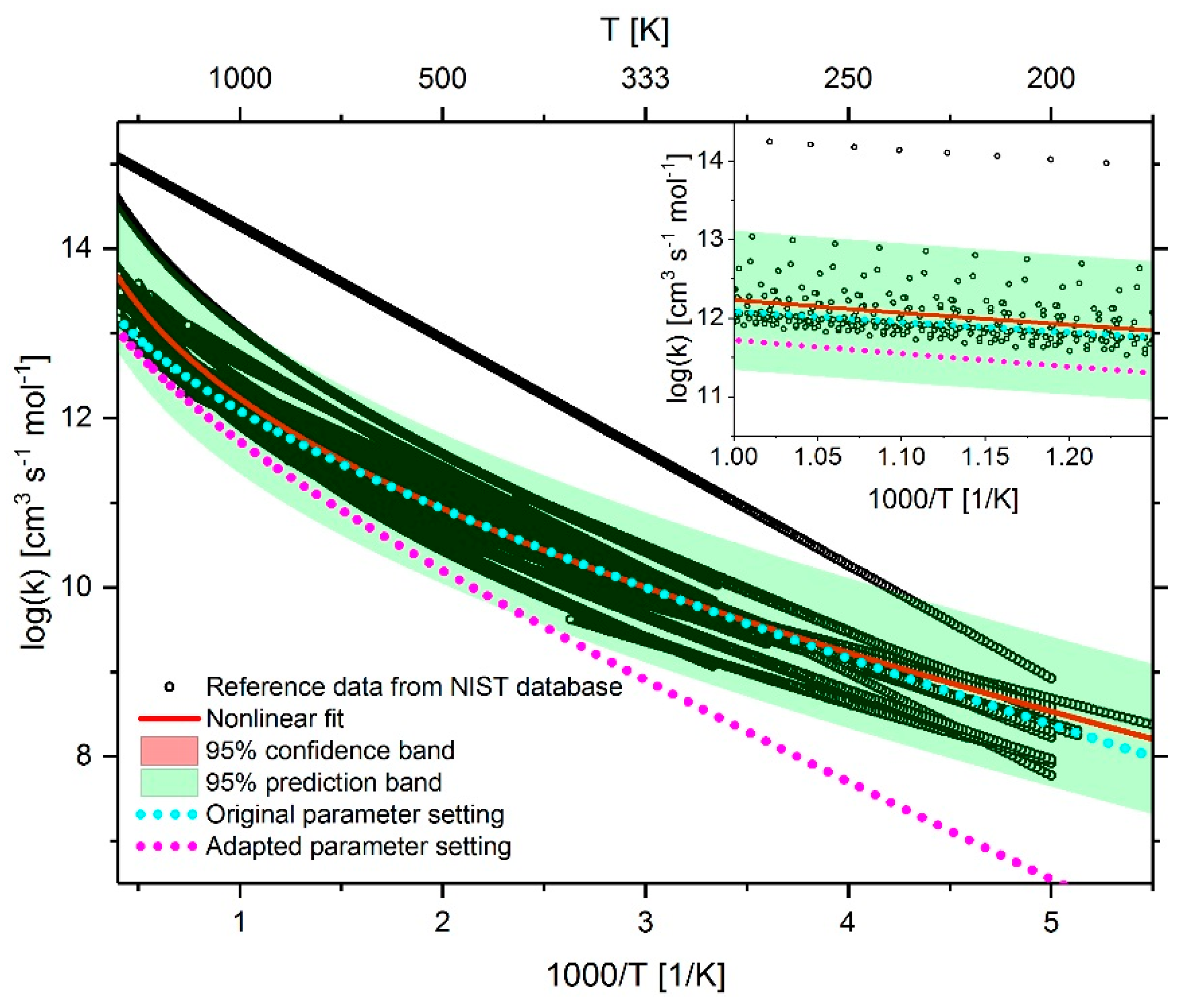
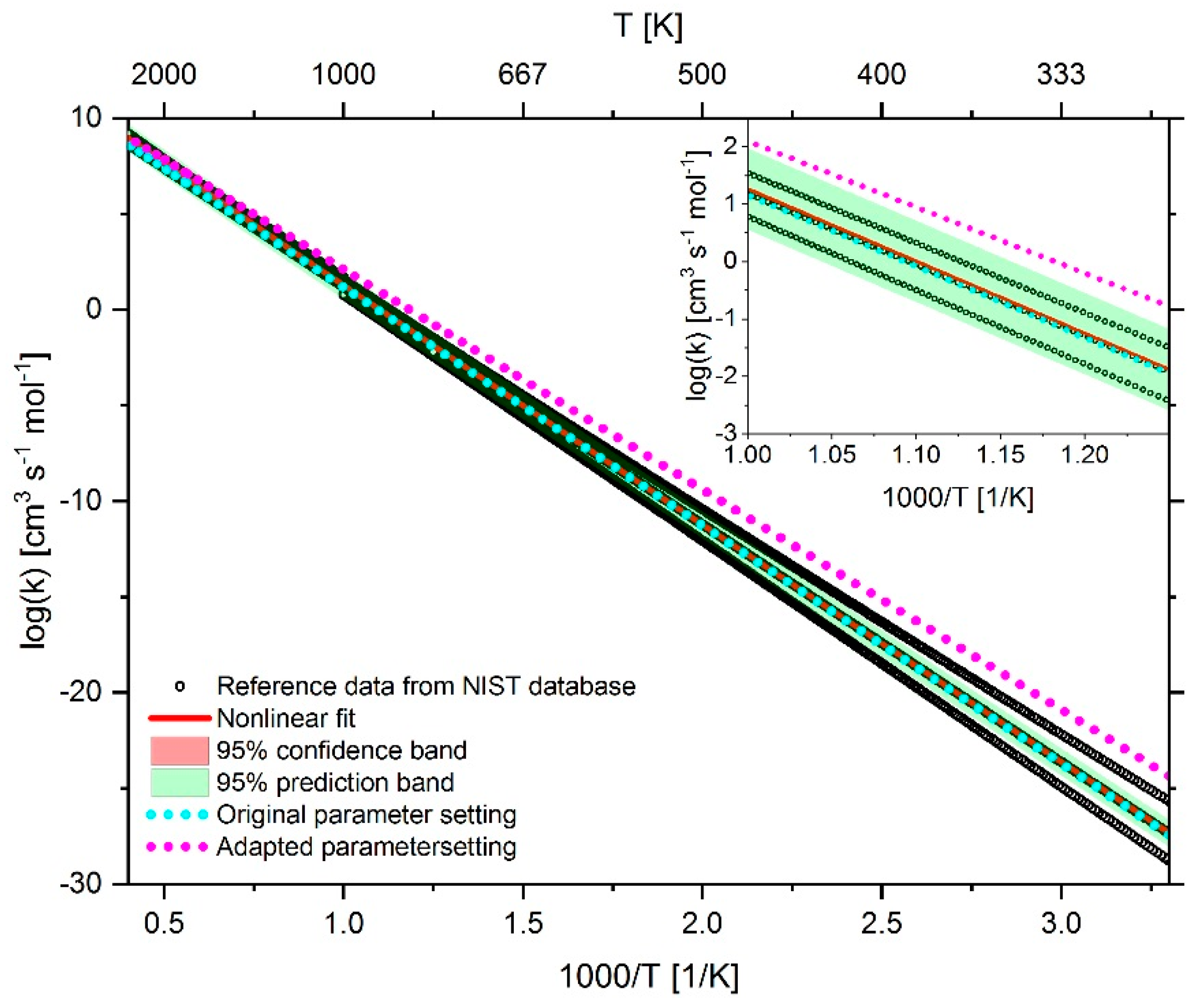
2.4. Adaption of Arrhenius Parameter
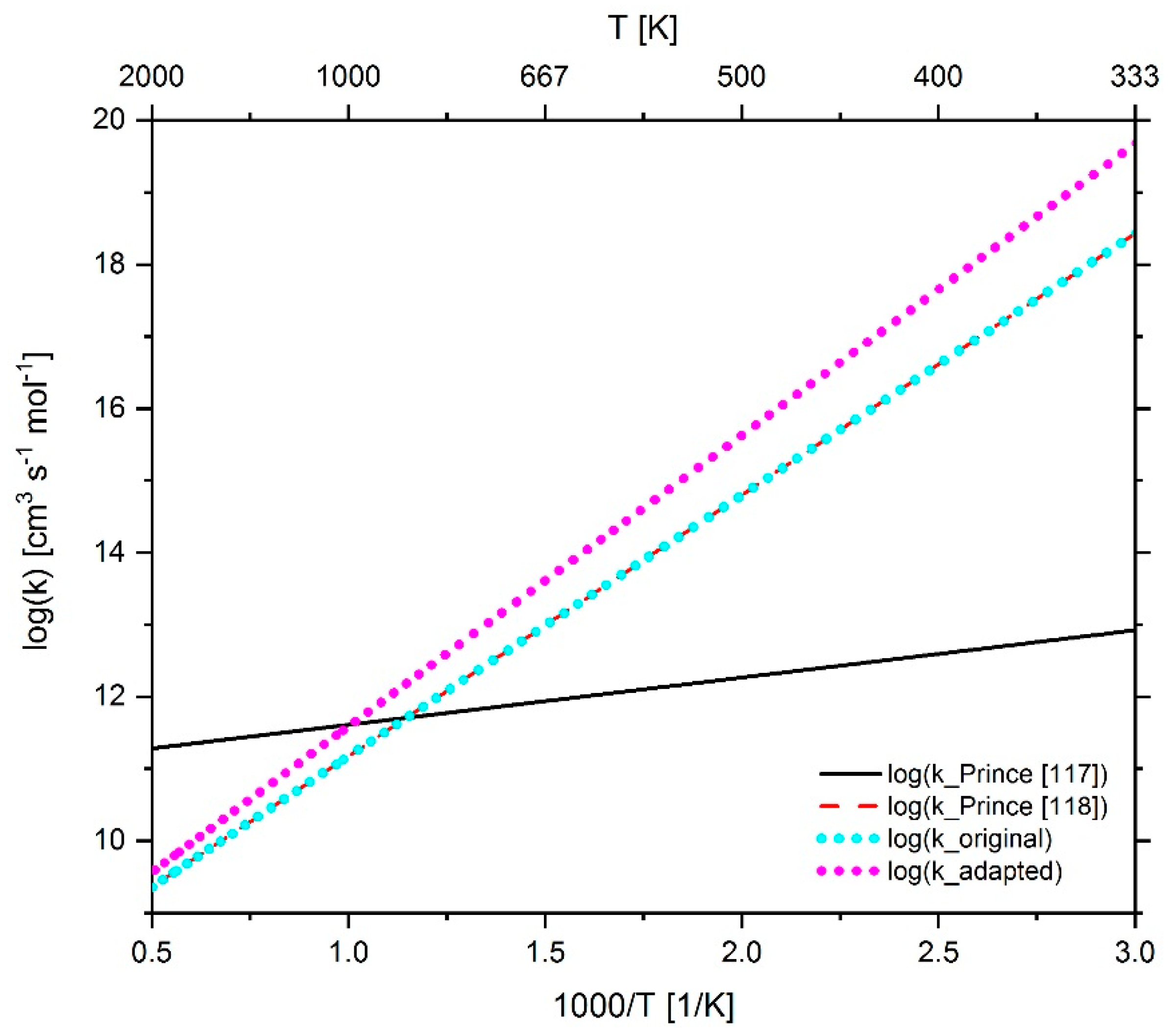
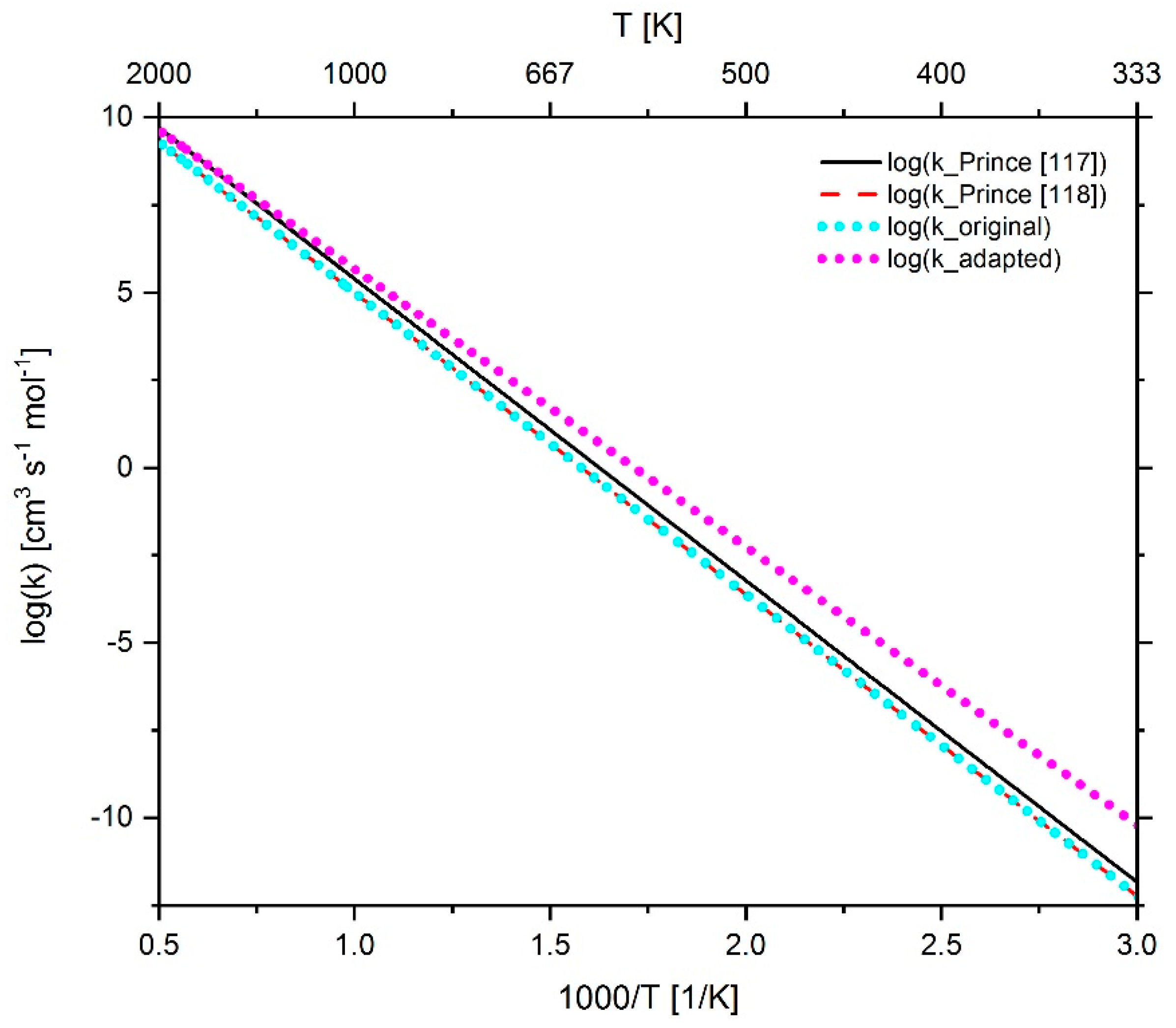
2.4.1. Adaption of Methane Chemistry
2.4.2. Adaption of n-heptane Chemistry
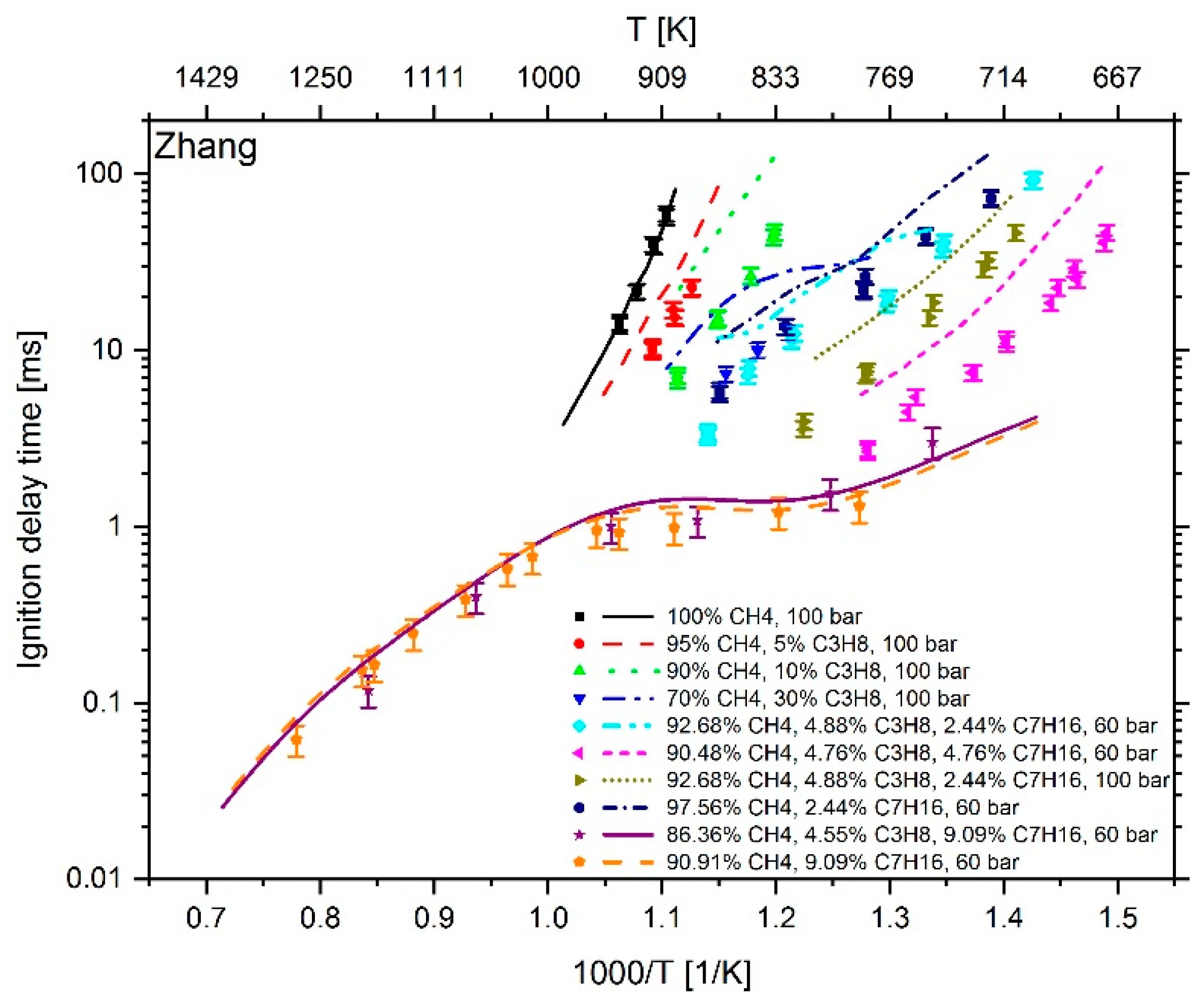
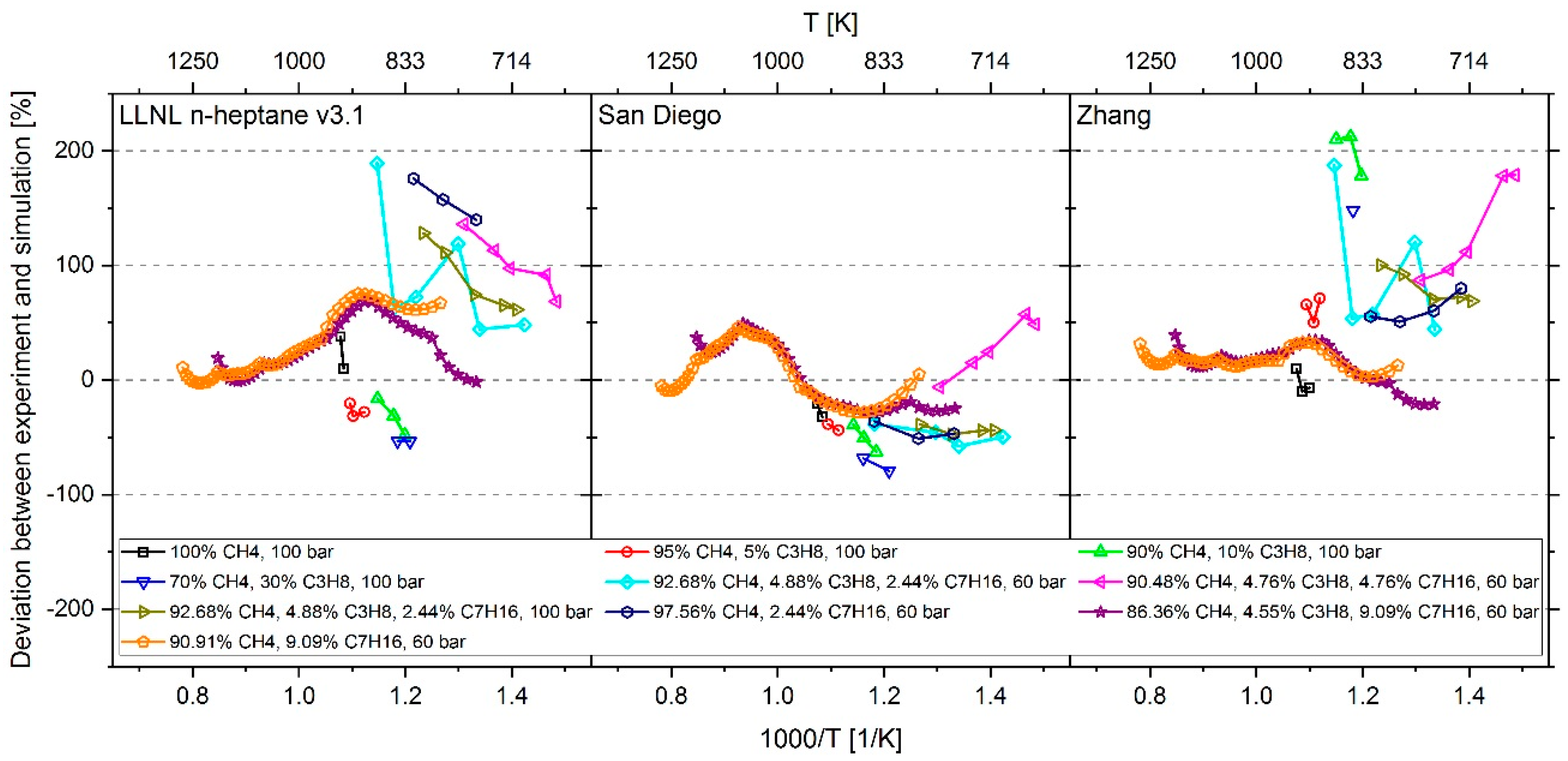
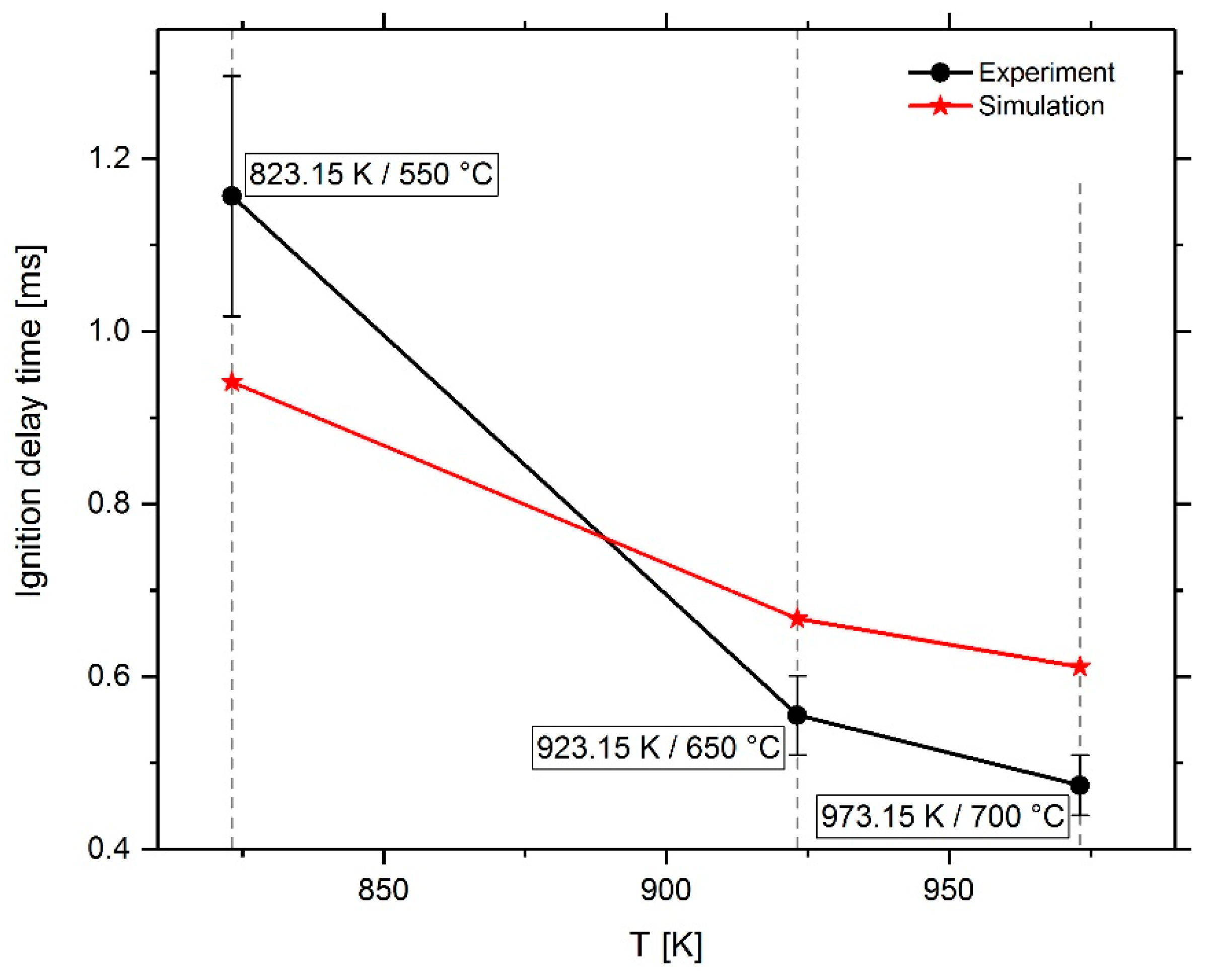
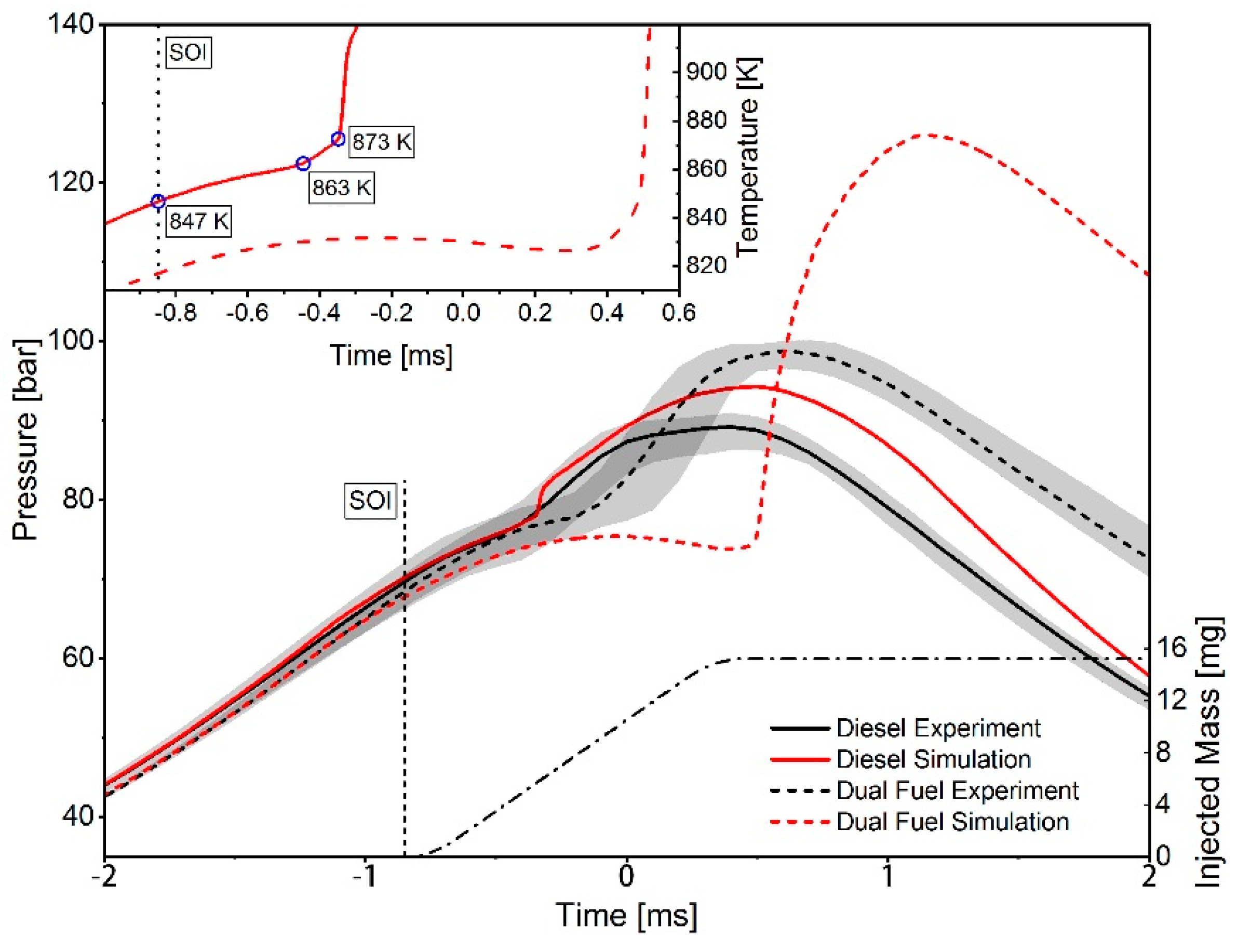
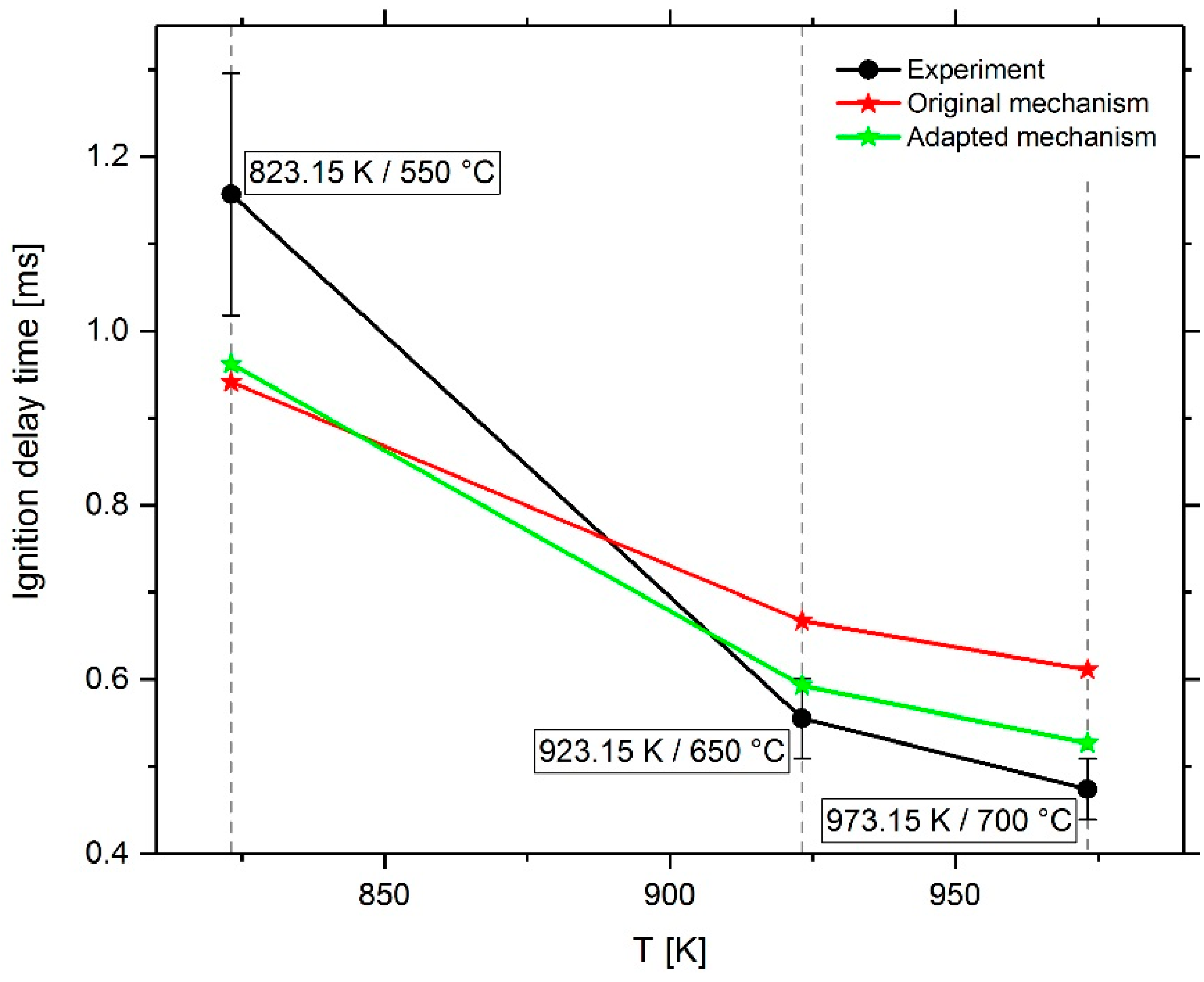
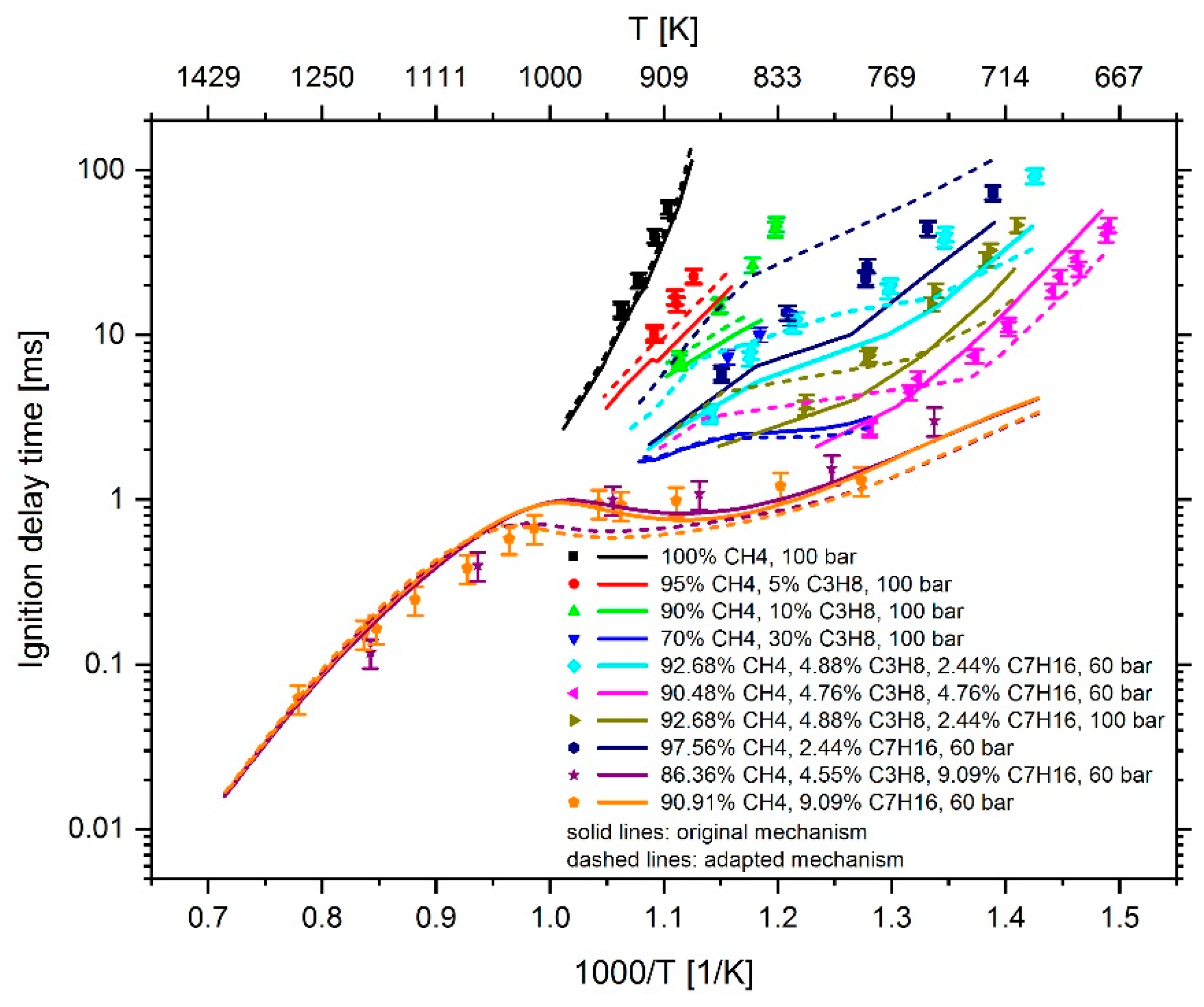
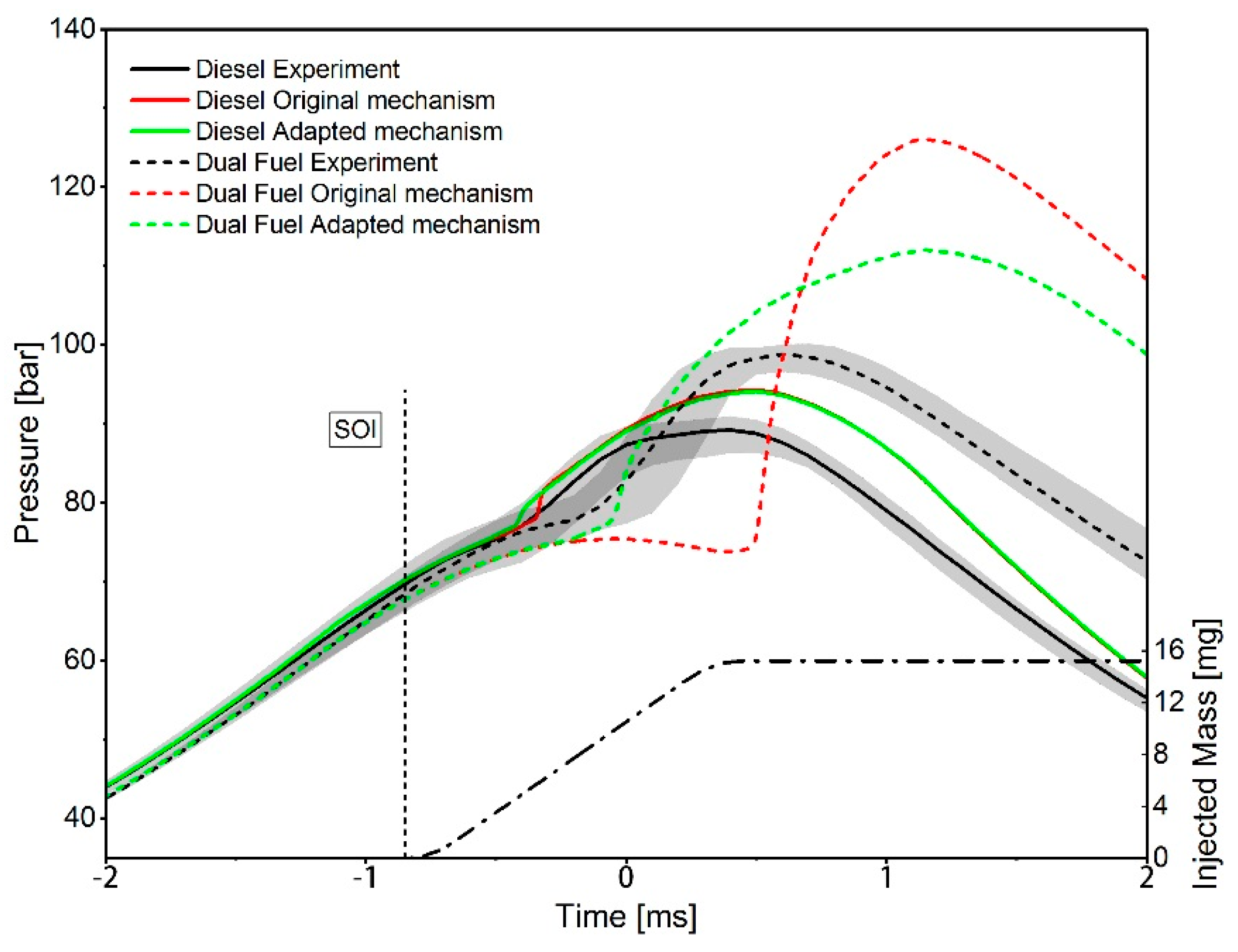
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heinemann, C.; Kasten, P.; Bauknecht, D.; Bracker, J.F.; Bürger, V.; Emele, L.; Hesse, T.; Kühnel, S.; Seebach, D.; Timpe, C. Die Bedeutung Strombasierter Stoffe für den Klimaschutz in Deutschland; Institute of Applied Ecology: Freiburg, Germany, 7 November 2019. [Google Scholar]
