Energy Storage Analysis of UIO-66 and Water Mixed Nanofluids: An Experimental and Theoretical Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental and Computational Methods
2.1. Experimental Methods
2.2. Simulation Details
2.2.1. MD Details
2.2.2. GCMC Details
3. Results and Discussion
3.1. Thermodynamic Energy of UIO-66
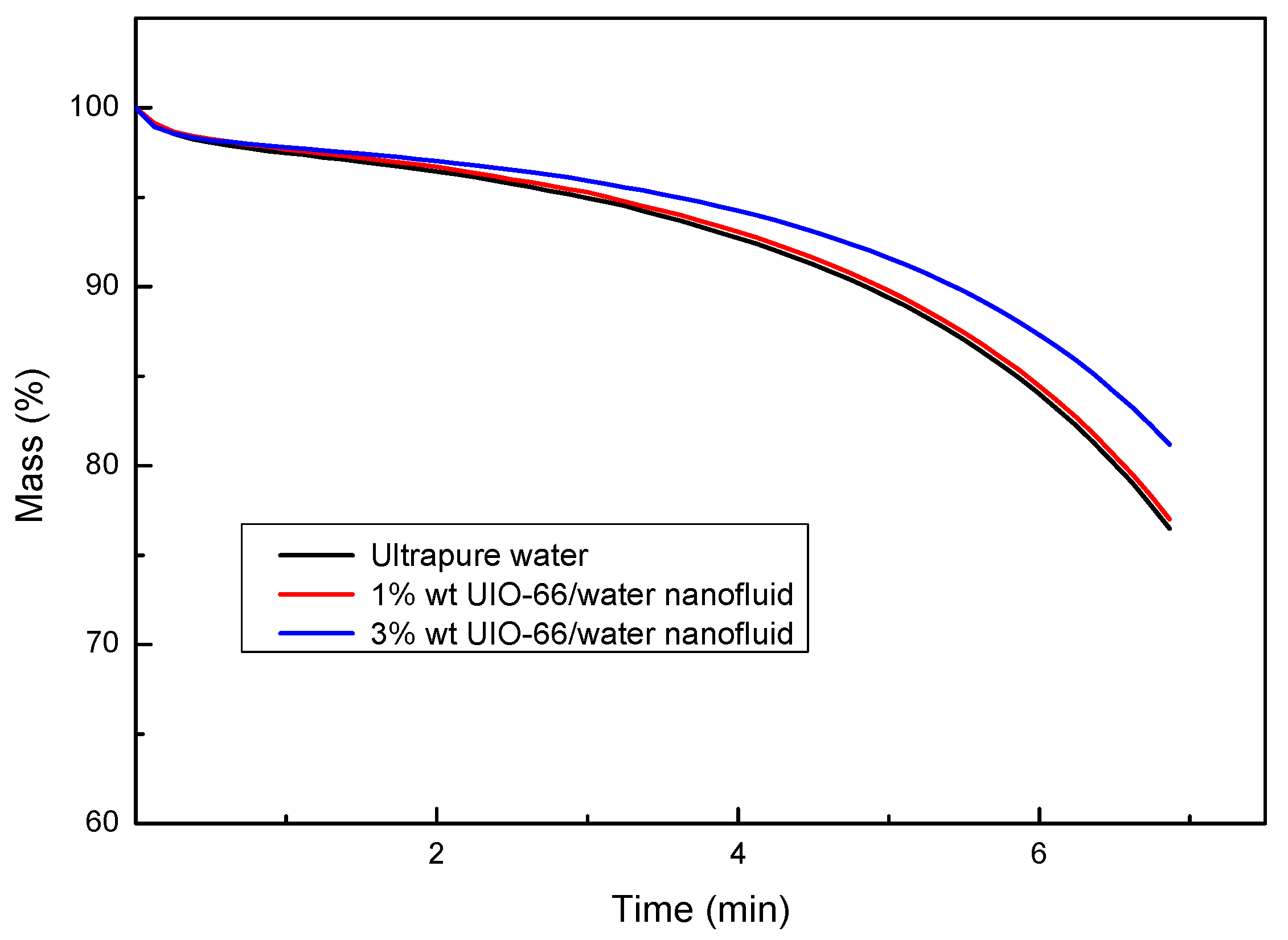
3.2. Thermal Energy Storage Properties of the H2O/UIO-66 Nanofluids
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morales-Rodriguez, R. Thermodynamics-Fundamentals and Its Application in Science; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Zhang, C.; Liu, C.; Xu, X.; Li, Q.; Wang, S.; Chen, X. Effects of superheat and internal heat exchanger on thermo-economic performance of organic Rankine cycle based on fluid type and heat sources. Energy 2018, 159, 482–495. [Google Scholar] [CrossRef]
- Liu, X.; Ye, Z.; Bai, L.; He, M. Performance comparison of two absorption-compression hybrid refrigeration systems using R1234yf/ionic liquid as working pair. Energy Convers. Manag. 2019, 181, 319–330. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, C.; Yang, F.; Su, C.; He, M. Experimental and correlational study of isobaric molar heat capacities of fatty acid esters: Ethyl nonanoate and ethyl dodecanoate. Fluid Phase Equilibria 2019, 479, 47–51. [Google Scholar] [CrossRef]
- Eastman, J.A.; Choi, S.U.S.; Li, S.; Yu, W.; Thompson, L.J. Anomalously increased effective thermal conductivities of ethylene glycol-based nanofluids containing copper nanoparticles. Appl. Phys. Lett. 2001, 78, 718–720. [Google Scholar] [CrossRef]
- Keblinski, P.; Phillpot, S.R.; Choi, S.U.S.; Eastman, J.A. Mechanisms of heat flow in suspensions of nano-sized particles (nanofluids). Int. J. Heat Mass Transf. 2002, 45, 855–863. [Google Scholar] [CrossRef]
- Ho, C.J.; Gao, J.Y. Preparation and thermophysical properties of nanoparticle-in-paraffin emulsion as phase change material. Int. Commun. Heat Mass Transf. 2009, 36, 467–470. [Google Scholar] [CrossRef]
- Yang, X.; Li, S.; Huang, H.; Li, J.; Kobayashi, N.; Kubota, M. Effect of carbon nanoadditives on lithium hydroxide monohydrate-based composite materials for low temperature chemical heat storage. Energies 2017, 10, 664. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, S.; Zhang, Z.; Yurchenko, D. High-performance piezoelectric wind energy harvester with Y-shaped attachments. Energy Convers. Manag. 2019, 181, 645–652. [Google Scholar] [CrossRef]
