1. Introduction
A 3–5 km deep borehole into granite is expected to encounter high salinity brines exceeding 200,000 mg/L total dissolved solids (TDS) such as those found in granites underlying the Canadian Shield, Fennoscandia, and elsewhere [
1,
2,
3]. High salinities aid in the safe disposal of deep boreholes because (1) they indicate isolation from fresh recharge, and (2) their greater density should prevent the subsequent circulation of fluids back to the surface. There are several explanations for why the brines are so concentrated, including:
Evaporative concentration and/or freezing of seawater followed by recharge [
4];
Fluid–rock interaction [
1] incorporation of water into hydrous phases;
Leakage of salt-rich fluid inclusions [
5];
Radiolytic dehydration of subsurface waters [
6].
Our purpose here is to test the second hypothesis and determine whether the fluid–rock interaction can dehydrate subsurface waters and produce brines similar in composition to those observed at depth. Deep brines tend to have a high chloride concentration, an elevated Ca/Na ratio, a diminished Mg, and a low Cl/Br ratio relative to seawater [
1,
3]. Bucher and Stober [
3] pointed to the potential ability and importance of the albite dissolution of zeolites to dehydrate passively and concentrate deep basement brines, but their primary focus was on granite alteration by fresh recharge waters collected from the Gotthard Rail Base Tunnel. We explore these reactions over longer time spans and at higher temperatures of deep granites to constrain the potential dehydration (brine formation) caused by silicate hydrolysis and the associated volume change of the rock itself. Silicate hydrolysis is modeled using reaction path calculations [
7,
8,
9]. Conceptually, the reaction path calculation titrates an assumed assemblage of minerals into an initial water volume and allows the resulting fluid to precipitate new minerals as they become thermodynamically saturated. The chemistry of the reacting fluid is tracked, as are the identity, mass, and volume of the newly formed minerals. In our case, the starting and ending mineral assemblages are constrained by geologic observations. The chemistry of the reacting fluid and absolute masses of newly formed minerals are key outputs.
Because the reaction path calculations do not include Cl- or Br-bearing salts, the calculated Cl/Br ratio does not change with reaction, though Cl- or Br-concentrations would increase indirectly as water is incorporated into hydrous alteration phases. The dissolution of existing salts at fracture surfaces would also affect Cl- or Br-concentrations (see
Section 5 below).
2. Materials and Methods
The overall reaction to be modeled is granite + H
2O → hydrous phases + brine. Chlorite and epidote are the most common hydrous silicates in deep granites; less common are mica, zeolite, laumontite, and prehnite [
10]. The ideal chemical formulae for each of these hydrous alteration products are listed in
Table 1 in order of decreasing hydration.
The volume of a typical granite consists of 20% quartz, 40% K-feldspar, 15% plagioclase (albite), 9% muscovite, 8% biotite, and 8% hornblende (see
Table 2). Using tabulated mineral molar volumes [
12] to convert a 10 kg (3.8 L) block gives a mixture of 33.3 moles of quartz, 14.4 moles of K-feldspar, 5.7 moles of albite, 2.2 moles of muscovite, 1.8 moles of biotite, and 0.9 moles of hornblende, which is “reacted” with 0.1 liter of seawater at 100 °C. This is a 38:1 rock–fluid volume ratio and is equivalent to a rock with a fluid-filled porosity of ~3%.
Reaction path calculations are done with the PHREEQC program [
9] using its llnl.dat (thermo.com.V8.R6.230) thermodynamic database. Biotite and hornblende end-members of phlogopite (KMg
3AlSi
3O
10(OH)
2) and pargasite (NaCa
2(Mg
4Al)(Si
6Al
2)O
22(OH)
2) are used in the calculation. Calcite, gypsum, and kaolinite are allowed to form if saturated. Seawater and groundwater were used in the calculations as generic reacting fluids.
The calculation was initially set up to allow the dissolution of all granite phases to reach equilibrium. In other words, we assumed sufficient time and access to water for the dissolution and growth of mineral surfaces. This is a reasonable assumption given the multi-million-year contact times of most brines with deep granites. Biotite was allowed to dissolve, but not to precipitate, which results in chlorite precipitation. The chlorite composition is set to the end-member Clinochlore 14Å, Mg5Al2Si3O10(OH)8 in the llnl.dat database. This was done in the PHREEQC input formalism by the sequential reaction of biotite and hornblende with the fluid and the equilibrium phase assemblage described above. The calculations were initially done using Mg end-member biotite and chlorite. Subsequent calculations considered Fe-rich end-member chlorite (daphnite) and biotite (annite) in the granite hydrolysis reaction. For our initial analysis, the anorthite component of plagioclase was set to zero, largely because of its small amount in most granites. Subsequent calculations considered the impact of anorthite on granite alteration. At the end of each simulated granite hydrolysis reaction, the molar amounts of primary dissolved and newly grown minerals from the PHREEQC output file make up the overall granite hydrolysis reaction.
