Hydro-Pyrolysis and Catalytic Upgrading of Biomass and Its Hydroxy Residue Fast Pyrolysis Vapors




Abstract
1. Introduction
2. Experiments
2.1. Catalyst Preparation
2.2. Catalyst Characterization
2.3. Pyrolysis-Gas Chromatography/Mass Spectrometry Technique
2.4. Feedstock Properties
3. Results and Discussion
3.1. Characterization of Catalysts
3.2. Py-GC/MS Results and Discussion
3.2.1. H2 and Catalyst Effects on the Overall Yield of M Pyrolysis Vapors
3.2.2. H2 and Catalyst Effects on Carboxylic Acids
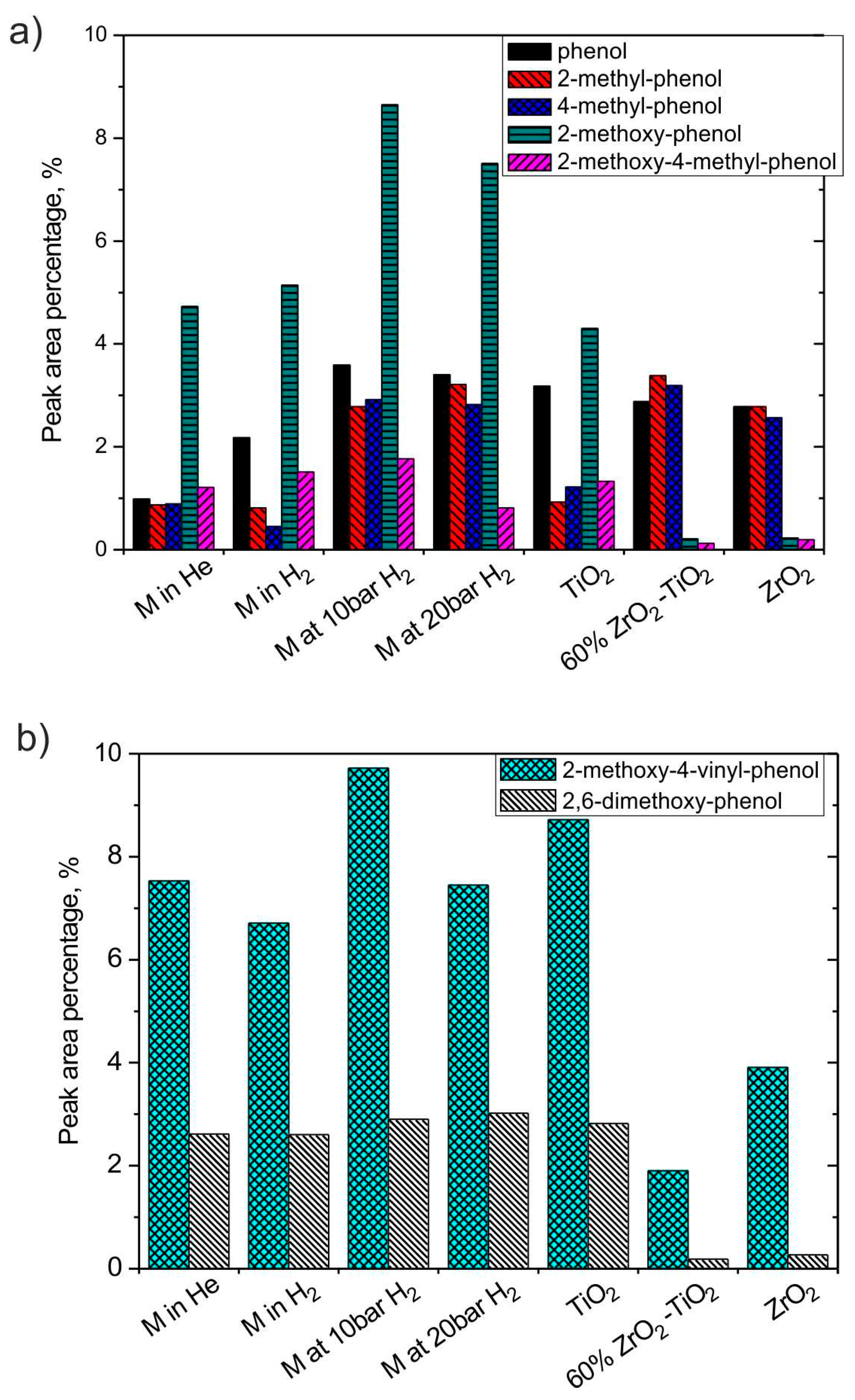
3.2.3. H2 and Catalyst Effects on Phenol Derivatives
3.2.4. The effect of H2 and Catalysts on Aromatic Hydrocarbons
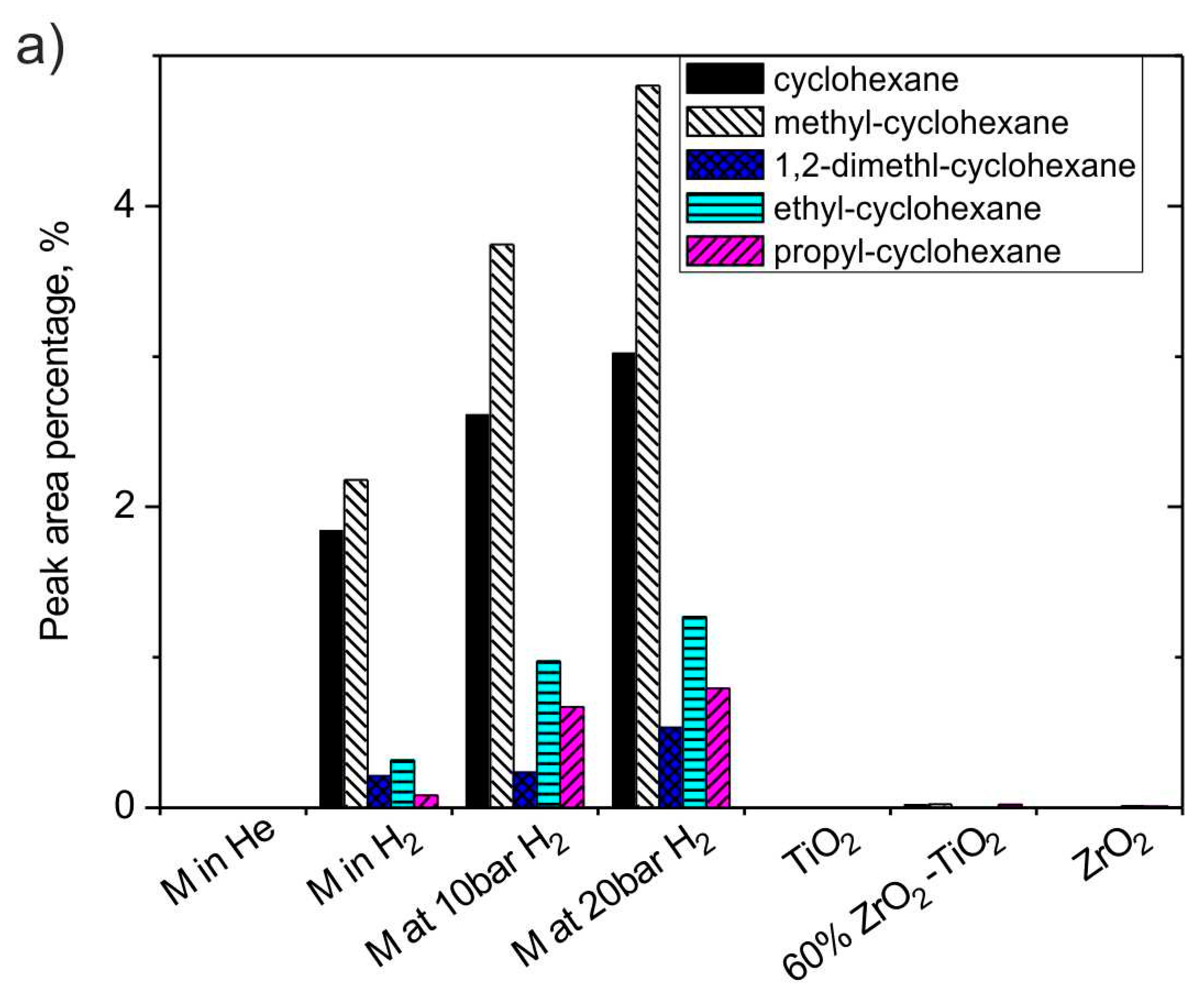
3.2.5. The Effect of H2 and Catalysts on Cyclo-Alkanes
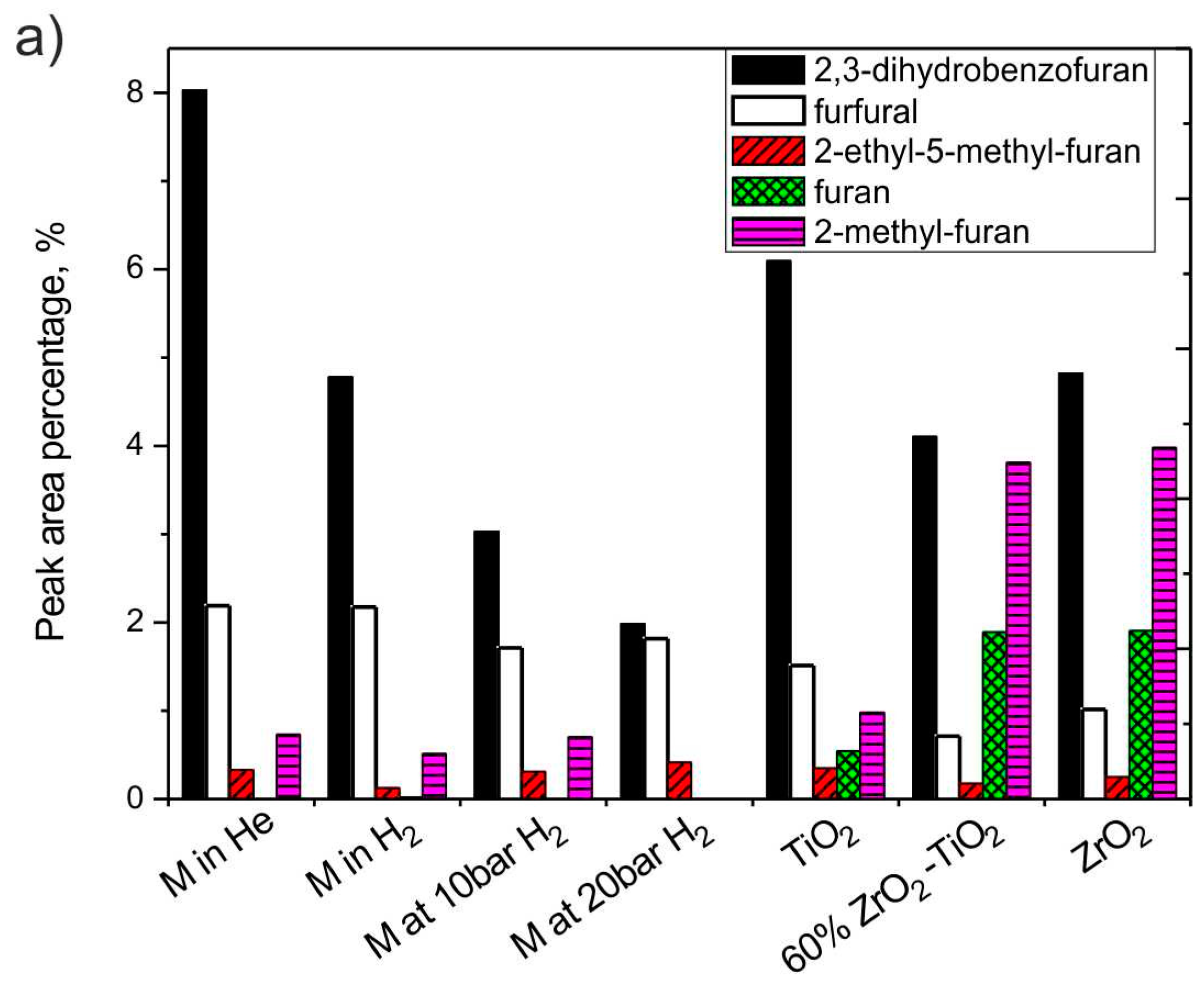
3.2.6. The Effect of H2 and Catalysts on Furan Derivatives and Alcohols
3.3. The Comparison of Catalytic Cracking and Hydrogenation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ahorsu, R.; Medina, F.; Constanti, M. Significance and Challenges of Biomass as a Suitable Feedstock for Bioenergy and Biochemical Production: A Review. Energies 2018, 11, 3366. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, Y.; Tang, Z.; Li, W.Z.; Zhu, X.F. Catalytic upgrading of biomass fast pyrolysis vapors with titania and zirconia/titania based catalysts. Fuel 2010, 89, 2096–2103. [Google Scholar] [CrossRef]
- Olazar, M.; Aguado, R.; Bilbao, J.; Barona, A. Pyrolysis of sawdust in a conical spouted-bed reactor with a HZSM-5 catalyst. Aiche J. 2000, 46, 1025–1033. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R.; Neuenschwander, G.G.; Rotness, L.J.; Olarte, M.V.; Zacher, A.H.; Solantausta, Y. Catalytic Hydroprocessing of Fast Pyrolysis Bio-oil from Pine Sawdust. Energy Fuels 2012, 26, 3891–3896. [Google Scholar] [CrossRef]
- Grams, J.N.M.; Ruppert, A.M.; Kwapinski, W. Catalytic performance of Ni catalyst supported on CeO2, ZrO2 and CeO2-ZrO2 in the upgrading of cellulose fast pyrolysis vapors. Comptes Rendus Chim. 2015, 18, 1223–1228. [Google Scholar] [CrossRef]