- Sohail, A.; Fatima, M.; Ellahi, R.; Akram, K.B. A video graphic assessment of ferrofluid during magnetic drug targeting: An application of artificial intelligence in nanomedicine. J. Mol. Liq. 2019, 285, 47–57. [Google Scholar] [CrossRef]
- Rashidi, S.; Akar, S.; Bovand, M.; Ellahi, R. Volume of fluid model to simulate the nanofluid flow and entropygeneration in a single slope solar still. Renew. Energy 2018, 115, 400–410. [Google Scholar] [CrossRef]
- Yang, X.; Xiong, T.; Dong, J.L.; Li, W.X.; Wang, Y. Investigation of the dynamic melting process in athermal energy storage unit using a helical coil heat exchanger. Energies 2017, 10, 1129. [Google Scholar] [CrossRef]
- Hussian, M.I.; Kim, J.; Kim, J. Nanofluid-powered dual-fluid photovoltaic/thermal (PV/T) system: Comparative numerical study. Energies 2019, 12, 775. [Google Scholar] [CrossRef]
- Cho, S.; Kim, J.; Kim, J. Optimal operation parameter estimation of energy storage for frequency regulation. Energies 2019, 12, 1782. [Google Scholar] [CrossRef]
- Zeeshan, A.; Shehzad, N.; Abbas, T.; Ellahi, R. Effects of radiative electro-magnetohydrodynamics diminishing internal energy of pressure-driven flowof titanium dioxide-water nanofluid due toentropy generation. Entropy 2019, 21, 236. [Google Scholar] [CrossRef]
- Chen, X.; Xu, B.; Liu, L. Nanoscale fluid mechanics and energy conversion. Appl. Mech. Rev. 2014, 66, 050803. [Google Scholar] [CrossRef]
- Xu, B.; Qiao, Y.; Chen, X. Mitigating impact/blast energy via a novel nanofluidic energy capture mechanism. J. Mech. Phys. Solids 2014, 62, 194–208. [Google Scholar] [CrossRef]
- Xu, B.; Chen, X. Liquid flow-induced energy harvesting in carbon nanotubes: A molecular dynamics study. Phys. Chem. Chem. Phys. 2013, 15, 1164–1168. [Google Scholar] [CrossRef]
- Xu, B.; Liu, L.; Lim, H.; Qiao, Y.; Chen, X. Harvesting energy from low-grade heat based on nanofluids. Nano Energy 2012, 1, 805–811. [Google Scholar] [CrossRef]
- McGrail, B.P.; Thallapally, P.K.; Blanchard, J.; Nune, S.K.; Jenks, J.J.; Dang, L.X. Metal-organic heat carrier nanofluids. Nano Energy 2013, 2, 845–855. [Google Scholar] [CrossRef]
- James, S.L. Metal-organic frameworks. Chem. Soc. Rev. 2003, 32, 276–288. [Google Scholar] [CrossRef]
- Murray, L.J.; Dincă, M.; Long, J.R. Hydrogen storage in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1294–1314. [Google Scholar] [CrossRef] [PubMed]