- Anto, S.; Mukherjee, S.S.; Muthappa, R.; Mathimani, T.; Deviram, G.; Kumar, S.S.; Verma, T.N.; Pugazhendhi, A. Algae as green energy reserve: Technological outlook on biofuel production. Chemosphere 2019, 242, 125079. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.C.; Kim, H.Y.; Yoon, S.K.; Choi, N.J. Reducing volatile organic compound emissions from diesel engines using canola oil biodiesel fuel and blends. Fuel 2018, 218, 266–274. [Google Scholar] [CrossRef]
- Schlick, H. Potentials and challenges of gas and dual-fuel engines for marine application. In Proceedings of the 5th CIMAC CASCADES, Busan, Korea, 23 October 2014. [Google Scholar]
- LOGEsoft. Available online: http://www.logesoft.com (accessed on 16 May 2019).
- Schuh, S.; Ramalingam, A.K.; Minwegen, H.; Heufer, K.A.; Winter, F. Experimental Investigation and Benchmark Study of Oxidation of Methane–Propane–n-Heptane Mixtures at Pressures up to 100 bar. Energies 2019, 12, 3410. [Google Scholar] [CrossRef]
- Pachler, R.F.; Ramalingam, A.K.; Heufer, K.A.; Winter, F. Reduction and validation of a chemical kinetic mechanism including necessity analysis and investigation of CH4/C3H8 oxidation at pressures up to 120 bar using a rapid compression machine. Fuel 2016, 172, 139–145. [Google Scholar] [CrossRef]