- French, R.; Czernik, S. Catalytic pyrolysis of biomass for biofuels production. Fuel Process. Technol. 2010, 91, 25–32. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Delichere, P.; Geantet, C. Hydrodeoxygenation of guaiacol Part II: Support effect for CoMoS catalysts on HDO activity and selectivity. Appl. Catal. B-Environ. 2011, 101, 246–255. [Google Scholar] [CrossRef]
- Kaewpengkrow, P.; Atong, D.; Sricharoenchaikul, V. Catalytic upgrading of pyrolysis vapors from Jatropha wastes using alumina, zirconia and titania based catalysts. Bioresour. Technol. 2014, 163, 262–269. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, Z.B.; Wang, X.Q.; Dong, C.Q.; Liu, Y.Q. Catalytic upgrading of biomass fast pyrolysis vapors using ordered mesoporous ZrO2, TiO2 and SiO2. In Proceedings of the International Conference on Applied Energy (Icae2014), Taipei, Taiwan, 30 May–2 June 2014; pp. 1937–1941. [Google Scholar]
- Wachala, M.; Grams, J.; Kwapinski, W.; Ruppert, A.M. Influence of ZrO2 on catalytic performance of Ru catalyst in hydrolytic hydrogenation of cellulose towards gamma-valerolactone. Int. J. Hydrogen Energy 2016, 41, 8688–8695. [Google Scholar] [CrossRef]
- Maity, S.K.; Rana, M.S.; Bej, S.K.; Ancheyta, J.; Dhar, G.M.; Rao, T. TiO2-ZrO2 mixed oxide as a support for hydrotreating catalyst. Catal. Lett. 2001, 72, 115–119. [Google Scholar] [CrossRef]
- Zhang, X.H.; Long, J.X.; Kong, W.; Zhang, Q.; Chen, L.G.; Wang, T.J.; Ma, L.L.; Li, Y.P. Catalytic Upgrading of Bio-oil over Ni-Based Catalysts Supported on Mixed Oxides. Energy Fuels 2014, 28, 2562–2570. [Google Scholar] [CrossRef]
- Ruppert, A.; Paryjczak, T. Pt/ZrO2/TiO2 catalysts for selective hydrogenation of crotonaldehyde: Tuning the SMSI effect for optimum performance. Appl. Catal. A-Gen. 2007, 320, 80–90. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Leahy, J.J.; Kwapinski, W. Catalytically upgrading bio-oil via esterification. Energy Fuel 2015, 29, 3691–3698. [Google Scholar] [CrossRef]
- Refaat, A.A. Biodiesel production using solid metal oxide catalysts. Int. J. Environ. Sci. Technol. 2011, 8, 203–221. [Google Scholar] [CrossRef]