- Li, J.R.; Sculley, J.; Zhou, H.C. Metal-organic frameworks for separations. Chem. Rev. 2012, 112, 869–932. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Thallapally, P.K.; McGrail, B.P.; Brown, D.R.; Liu, J. Progress in adsorption-based CO2 capture by metal-organic frameworks. Chem. Soc. Rev. 2012, 41, 2308–2322. [Google Scholar] [CrossRef] [PubMed]
- Henninger, S.K.; Habib, H.A.; Janiak, C. MOFs as adsorbents for low temperature heating and cooling applications. J. Am. Chem. Soc. 2009, 131, 2776–2777. [Google Scholar] [CrossRef] [PubMed]
- Rezk, A.; Al-Dadah, R.; Mahmoud, S.; Elsayed, A. Characterisation of metal organic frameworks for adsorption cooling. Int. J. Heat Mass Transf. 2012, 55, 7366–7374. [Google Scholar] [CrossRef]
- Elsayed, A.; Elsayed, E.; Al-Dadah, R.; Mahmoud, S.; Elshaer, A.; Kaialy, W. Thermal energy storage using metal–organic framework materials. Appl. Energy 2017, 186, 509–519. [Google Scholar] [CrossRef]
- Zheng, J.; Vemuri, R.S.; Estevez, L.; Koech, P.K.; Varga, T.; Camaioni, D.M.; Blake, T.A.; McGrail, B.P.; Motkuri, R.K. Pore-engineered metal–organic frameworks with excellent adsorption of water and fluorocarbon refrigerant for cooling applications. J. Am. Chem. Soc. 2017, 139, 10601–10604. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: New York, NY, USA, 2002. [Google Scholar]
- Zhang, N.; Huo, J.; Yang, B.; Ruan, X.; Zhang, X.; Bao, J.; Qi, W.; He, G. Understanding of imidazolium group hydration and polymer structure for hydroxide anion conduction in hydrated imidazolium-g-PPO membrane by molecular dynamics simulations. Chem. Eng. Sci. 2018, 192, 1167–1176. [Google Scholar] [CrossRef]
- Sun, X.; Wick, C.D.; Thallapally, P.K.; McGrail, B.P.; Dang, L.X. Computational study of hydrocarbon adsorption in metal−organic framework Ni2(dhtp). J. Phys. Chem. B 2011, 115, 2842–2849. [Google Scholar] [CrossRef]
- Annapureddy, H.V.R.; Motkuri, R.K.; Nguyen, P.T.M.; Truong, T.B.; Thallapally, P.K.; McGrail, B.P.; Dang, L.X. Computational studies of adsorption in metal organic frameworks and interaction of nanoparticles in condensed phases. Mol. Simul. 2014, 40, 571–584. [Google Scholar] [CrossRef]
- Cmarik, G.E.; Kim, M.; Cohen, S.M.; Walton, K.S. Tuning the adsorption properties of UiO-66 via ligand functionalization. Langmuir 2012, 28, 15606–15613. [Google Scholar] [CrossRef] [PubMed]
- Chaemchuen, S.; Xiao, X.; Klomkliang, N.; Yusubov, M.S.; Verpoort, F. Tunable Metal—Organic Frameworks for Heat Transformation Applications. Nanomaterials 2018, 8, 661. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.J.; Brown, Z.J.; Colon, Y.J.; Siu, P.W.; Scheidt, K.A.; Snurr, R.Q.; Hupp, J.T.; Farha, O.K. A facile synthesis of UiO-66, UiO-67 and their derivatives. Chem. Commun. 2013, 49, 9449–9451. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhao, Y.; Lv, Z.; Song, F.; Zhong, Q. Preparation and enhanced CO2 adsorption capacity of UiO-66/graphene oxide composites. J. Ind. Eng. Chem. 2015, 27, 102–107. [Google Scholar] [CrossRef]