- Frühhaber, J.; Peter, A.; Schuh, S.; Lauer, T.; Wensing, M.; Winter, F.; Priesching, P.; Pachler, K. Modeling the Pilot Injection and the Ignition Process of a Dual Fuel Injector with Experimental Data from a Combustion Chamber Using Detailed Reaction Kinetics. In Proceedings of the International Powertrains, Fuels & Lubricants Meeting, Heidelberg, Germany, 17–19 September 2018. SAE Technical Paper 2018-01-1724. [Google Scholar] [CrossRef]
- Fischer, M. Modellierung der Multikomponenten-Verdampfung im Homogenisierten Dieselmotorischen Brennverfahren; Cuvillier Verlag: Göttingen, Germany, 2006; Volume 1. [Google Scholar]
- Jing, B.; Wandel, A.P. Numerical investigation into natural gas–diesel dual-fuel engine configuration. In Proceedings of the 11th Asia-Pacific Conference on Combustion, ASPACC 2017, The University of Sydney, Sydney, NSW, Australia, 10–14 December 2017. [Google Scholar]
- Li, Y.; Li, H.; Guo, H.; Li, Y.; Yao, M. A numerical investigation on methane combustion and emissions from a natural gas-diesel dual fuel engine using CFD model. Appl. Energy 2017, 205, 153–162. [Google Scholar] [CrossRef]
- Zhao, W.; Yang, W.; Fan, L.; Zhou, D.; Ma, X. Development of a skeletal mechanism for heavy-duty engines fuelled by diesel and natural gas. Appl. Therm. Eng. 2017, 123, 1060–1071. [Google Scholar] [CrossRef]
- Eder, L.; Ban, M.; Pirker, G.; Vujanovic, M.; Priesching, P.; Wimmer, A. Development and Validation of 3D-CFD Injection and Combustion Models for Dual Fuel Combustion in Diesel Ignited Large Gas Engines. Energies 2018, 11, 643. [Google Scholar] [CrossRef]
- Mansor, W.N.W.; Olsen, D.B. Computational modeling of diesel and dual fuel combustion using CONVERGE CFD software. ARPN J. Eng. Appl. Sci. 2016, 11, 13697–13707. [Google Scholar]
- Tschöke, H. Diesel- und Benzindirekteinspritzung V: Spraybildung, Simulation, Applikation, Messtechnik: Mit 29 Tabellen; Expert-Verlag: Renningen, Germany, 2009. [Google Scholar]
- Reif, K. Dieselmotor-Management: Systeme, Komponenten, Steuerung und Regelung; Vieweg+Teubner Verlag: Wiesbaden, Germany, 2012. [Google Scholar]
- Hockett, A.; Hampson, G.; Marchese, A.J. Development and Validation of a Reduced Chemical Kinetic Mechanism for Computational Fluid Dynamics Simulations of Natural Gas/Diesel Dual-Fuel Engines. Energy Fuels 2016, 30, 2414–2427. [Google Scholar] [CrossRef]
- Huang, H.; Lv, D.; Zhu, J.; Zhu, Z.; Chen, Y.; Pan, Y.; Pan, M. Development of a new reduced diesel/natural gas mechanism for dual-fuel engine combustion and emission prediction. Fuel 2019, 236, 30–42. [Google Scholar] [CrossRef]
- Bierschenk, D.M.; Pillai, M.R.; Lin, Y.; Barnett, S.A. Effect of Ethane and Propane in Simulated Natural Gas on the Operation of Ni-YSZ Anode Supported Solid Oxide Fuel Cells. Fuel Cells 2010, 10, 1129–1134. [Google Scholar] [CrossRef]
- Chemical-Kinetic Mechanisms for Combustion Applications; San Diego Mechanism Web Page, Mechanical and Aerospace Engineering (Combustion Research), University of California at San Diego. Available online: http://combustion.ucsd.edu (accessed on 17 March 2017).