- Zhang, S.P.; Yan, Y.J.; Li, T.C.; Ren, Z.W. Upgrading of liquid fuel from the pyrolysis of biomass. Bioresour. Technol. 2005, 96, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, T.J.; Ma, L.L.; Zhang, Q.; Liang, W. Upgrading of the liquid fuel from fast pyrolysis of biomass over MoNi/gamma-Al2O3 catalysts. Appl. Energy 2010, 87, 2886–2891. [Google Scholar] [CrossRef]
- Li, H.; Riisager, A.; Saravanamurugan, S.; Pandey, A.; Sangwan, R.S.; Yang, S.; Luque, R. Carbon-Increasing Catalytic Strategies for Upgrading Biomass into Energy-Intensive Fuels and Chemicals. ACS Catal. 2018, 8, 148–187. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A-Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Mendes, M.J.; Santos, O.A.A.; Jordao, E.; Silva, A.M. Hydrogenation of oleic acid over ruthenium catalysts. Appl. Catal. A-Gen. 2001, 217, 253–262. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.W.; Zacher, A.H.; Wang, L.; Ren, S.J.; Liang, J.; Wei, Y.; Liu, Y.P.; Tang, J.M.; Zhang, Q.; et al. A review of catalytic hydrodeoxygenation of lignin-derived phenols from biomass pyrolysis. Bioresour. Technol. 2012, 124, 470–477. [Google Scholar] [CrossRef]
- Pandey, M.P.; Kim, C.S. Lignin Depolymerization and Conversion: A Review of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Danalatos, N.G.; Archontoulis, S.V.; Mitsios, I. Potential growth and biomass productivity of Miscanthus x giganteus as affected by plant density and N-fertilization in central Greece. Biomass Bioenergy 2007, 31, 145–152. [Google Scholar] [CrossRef]
- Dussan, K.; Girisuta, B.; Haverty, D.; Leahy, J.J.; Hayes, M.H. Kinetics of levulinic acid and furfural production from Miscanthus x giganteus. Bioresour Technol. 2013, 149, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Kwapinska, M.; Kwapinski, W.; Czajka, K.M.; Kennedy, J.; Leahy, J.J. Impact of torrefaction on properties of Miscanthus x giganteus relevant to gasification. Fuel 2014, 121, 189–197. [Google Scholar] [CrossRef]
- Wang, X.H.; Li, J.G.; Kamiyama, H.; Ishigaki, T. Fe-doped TiO2 nanopowders by oxidative pyrolysis of organometallic precursors in induction thermal plasma: Synthesis and structural characterization. Thin Solid Films 2006, 506–507, 278–282. [Google Scholar] [CrossRef]