- Yang, J.J.; Ding, Y.D.; Liao, Q.; Zhu, X.; Liu, Q.W. Influence of processing conditions on compressive strength and CO2 adsorption of UiO-66 pellets, IHTC16-22457. In Proceedings of the International Heat Transfer Conference, Beijing, China, 10–15 August 2018. [Google Scholar]
- NIST. Available online: http://webbook.nist.gov/chemistry/fluid/ (accessed on 1 June 2018).
- Accelrys, I. Materials Studio; Accelrys Software Inc.: San Diego, CA, USA, 2010. [Google Scholar]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Hu, J.; Liu, C.; Li, Q.; Shi, X. Molecular simulation of thermal energy storage of mixed CO2/IRMOF-1 nanoparticle nanofluid. Int. J. Heat Mass Transf. 2018, 125, 1345–1348. [Google Scholar] [CrossRef]
- Jiang, C.; Song, L.; Zhang, J.; Sun, L.; Xu, F.; Li, F.; Jiao, Q.; Sun, Z.; Xing, Y.; Du, Y.; et al. Thermodynamic properties and heat capacities of Co (BTC)1/3 (DMF) (HCOO). J. Therm. Anal. Calorim. 2010, 102, 1087–1093. [Google Scholar] [CrossRef]
- Lv, X.; Tan, Z.; Gao, X.; Zhang, Z.; Yang, L.; Zhao, J.; Sun, L.; Zhang, T. Synthesis and thermodynamic properties of a metal-organic framework: [LaCu6(μ-OH)3(Gly)6im6](ClO4)6. Thermochimica Acta 2006, 450, 102–104. [Google Scholar] [CrossRef]
- Mu, B.; Walton, K.S. Thermal Analysis and Heat Capacity Study of Metal–Organic Frameworks. J. Phys. Chem. C 2011, 115, 22748–22754. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Li, Q.; Wang, Q. Energy Storage Analysis of UIO-66 and Water Mixed Nanofluids: An Experimental and Theoretical Study. Energies 2019, 12, 2521. https://doi.org/10.3390/en12132521
Zhou Y, Li Q, Wang Q. Energy Storage Analysis of UIO-66 and Water Mixed Nanofluids: An Experimental and Theoretical Study. Energies. 2019; 12(13):2521. https://doi.org/10.3390/en12132521
Chicago/Turabian StyleZhou, Yingjie, Qibin Li, and Qiang Wang. 2019. "Energy Storage Analysis of UIO-66 and Water Mixed Nanofluids: An Experimental and Theoretical Study" Energies 12, no. 13: 2521. https://doi.org/10.3390/en12132521
APA StyleZhou, Y., Li, Q., & Wang, Q. (2019). Energy Storage Analysis of UIO-66 and Water Mixed Nanofluids: An Experimental and Theoretical Study. Energies, 12(13), 2521. https://doi.org/10.3390/en12132521