- Zhang, K.; Banyon, C.; Bugler, J.; Curran, H.J.; Rodriguez, A.; Herbinet, O.; Battin-Leclerc, F.; B’Chir, C.; Heufer, K.A. An updated experimental and kinetic modeling study of n-heptane oxidation. Combust. Flame 2016, 172, 116–135. [Google Scholar] [CrossRef]
- Curran, H.J.; Gaffuri, P.; Pitz, W.J.; Westbrook, C.K. A comprehensive modeling study of iso-octane oxidation. Combust. Flame 2002, 129, 253–280. [Google Scholar] [CrossRef]
- Mehl, M.; Pitz, W.J.; Westbrook, C.K.; Curran, H.J. Kinetic modeling of gasoline surrogate components and mixtures under engine conditions. Proc. Combust. Inst. 2011, 33, 193–200. [Google Scholar] [CrossRef]
- Cai, L.; Pitsch, H. Optimized chemical mechanism for combustion of gasoline surrogate fuels. Combust. Flame 2015, 162, 1623–1637. [Google Scholar] [CrossRef]
- Zeuch, T.; Moréac, G.; Ahmed, S.S.; Mauss, F. A comprehensive skeletal mechanism for the oxidation of n-heptane generated by chemistry-guided reduction. Combust. Flame 2008, 155, 651–674. [Google Scholar] [CrossRef]
- Karavalakis, G.; Hajbabaei, M.; Durbin, T.D.; Johnson, K.C.; Zheng, Z.; Miller, W.J. The effect of natural gas composition on the regulated emissions, gaseous toxic pollutants, and ultrafine particle number emissions from a refuse hauler vehicle. Energy 2013, 50, 280–291. [Google Scholar] [CrossRef]
- Van Basshuysen, R. Erdgas und Erneuerbares Methan für den Fahrzeugantrieb: Wege zur Klimaneutralen Mobilität; Springer Fachmedien Wiesbaden: Wiesbaden, Germany, 2015. [Google Scholar] [CrossRef]
- Kidnay, A.J.; Parrish, W.R. Fundamentals of Natural Gas Processing; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Al-Saleh, M.A.; Duffuaa, S.O.; Al-Marhoun, M.A.; Al-Zayer, J.A. Impact of crude oil production on the petrochemical industry in Saudi Arabia. Energy 1991, 16, 1089–1099. [Google Scholar] [CrossRef]
- Cummins Westport Fuel Quality Calculator. Available online: https://www.cumminswestport.com/fuel-quality-calculator (accessed on 12 November 2019).
- Sung, C.-J.; Curran, H.J. Using rapid compression machines for chemical kinetics studies. Prog. Energy Combust. Sci. 2014, 44, 1–18. [Google Scholar] [CrossRef]
- Petersen, E.L.; Hanson, R.K. Nonideal effects behind reflected shock waves in a high-pressure shock tube. Shock Waves 2001, 10, 405–420. [Google Scholar] [CrossRef]
- Heufer, K.A.; Olivier, H. Determination of ignition delay times of different hydrocarbons in a new high pressure shock tube. Shock Waves 2010, 20, 307–316. [Google Scholar] [CrossRef]
- LOGEresearch Manual, Book 5; LOGE AB: Lund, Sweden, 2017.
- Ji, W.; Ren, Z.; Law, C.K. Evolution of sensitivity directions during autoignition. Proc. Combust. Inst. 2019, 37, 807–815. [Google Scholar] [CrossRef]
- Manion, J.A.; Huie, R.E.; Levin, R.D.; Burgess, D.R., Jr.; Orkin, V.L.; Tsang, W.; McGivern, W.S.; Hudgens, J.W.; Knyazev, V.D.; Atkinson, D.B.; et al. NIST Chemical Kinetics Database, NIST Standard Reference Database 17; Version 7.0 (Web Version), Data Version 2015.09; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2019. Available online: https://kinetics.nist.gov/kinetics/citation.jsp (accessed on 2 October 2019).
- Srinivasan, N.K.; Su, M.C.; Sutherland, J.W.; Michael, J.V. Reflected Shock Tube Studies of High-Temperature Rate Constants for OH + CH4 → CH3 + H2O and CH3 + NO2 → CH3O + NO. J. Phys. Chem. A 2005, 109, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Bonard, A.; Daële, V.; Delfau, J.-L.; Vovelle, C. Kinetics of OH Radical Reactions with Methane in the Temperature Range 295−660 K and with Dimethyl Ether and Methyl-tert-butyl Ether in the Temperature Range 295−618 K. J. Phys. Chem. A 2002, 106, 4384–4389. [Google Scholar] [CrossRef]
- DeMore, W.B.; Sander, S.P.; Golden, D.M.; Hampson, R.F.; Kurylo, M.J.; Howard, C.J.; Ravishankara, A.R.; Kolb, C.E.; Molina, M.J. Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling: Evaluation Number 12; JPL Publication 97-4; Jet Propulsion Laboratory, California Institute of Technology: Pasadena, CA, USA, 1997; pp. 1–266. [Google Scholar]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Hampson, R.F.; Kerr, J.A.; Rossi, M.J.; Troe, J. Evaluated Kinetic, Photochemical and Heterogeneous Data for Atmospheric Chemistry: Supplement V. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry. J. Phys. Chem. Ref. Data 1997, 26, 521–1011. [Google Scholar] [CrossRef]
- Demore, W.; Sander, S.; Golden, D.; Hampson, R.; Kurylo, M.; Howard, C.; Ravishankara, A.R.; Kolb, C.; Molina, M. Chemical Kinetic and Photochemical Data for Use in Stratospheric Modeling: Evaluation No. 11 of the NASA Panel for Data Evaluation; JPL Publication 94-26; Jet Propulsion Laboratory, California Institute of Technology: Pasadena, CA, USA, 1994; pp. 1–2. [Google Scholar]
- Baulch, D.L.; Cobos, C.J.; Cox, R.A.; Esser, C.; Frank, P.; Just, T.; Kerr, J.A.; Pilling, M.J.; Troe, J.; Walker, R.W.; et al. Evaluated Kinetic Data for Combustion Modelling. J. Phys. Chem. Ref. Data 1992, 21, 411–734. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Hampson, R.F.; Kerr, J.A.; Troe, J. Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Supplement IV. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry. J. Phys. Chem. Ref. Data 1992, 21, 1125–1568. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Hampson, R.F.; Kerr, J.A.; Troe, J. Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Supplement III. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry. J. Phys. Chem. Ref. Data 1989, 18, 881–1097. [Google Scholar] [CrossRef]
- Tsang, W.; Hampson, R.F. Chemical Kinetic Data Base for Combustion Chemistry. Part I. Methane and Related Compounds. J. Phys. Chem. Ref. Data 1986, 15, 1087–1279. [Google Scholar] [CrossRef]
- Felder, W.; Madronich, S. High Temperature Photochemistry (HTP): Kinetics and Mechanism Studies of Elementary Combustion Reactions over 300-1700 K. Combust. Sci. Technol. 1986, 50, 135–150. [Google Scholar] [CrossRef]
- Baulch, D.L.; Bowers, M.; Malcolm, D.G.; Tuckerman, R.T. Evaluated Kinetic Data for High-Temperature Reactions. Volume 5. Part 1. Homogeneous Gas Phase Reactions of the Hydroxyl Radical with Alkanes. J. Phys. Chem. Ref. Data 1986, 15, 465–592. [Google Scholar] [CrossRef]
- Atkinson, R. Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions. Chem. Rev. 1986, 86, 69–201. [Google Scholar] [CrossRef]
- Warnatz, J. Rate Coefficients in the C/H/O System. In Combustion Chemistry; Gardiner, W.C., Ed.; Springer: New York, NY, USA, 1984; pp. 197–360. [Google Scholar] [CrossRef]
- Cohen, N.; Westberg, K.R. Chemical Kinetic Data Sheets for High-Temperature Chemical Reactions. J. Phys. Chem. Ref. Data 1983, 12, 531–590. [Google Scholar] [CrossRef]
- Zellner, R. Non-Arrhenius behavior in bimolecular reactions of the hydroxyl radical. J. Phys. Chem. 1979, 83, 18–23. [Google Scholar] [CrossRef]
- Ernst, J.; Wagner, H.G.; Zellner, R. A Combined Flash Photolysis/Shock-Tube Study of the Absolute Rate Constants for Reactions of the Hydroxyl Radical with CH4 and CF3H around 1300 K. Ber. Der Bunsenges. Für Phys. Chem. 1978, 82, 409–414. [Google Scholar] [CrossRef]
- Wilson, W.E. A Critical Review of the Gas-Phase Reaction Kinetics of the Hydroxyl Radical. J. Phys. Chem. Ref. Data 1972, 1, 535–573. [Google Scholar] [CrossRef]
- Wilson, W.E.; Westenberg, A.A. Study of the reaction of hydroxyl radical with methane by quantitative ESR. Symp. (Int.) Combust. 1967, 11, 1143–1150. [Google Scholar] [CrossRef]
- Dixon-Lewis, G.; Williams, A. Some observations on the combustion of methane in premixed flames. Symp. (Int.) Combust. 1967, 11, 951–958. [Google Scholar] [CrossRef]
- Westenberg, A.A.; Fristrom, R.M. METHANE-OXYGEN FLAME STRUCTURE. IV. CHEMICAL KINETIC CONSIDERATIONS. J. Phys. Chem. 1961, 65, 591–601. [Google Scholar] [CrossRef]