- Peng, L.; Zhuang, J.; Lin, L. Effects of Zr/Ti molar ratio in SO2−4/ZrO2-TiO2 calcined at different temperatures on its surface properties and glucose reactivity in near-critical methanol. J. Nat. Gas Chem. 2012, 21, 138–147. [Google Scholar] [CrossRef]
- Noda, L.K.; de Almeida, R.M.; Probst, L.F.D.; Gonçalves, N.S. Characterization of sulfated TiO2 prepared by the sol–gel method and its catalytic activity in the n-hexane isomerization reaction. J. Mol. Catal. A Chem. 2005, 225, 39–46. [Google Scholar] [CrossRef]
- Babou, F.; Coudurier, G.; Vedrine, J.C. Acidic Properties of Sulfated Zirconia—An Infrared Spectroscopic Study. J. Catal. 1995, 152, 341–349. [Google Scholar] [CrossRef]
- Ramadan, A.R.; Yacoub, N.; Bahgat, S.; Ragai, J. Surface and acidic properties of mixed titanium and zirconium sulfated oxides. Colloids Surf. A Physicochem. Eng. Asp. 2007, 302, 36–43. [Google Scholar] [CrossRef]
- Ward, D.A.; Ko, E.I. One-step synthesis and characterization of zirconia-sulfate aerogels as solid superacids. J. Catal. 1994, 150, 18–33. [Google Scholar] [CrossRef]
- Azeez, A.M.; Meier, D.; Odermatt, J.; Willner, T. Fast Pyrolysis of African and European Lignocellulosic Biomasses Using Py-GC/MS and Fluidized Bed Reactor. Energy Fuels 2010, 24, 2078–2085. [Google Scholar] [CrossRef]
- Siddiqui, M.N. Catalytic pyrolysis of Arab Heavy residue and effects on the chemistry of asphaltene. J. Anal. Appl. Pyrolysis 2010, 89, 278–285. [Google Scholar] [CrossRef]
- Iliopoulou, E.F.; Stefanidis, S.D.; Kalogiannis, K.G.; Delimitis, A.; Lappas, A.A.; Triantafyllidis, K.S. Catalytic upgrading of biomass pyrolysis vapors using transition metal-modified ZSM-5 zeolite. Appl. Catal. B Environ. 2012, 127, 281–290. [Google Scholar] [CrossRef]
- Mante, O.D.; Agblevor, F.A.; Oyama, S.T.; McClung, R. Catalytic pyrolysis with ZSM-5 based additive as co-catalyst to Y-zeolite in two reactor configurations. Fuel 2014, 117, 649–659. [Google Scholar] [CrossRef]
- Zheng, A.; Zhao, Z.; Chang, S.; Huang, Z.; Wu, H.; Wang, X.; He, F.; Li, H. Effect of crystal size of ZSM-5 on the aromatic yield and selectivity from catalytic fast pyrolysis of biomass. J. Mol. Catal. A Chem. 2014, 383–384, 23–30. [Google Scholar] [CrossRef]
- Qiang, L.; Wen-Zhi, L.; Dong, Z.; Xi-Feng, Z. Analytical pyrolysis gas chromatography mass spectrometry Py-GC/MS of sawdust with Al/SBA-15 catalysts. J. Anal. Appl. Pyrolysis 2009, 84, 131–138. [Google Scholar] [CrossRef]
- Wang, D.; Xiao, R.; Zhang, H.; He, G. Comparison of catalytic pyrolysis of biomass with MCM-41 and CaO catalysts by using TGA–FTIR analysis. J. Anal. Appl. Pyrolysis 2010, 89, 171–177. [Google Scholar] [CrossRef]
- Pattiya, A.; Titiloye, J.O.; Bridgwater, A.V. Fast pyrolysis of cassava rhizome in the presence of catalysts. J. Anal. Appl. Pyrolysis 2008, 81, 72–79. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, Z.F.; Dong, C.Q.; Zhu, X.F. Catalytic Upgrading of Biomass Fast Pyrolysis Vapors with Nano Metal Oxides: An Analytical Py-GC/MS Study. Energies 2010, 3, 1805–1820. [Google Scholar] [CrossRef]
- Grams, J.; Niewiadomski, M.; Ruppert, A.M.; Kwapiński, W. Influence of Ni catalyst support on the product distribution of cellulose fast pyrolysis vapors upgrading. J. Anal. Appl. Pyrolysis 2015, 113, 557–563. [Google Scholar] [CrossRef]
- Dong, C.; Zhang, Z.; Lu, Q.; Yang, Y. Characteristics and mechanism study of analytical fast pyrolysis of poplar wood. Energy Convers. Manag. 2012, 57, 49–59. [Google Scholar] [CrossRef]
- Jiang, G.; Nowakowski, D.J.; Bridgwater, A.V. Effect of the temperature on the composition of lignin pyrolysis products. Energy Fuels 2010, 24, 4470–4475. [Google Scholar] [CrossRef]
- Hosoya, T.; Kawamoto, H.; Saka, S. Secondary reactions of lignin-derived primary tar components. J. Anal. Appl. Pyrolysis 2008, 83, 78–87. [Google Scholar] [CrossRef]
- Melligan, F.; Hayes, M.H.B.; Kwapinski, W.; Leahy, J.J. A study of hydrogen pressure during hydropyrolysis of Miscanthus x giganteus and online catalytic vapour upgrading with Ni on ZSM-5. J. Anal. Appl. Pyrolysis 2013, 103, 369–377. [Google Scholar] [CrossRef]