- Hong, Z.; Davidson, D.F.; Lam, K.-Y.; Hanson, R.K. A shock tube study of the rate constants of HO2 and CH3 reactions. Combust. Flame 2012, 159, 3007–3013. [Google Scholar] [CrossRef]
- Bryukov, M.G.; Knyazev, V.D.; Lomnicki, S.M.; McFerrin, C.A.; Dellinger, B. Temperature-Dependent Kinetics of the Gas-Phase Reactions of OH with Cl2, CH4, and C3H8. J. Phys. Chem. A 2004, 108, 10464–10472. [Google Scholar] [CrossRef]
- Calpini, B.; Jeanneret, F.; Bourqui, M.; Clappier, A.; Vajtai, R.; van den Bergh, H. Direct measurement of the total reaction rate of OH in the atmosphere. Analusis 1999, 27, 328–336. [Google Scholar] [CrossRef]
- Gierczak, T.; Talukdar, R.K.; Herndon, S.C.; Vaghjiani, G.L.; Ravishankara, A.R. Rate Coefficients for the Reactions of Hydroxyl Radicals with Methane and Deuterated Methanes. J. Phys. Chem. A 1997, 101, 3125–3134. [Google Scholar] [CrossRef]
- Mellouki, A.; Teton, S.; Laverdet, G.; Quilgars, A.; Le Bras, G. Kinetic studies of OH reactions with H2O, C3H8 and CH4 using the pulsed laser photolysis - laser induced fluorescence method. J. Chim. Phys. 1994, 91, 473–487. [Google Scholar] [CrossRef]
- Sharkey, P.; Smith, I.W.M. Kinetics of elementary reactions at low temperatures: Rate constants for the reactions of OH with HCl (298 ≥ T/K ≥ 138), CH4(298 ≥ T/K ≥ 178) and C2H6(298 ≥ T/K ≥ 138). J. Chem. Soc. Faraday Trans. 1993, 89, 631–637. [Google Scholar] [CrossRef]
- Dunlop, J.R.; Tully, F.P. A kinetic study of hydroxyl radical reactions with methane and perdeuterated methane. J. Phys. Chem. 1993, 97, 11148–11150. [Google Scholar] [CrossRef]
- Yetter, R.A.; Dryer, F.L. Inhibition of moist carbon monoxide oxidation by trace amounts of hydrocarbons. Symp. (Int.) Combust. 1992, 24, 757–767. [Google Scholar] [CrossRef]
- Lancar, I.T.; LeBras, G.; Poulet, G. Redetermination de la constante de vitesse de la reaction CH4 + OH et son implication atmospherique. C. R. Acad. Sci. Paris 1992, 315, 1487–1492. [Google Scholar]
- Finlayson-Pitts, B.J.; Ezell, M.J.; Jayaweera, T.M.; Berko, H.N.; Lai, C.C. Kinetics of the reactions of OH with methyl chloroform and methane: Implications for global tropospheric OH and the methane budget. Geophys. Res. Lett. 1992, 19, 1371–1374. [Google Scholar] [CrossRef]
- Vaghjiani, G.L.; Ravishankara, A.R. New measurement of the rate coefficient for the reaction of OH with methane. Nature 1991, 350, 406–409. [Google Scholar] [CrossRef]
- Bott, J.F.; Cohen, N. A shock tube study of the reaction of the hydroxyl radical with H2, CH4, c-C5H10, and i-C4H10. Int. J. Chem. Kinet. 1989, 21, 485–498. [Google Scholar] [CrossRef]
- Smith, G.P.; Fairchild, P.W.; Jeffries, J.B.; Crosley, D.R. Laser pyrolysis/laser fluorescence studies of high-temperature reaction rates: Description of the method and results for OH + CH4, C3H8, and C3H6. J. Phys. Chem. 1985, 89, 1269–1278. [Google Scholar] [CrossRef]
- Madronich, S.; Felder, W. Direct measurements of the rate coefficient for the reaction OH + CH4 → CH3 + H2O over 300–1500 K. Symp. (Int.) Combust. 1985, 20, 703–713. [Google Scholar] [CrossRef]
- Jonah, C.D.; Mulac, W.A.; Zeglinski, P. Rate Constants for the Reaction of OH + CO, OD + CO, and OH + Methane as a Function of Temperature. J. Phys. Chem. 1984, 88, 4100–4104. [Google Scholar] [CrossRef]
- Baulch, D.L.; Craven, R.J.B.; Din, M.; Drysdale, D.D.; Grant, S.; Richardson, D.J.; Walker, A.; Watling, G. Rates of hydroxy radical reactions with methane, ethane and propane over the temperature range 403–696 K. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1983, 79, 689–698. [Google Scholar] [CrossRef]
- Jeong, K.M.; Kaufman, F. Kinetics of the reaction of hydroxyl radical with methane and with nine chlorine- and fluorine-substituted methanes. 1. Experimental results, comparisons, and applications. J. Phys. Chem. 1982, 86, 1808–1815. [Google Scholar] [CrossRef]
- Fairchild, P.W.; Smith, G.P.; Crosley, D.R. A laser pyrolysis/laser fluorescence technique for combustion chemical kinetics. Symp. (Int.) Combust. 1982, 19, 107–115. [Google Scholar] [CrossRef]
- Husain, D.; Plane, J.M.; Slater, N.K.H. Kinetic investigation of the reactions of OH(X2π) with the hydrogen halides, HCl, DCl, HBr and DBr by time-resolved resonance fluorescence (A2Σ+-X2π). J. Chem. Soc. Faraday Trans. 2 1981, 77, 1949–1962. [Google Scholar] [CrossRef]
- Tully, F.P.; Ravishankara, A.R. Flash photolysis-resonance fluorescence kinetic study of the reactions hydroxyl + molecular hydrogen → water + atomic hydrogen and hydroxyl + methane → water + methyl from 298 to 1020 K. J. Phys. Chem. 1980, 84, 3126–3130. [Google Scholar] [CrossRef]
- Sworski, T.J.; Hochanadel, C.J.; Ogren, P.J. Flash photolysis of water vapor in methane. Hydrogen and hydroxyl yields and rate constants for methyl reactions with hydrogen and hydroxyl. J. Phys. Chem. 1980, 84, 129–134. [Google Scholar] [CrossRef]
- Zellner, R.; Steinert, W. A flash photolysis study of the rate of the reaction OH + CH4 → CH3 + H2O over an extended temperature range. Int. J. Chem. Kinet. 1976, 8, 397–409. [Google Scholar] [CrossRef]
- Howard, C.J.; Evenson, K.M. Rate constants for the reactions of OH with CH4 and fluorine, chlorine, and bromine substituted methanes at 296 K. J. Chem. Phys. 1976, 64, 197–202. [Google Scholar] [CrossRef]
- Cox, R.A.; Derwent, R.G.; Holt, P.M. Relative rate constants for the reactions of OH radicals with H2, CH4, CO, NO and HONO at atmospheric pressure and 296 K. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1976, 72, 2031–2043. [Google Scholar] [CrossRef]
- Cox, R.A.; Derwent, R.G.; Eggleton, A.E.J.; Lovelock, J.E. Photochemical oxidation of halocarbons in the troposphere. Atmos. Environ. (1967) 1976, 10, 305–308. [Google Scholar] [CrossRef]
- Zellner, R. Rate Measurements of Some Bimolecular Reactions of the Hydroxyl Radical over an Extended Temperature Range. Mol. Rate Process. Pap. Symp. 1975, 7. [Google Scholar]
- Steinert, W.; Zellner, R. Rates of Reaction of OH with CO and CH4 over an Extended Temperature Range. Deuxiene Symp. Eur. Sur La Combust. 1975, 2. [Google Scholar]
- Overend, R.P.; Paraskevopoulos, G.; Cvetanović, R.J. Rates of OH Radical Reactions. I. Reactions with H2, CH4, C2H6, and C3H8 at 295 K. Can. J. Chem. 1975, 53, 3374–3382. [Google Scholar] [CrossRef]
- Gordon, S.; Mulac, W.A. Reaction of the OH(X2π) Radical Produced by the Pulse Radiolysis of Water Vapor. Proc. Symp. Chem. Kinet. Data Up. Low. Atmos. 1975, 289, 299. [Google Scholar]
- Margitan, J.J.; Kaufman, F.; Anderson, J.G. The reaction of OH with CH4. Geophys. Res. Lett. 1974, 1, 80–81. [Google Scholar] [CrossRef]
- Davis, D.D.; Fischer, S.; Schiff, R. Flash photolysis-resonance fluorescence kinetics study: Temperature dependence of the reactions OH + CO → CO2 + H and OH + CH4 → H2O + CH3. J. Chem. Phys. 1974, 61, 2213–2219. [Google Scholar] [CrossRef]
- Peeters, J.; Mahnen, G. Reaction mechanisms and rate constants ofelementary steps in methane-oxygen flames. Symp. (Int.) Combust. 1973, 14, 133–146. [Google Scholar] [CrossRef]
- Baker, R.R.; Baldwin, R.R.; Walker, R.W. The use of the H2+O2 reaction in determining the velocity constants of elementary reaction in hydrocarbon oxidation. Symp. (Int.) Combust. 1971, 13, 291–299. [Google Scholar] [CrossRef]
- Greiner, N.R. Hydroxyl Radical Kinetics by Kinetic Spectroscopy. VI. Reactions with Alkanes in the Range 300–500°K. J. Chem. Phys. 1970, 53, 1070–1076. [Google Scholar] [CrossRef]
- Greiner, N.R. Hydroxyl Radical Kinetics by Kinetic Spectroscopy. IV. Some Deuterium Isotope Effects. J. Chem. Phys. 1968, 48, 1413. [Google Scholar] [CrossRef]
- Horne, D.G.; Norrish, R.G.W. Rate of H-abstraction by OH from Hydrocarbons. Nature 1967, 215, 1373–1374. [Google Scholar] [CrossRef]
- Greiner, N.R. Hydroxyl-Radical Kinetics by Kinetic Spectroscopy. I. Reactions with H2, CO, and CH4 at 300°K. J. Chem. Phys. 1967, 46, 2795–2799. [Google Scholar] [CrossRef]
- Fristrom, R.M. Radical concentrations and reactions in a methane-oxygen flame. Symp. (Int.) Combust. 1963, 9, 560–575. [Google Scholar] [CrossRef]
- Fenimore, C.P.; Jones, G.W. Rate of Reaction of Methane with H Atoms and Oh Radicals in Flames. J. Phys. Chem. 1961, 65, 2200–2203. [Google Scholar] [CrossRef]
- Bravo-Pérez, G.; Alvarez-Idaboy, J.R.; Jiménez, A.G.; Cruz-Torres, A. Quantum chemical and conventional TST calculations of rate constants for the OH+alkane reaction. Chem. Phys. 2005, 310, 213–223. [Google Scholar] [CrossRef]
- Masgrau, L.; González-Lafont, À.; Lluch, J.M. The reactions CHnD4−n+OH→P and CH4+OD→CH3+HOD as a test of current direct dynamics computational methods to determine variational transition-state rate constants. I. J. Chem. Phys. 2001, 114, 2154–2165. [Google Scholar] [CrossRef]
- Masgrau, L.; González-Lafont, À.; Lluch, J.M. The reactions CHnD4−n+OH→P and CH4+OD→CH3+HOD as a test of current direct dynamics multicoefficient methods to determine variational transition state rate constants. II. J. Chem. Phys. 2001, 115, 4515–4526. [Google Scholar] [CrossRef]
- Schwartz, M.; Marshall, P.; Berry, R.J.; Ehlers, C.J.; Petersson, G.A. Computational Study of the Kinetics of Hydrogen Abstraction from Fluoromethanes by the Hydroxyl Radical. J. Phys. Chem. A 1998, 102, 10074–10081. [Google Scholar] [CrossRef][Green Version]
- Melissas, V.S.; Truhlar, D.G. Interpolated variational transition state theory and tunneling calculations of the rate constant of the reaction OH+CH4 at 223–2400 K. J. Chem. Phys. 1993, 99, 1013–1027. [Google Scholar] [CrossRef]
- Dobbs, K.D.; Dixon, D.A.; Komornicki, A. Ab initio prediction of the barrier height for abstraction of H from CH4 by OH. J. Chem. Phys. 1993, 98, 8852–8858. [Google Scholar] [CrossRef]
- Cohen, N. Are reaction rate coefficients additive? Revised transition state theory calculations for OH + alkane reactions. Int. J. Chem. Kinet. 1991, 23, 397–417. [Google Scholar] [CrossRef]
- Truong, T.N.; Truhlar, D.G. Ab initio transition state theory calculations of the reaction rate for OH+CH4→H2O+CH3. Ber. Bunsenges. Phys. Chem. 1990, 93, 1761–1769. [Google Scholar] [CrossRef]
- Gonzalez, C.; McDouall, J.J.W.; Schlegel, H.B. Ab initio study of the reactions between methane and hydroxyl, hydrogen atom, and triplet oxygen atom. J. Phys. Chem. 1990, 94, 7467–7471. [Google Scholar] [CrossRef]
- Cohen, N.; Benson, S.W. Transition-state-theory calculations for reactions of hydroxyl radicals with haloalkanes. J. Phys. Chem. 1987, 91, 162–170. [Google Scholar] [CrossRef]
- Leroy, G.; Sana, M.; Tinant, A. Etude theorique des reactions d’abstraction d’hydrogene RH + X = R + HX, avec R, X≡H, CH3, NH2, OH et F. Can. J. Chem. 1985, 63, 1447–1456. [Google Scholar] [CrossRef]
- Cohen, N. The use of transition-state theory to extrapolate rate coefficients for reactions of oh with alkanes. Int. J. Chem. Kinet. 1982, 14, 1339–1362. [Google Scholar] [CrossRef]
- Shaw, R. Semi-empirical extrapolation and estimation of rate constants for abstraction of H from methane by H, O, HO, and O2. J. Phys. Chem. Ref. Data 1978, 7, 1179–1190. [Google Scholar] [CrossRef]
- Gaillard-Cusin, F.; James, H.; Rouan, J.-P. No. 105. - Le phenomene d’inhibition-promotion par le methane de la combustion de hautes pressions de l’oxyde de carbone. J. Chim. Phys. 1969, 66, 751–756. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Teng, Y.; Shimomura, S.; Tabata, K.; Suzuki, E. Ab Initio Study for Selective Oxidation of Methane with NOx (x = 1, 2). J. Phys. Chem. A 1999, 103, 8272–8278. [Google Scholar] [CrossRef]
- Skinner, G.B.; Lifshitz, A.; Scheller, K.; Burcat, A. Kinetics of Methane Oxidation. J. Chem. Phys. 1972, 56, 3853–3861. [Google Scholar] [CrossRef]
- Mayer, S.W.; Schieler, L. Activation energies and rate constants computed for reactions of oxygen with hydrocarbons. J. Phys. Chem. 1968, 72, 2628–2631. [Google Scholar] [CrossRef]
- Baldwin, R.R.; Jones, P.N.; Walker, R.W. Determination of the rate constant for HO2 + CH4 →H2O2 → H2O2 + CH3 at 443 °C. J. Chem. Soc. Faraday Trans. 2 1988, 84, 199–207. [Google Scholar] [CrossRef]
- Fu, X.; Aggarwal, S.K. Two-stage ignition and NTC phenomenon in diesel engines. Fuel 2015, 144, 188–196. [Google Scholar] [CrossRef]
- OriginPro; Version 2017; OriginLab Corporation: Northampton, MA, USA, 2017.
- Ranzi, E.; Frassoldati, A.; Grana, R.; Cuoci, A.; Faravelli, T.; Kelley, A.P.; Law, C.K. Hierarchical and comparative kinetic modeling of laminar flame speeds of hydrocarbon and oxygenated fuels. Prog. Energy Combust. Sci. 2012, 38, 468–501. [Google Scholar] [CrossRef]
- Prince, J.C.; Williams, F.A.; Ovando, G.E. A short mechanism for the low-temperature ignition of n-heptane at high pressures. Fuel 2015, 149, 138–142. [Google Scholar] [CrossRef]
- Prince, J.C.; Williams, F.A. Revised short mechanism for the low-temperature ignition of n-heptane for a wider pressure range. Fuel 2015, 150, 730–731. [Google Scholar] [CrossRef]
- Stagni, A.; Cuoci, A.; Frassoldati, A.; Faravelli, T.; Ranzi, E. Lumping and Reduction of Detailed Kinetic Schemes: An Effective Coupling. Ind. Eng. Chem. Res. 2013, 53, 9004–9016. [Google Scholar] [CrossRef]
- Turányi, T.; Tomlin, A.S. Analysis of Kinetic Reaction Mechanisms; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]

































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Facility | Fuel Composition | Pressure | Temp. | Ref. | |||
|---|---|---|---|---|---|---|---|
| CH4 (mol %) | C3H8 (mol %) | C7H16 (mol %) | λ (−) | (bar) | (K) | ||
| Rapid compression machine | 100 | 0 | 0 | 1.9 | 100 | 906–941 | [7] |
| 95 | 5 | 0 | 1.9 | 100 | 888–916 | [7] | |
| 90 | 10 | 0 | 1.9 | 100 | 803–898 | [6] | |
| 70 | 30 | 0 | 1.9 | 100 | 826–865 | [7] | |
| 92.68 | 4.88 | 2.44 | 1.685 | 60 | 701–877 | [6] | |
| 90.48 | 4.76 | 4.76 | 1.513 | 60 | 671–781 | [6] | |
| 92.68 | 4.88 | 2.44 | 1.685 | 100 | 709–817 | [6] | |
| 97.56 | 0 | 2.44 | 1.67 | 60 | 720–869 | [6] | |
| Shock tube | 86.36 | 4.55 | 9.09 | 1.257 | 60 | 748–1187 | [6] |
| 90.91 | 0 | 9.09 | 1.226 | 60 | 785–1284 | [6] | |
| Reaction Number | Reaction Equation |
|---|---|
| 273 | C7H16 + HO2 ↔ H2O2 + n-C7H15 |
| 296 | n-C7H15 + O2 ↔ C7H14 + HO2 |
| 297 | n-C7H15 + O2 ↔ n-C7-QOOH |
| 298 | n-C7-QOOH ↔ HO2 + C7H14 |
| 299 | n-C7-QOOH + O2 ↔ n-C7-OQOOH + OH |
| 300 | n-C7-OQOOH ↔ OH + CH2O + CO + C2H4 + n-C3H7 |
| Reaction Number | Reaction Equation |
|---|---|
| 17 | 2 HO2 ↔ H2O2 + O2# |
| 18 | 2 HO2 ↔ H2O2 + O2## |
| 38 | CH2O + OH ↔ HCO + H2O |
| 40 | CH2O + HO2 ↔ HCO + H2O2 |
| 42 | CH4 + OH ↔ H2O + CH3 |
| 44 | CH4 + O2 ↔ CH3 + HO2 |
| 45 | CH4 + HO2 ↔ CH3 + H2O2 |
| 51 | CH3 + HO2 ↔ CH3O + OH |
| 52 | CH3 + O2 ↔ CH2O + OH |
| 242 | n-C3H7 + O2 ↔ C3H6 + HO2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuh, S.; Frühhaber, J.; Lauer, T.; Winter, F. A Novel Dual Fuel Reaction Mechanism for Ignition in Natural Gas–Diesel Combustion. Energies 2019, 12, 4396. https://doi.org/10.3390/en12224396
Schuh S, Frühhaber J, Lauer T, Winter F. A Novel Dual Fuel Reaction Mechanism for Ignition in Natural Gas–Diesel Combustion. Energies. 2019; 12(22):4396. https://doi.org/10.3390/en12224396
Chicago/Turabian StyleSchuh, Sebastian, Jens Frühhaber, Thomas Lauer, and Franz Winter. 2019. "A Novel Dual Fuel Reaction Mechanism for Ignition in Natural Gas–Diesel Combustion" Energies 12, no. 22: 4396. https://doi.org/10.3390/en12224396
APA StyleSchuh, S., Frühhaber, J., Lauer, T., & Winter, F. (2019). A Novel Dual Fuel Reaction Mechanism for Ignition in Natural Gas–Diesel Combustion. Energies, 12(22), 4396. https://doi.org/10.3390/en12224396