- Şenol, O.İ.; Ryymin, E.-M.; Viljava, T.-R.; Krause, A.O.I. Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts. J. Mol. Catal. A Chem. 2007, 277, 107–112. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of guaiacol with CoMo catalysts. Part I: Promoting effect of cobalt on HDO selectivity and activity. Appl. Catal. B Environ. 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Elliott, D.C. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21, 1792–1815. [Google Scholar] [CrossRef]
- Ruddy, D.A.; Schaidle, J.A.; Ferrell, J.R., III; Wang, J.; Moens, L.; Hensley, J.S. Recent advances in heterogeneous catalysts for bio-oil upgrading via “ex situ catalytic fast pyrolysis”: Catalyst development through the study of model compounds. Green Chem. 2014, 16, 454–490. [Google Scholar] [CrossRef]
- Adam, J.A.E.; Lappas, A.; Stocker, M.; Nilsen, M.H.; Bouzga, A. Investigation of the effect of metal sites in Me-Al-MCM-41 (Me = Fe, Cu or Zn) on the catalytic upgrading of biomass derived fast pyrolysis vapours in a fixed bed reactor using mesoporous materials. Microporous Mesoporous Mater. 2007, 96, 93–101. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Chen, L.; Zhao, B.; Yang, S.; Xie, X. Effects of Fe contents on fast pyrolysis of biomass with Fe/CaO catalysts. J. Anal. Appl. Pyrol. 2016, 119, 133–138. [Google Scholar] [CrossRef]
- Karnjanakom, S.; Bayu, A.; Xiaoketi, P.; Hao, X.; Kongparakul, S.; Samart, C.; Abudula, A.; Guan, G. Selective production of aromatic hydrocarbons from catalytic pyrolysis of biomass over Cu or Fe loaded mesoporous rod-like alumina. RSC Adv. 2016, 6, 50618–50629. [Google Scholar] [CrossRef]
- Karagoz, S.; Kawakami, T.; Kako, A.; Iiguni, Y.; Ohtani, H. Single shot pyrolysis and on-line conversion of lignocellulosic biomass with HZSM-5 catalyst using tandem micro-reactor-GC-MS. RSC Adv. 2016, 6, 46108–46115. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Production of aromatic hydrocarbons via catalytic pyrolysis of biomass over Fe-modified HZSM-5 zeolites. ACS Sustain. Chem. Eng. 2015, 3, 1623–1631. [Google Scholar] [CrossRef]
- Stefanidis, S.D.; Karakoulia, S.A.; Kalogiannis, K.G.; Iliopoulou, E.F.; Delimitis, A.; Yiannoulakis, H.; Zampetakis, T.; Lappas, A.A.; Triantafyllidis, K.S. Natural magnesium oxide (MgO) catalysts: A cost-effective sustainable alternative to acid zeolites for the in situ upgrading ofbiomass fast pyrolysis oil. Appl. Catal. B Environ. 2016, 196, 155–173. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | M | HR | Lignin |
|---|---|---|---|
| Moisture, wt.% | 8.82 ± 0.22 | 6.21 ± 0.20 | - |
| Cellulose, wt.% | 37.1 ± 2.2 | 0 | - |
| Hemicellulose, wt.% | 18.0 ± 1.2 | 0 | - |
| Lignin, wt.% | 20.1 ± 3.4 | 44.2 ± 4.4 | 100 |
| Ash, wt.% | 3.55 ± 0.42 | 18.1 ± 1.31 | - |
| C, wt.% | 46.6 | 55.7 | 54.1 |
| H, wt.% | 6.36 | 4.75 | 5.39 |
| N, wt.% | 0.41 | 0.37 | 0.62 |
| S, wt.% | - | - | 4 |
| O *, wt.% | 46.6 | 39.2 | 35.9 |
| HHV, kJ/g | 18.7 | 20.2 | 22.3 |
| Catalysts | Surface Area m2/g | Surface Acidity µmol/g | Surface Acidity µmol/m2 | Pore Volume cm3/g | Pore Diameter nm |
|---|---|---|---|---|---|
| pure TiO2 | 14.4 | 58.2 | 4.04 | 0.112 | 8.3 |
| 60 wt.% ZrO2-TiO2 | 103 | 493 | 4.81 | 0.262 | 4.9 |
| pure ZrO2 | 97.3 | 469 | 4.82 | 0.255 | 6.2 |
| Components wt.% | Miscanthus | Lignin | Hydrolysis Residue | ||||||
|---|---|---|---|---|---|---|---|---|---|
| He | H2 | cat | He | H2 | cat | He | H2 | cat | |
| acetic acid | 17.0 | 9.98 | 18.28 | 0 | 0 | 0 | 5.99 | 3.81 | 5.72 |
| Phenol | 0.99 | 2.18 | 2.88 | 3.09 | 7.26 | 6.32 | 7.10 | 10.9 | 11.9 |
| 2-methyl-phenol | 0.87 | 0.81 | 3.38 | 2.27 | 1.81 | 3.21 | 1.37 | 1.89 | 1.09 |
| 4-methyl-phenol | 0.89 | 0.45 | 3.19 | 1.73 | 4.32 | 3.12 | 3.46 | 3.82 | 3.01 |
| 4-ethyl phenol | 0.12 | 0.18 | 0.67 | 0.04 | 0.00 | 1.89 | 2.70 | 2.18 | 3.90 |
| 2-methoxy-pheonl | 4.73 | 5.14 | 0.21 | 18.57 | 21.1 | 18.9 | 3.21 | 2.98 | 3.91 |
| 2-methoxy-4-methyl-pheonl | 1.21 | 1.51 | 0.12 | 0.13 | 0.29 | 1.29 | 2.70 | 4.29 | 2.78 |
| 2-methoxy-4-ethyl-pheonl | 1.16 | 1.00 | 0.91 | 0.76 | 0.19 | 1.09 | 1.55 | 1.52 | 1.67 |
| low MW phenols (MW < 150) | 9.97 | 11.2 | 11.3 | 26.59 | 35.0 | 35.9 | 22.09 | 27.6 | 28.3 |
| 2-methoxy-4-vinyl-phenol | 7.53 | 6.71 | 1.90 | 5.37 | 5.21 | 3.21 | 2.99 | 3.90 | 1.18 |
| 2,6-dimethoxy-phenol | 2.30 | 2.60 | 0.19 | 5.67 | 5.91 | 4.91 | 2.62 | 3.67 | 1.90 |
| high low MW phenols (MW > 150) | 9.83 | 9.31 | 2.09 | 11.04 | 11.1 | 8.12 | 5.61 | 7.57 | 3.08 |
| Total phenols | 19.8 | 20.5 | 13.4 | 37.63 | 46.1 | 44.0 | 27.69 | 35.1 | 31.4 |
| benzene | 0.00 | 3.90 | 2.89 | 0.21 | 2.02 | 1.91 | 0.11 | 2.02 | 2.02 |
| toluene | 0.29 | 1.29 | 5.38 | 1.04 | 3.12 | 4.80 | 0.70 | 2.88 | 3.00 |
| p-xylene | 0.00 | 1.21 | 2.78 | 0.37 | 1.08 | 3.09 | 0.00 | 0.80 | 1.71 |
| 1-ethyl-methyl-benzene | 0.04 | 0.00 | 1.02 | 0.32 | 1.09 | 1.11 | 0.08 | 0.19 | 0.79 |
| aromatic hydrocarbons | 0.32 | 6.40 | 12.0 | 1.93 | 7.31 | 10.9 | 0.89 | 5.89 | 7.52 |
| cyclohexane | 0 | 1.84 | 0.02 | 0 | 2.20 | 0.00 | 0 | 2.53 | 0.11 |
| methyl-cyclohexane | 0 | 2.18 | 0.02 | 0 | 3.29 | 0.00 | 0 | 1.24 | 0.02 |
| 1,2-dimethl-cyclohexane | 0 | 0.21 | 0 | 0 | 1.02 | 0.09 | 0 | 0.01 | 0.07 |
| ethyl-cyclohexane | 0 | 0.32 | 0 | 0 | 0 | 0 | 0 | 0.61 | 0.02 |
| propyl-cyclohexane | 0 | 0.08 | 0.02 | 0 | 1.90 | 0.39 | 0.25 | 0.19 | 0 |
| methyl-cyclopentane | 0 | 0.87 | 0.32 | 0 | 2.10 | 0.19 | 0 | 0.67 | 0.52 |
| 1,2-dimethyl-cyclopentane | 0 | 0.92 | 0.13 | 0 | 0 | 0.28 | 0 | 0.82 | 0 |
| cyclo alkanes | 0 | 6.42 | 0.51 | 0.00 | 10.5 | 0.95 | 0.25 | 6.07 | 0.74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Leahy, J.J.; Grams, J.; Kwapinski, W. Hydro-Pyrolysis and Catalytic Upgrading of Biomass and Its Hydroxy Residue Fast Pyrolysis Vapors. Energies 2019, 12, 3474. https://doi.org/10.3390/en12183474
Liu Y, Leahy JJ, Grams J, Kwapinski W. Hydro-Pyrolysis and Catalytic Upgrading of Biomass and Its Hydroxy Residue Fast Pyrolysis Vapors. Energies. 2019; 12(18):3474. https://doi.org/10.3390/en12183474
Chicago/Turabian StyleLiu, Yichen, James J. Leahy, Jacek Grams, and Witold Kwapinski. 2019. "Hydro-Pyrolysis and Catalytic Upgrading of Biomass and Its Hydroxy Residue Fast Pyrolysis Vapors" Energies 12, no. 18: 3474. https://doi.org/10.3390/en12183474
APA StyleLiu, Y., Leahy, J. J., Grams, J., & Kwapinski, W. (2019). Hydro-Pyrolysis and Catalytic Upgrading of Biomass and Its Hydroxy Residue Fast Pyrolysis Vapors. Energies, 12(18), 3474. https://doi.org/10.3390/en12183474