3. Results
At 100 °C, the overall granite hydrolysis reaction with seawater is:
which produces a residual Ca–Na–Cl brine with a pH ~6.7. Minor amounts (<0.02 moles) of epidote, calcite, and gypsum form as well. Calculations indicate that the albite and K-feldspar masses increase substantially. Almost all of the original quartz is dissolved. There is a net loss of 4.9 moles of liquid H
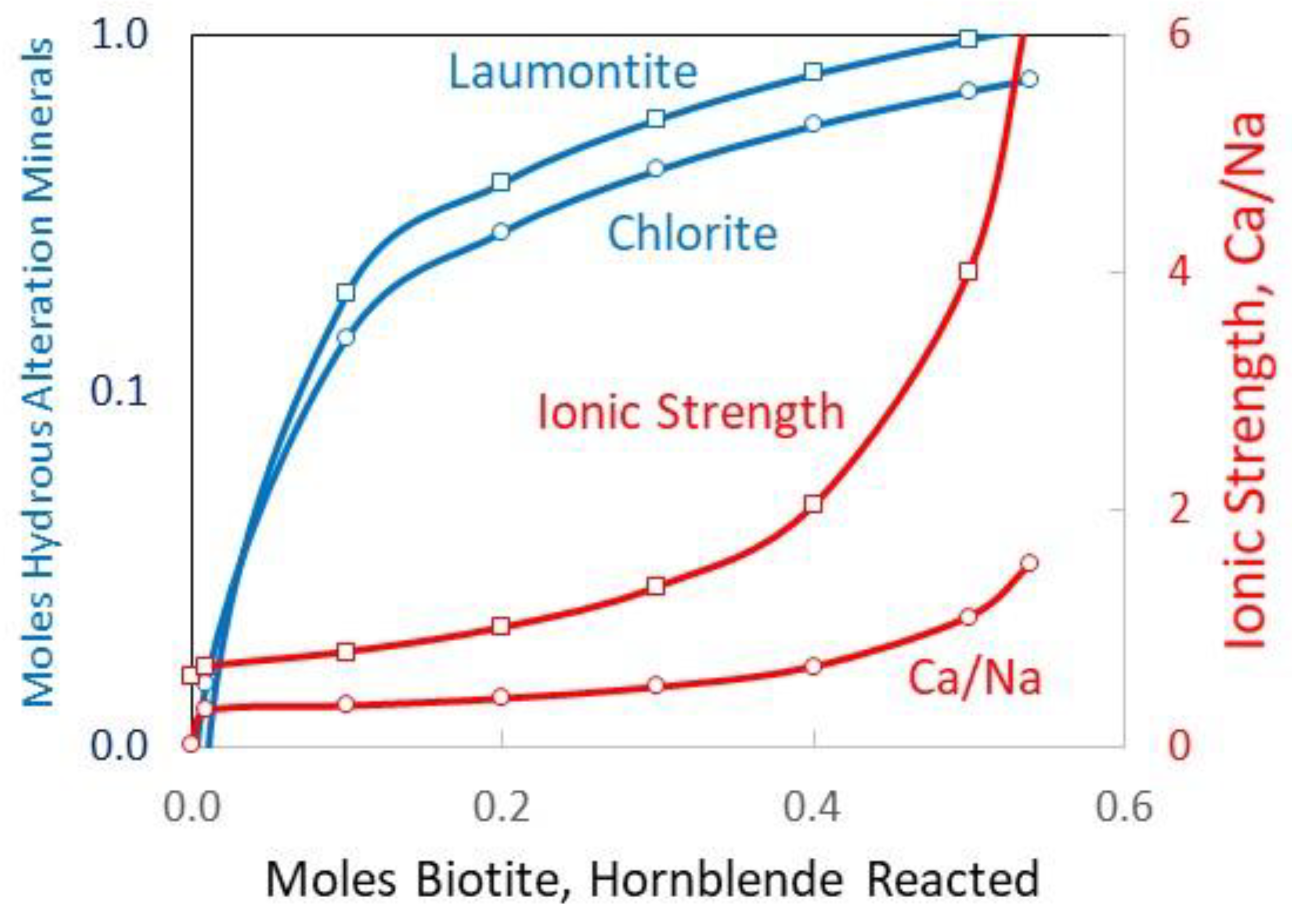
2O that causes the ionic strength of the solution to increase from an initial starting ionic strength of 0.5 M to >6 M (
Figure 1). Typically, PHREEQC runs stopped or crashed at high ionic strengths. Stoichiometrically, there is an addition of 0.54 moles of H
2O from hornblende dissolution, 0.54 moles from biotite dissolution, and 1.29 moles from muscovite dissolution, followed by a free water loss of 7.23 moles by incorporating H
2O into laumontite and chlorite, hence a net loss of 4.86 moles of free water from granite hydrolysis. This means that the initial 0.1 L of water (5.55 mol) is reduced to 0.012 L, which results in a much higher level of salinity. Note that the higher ionic strength absolute values are uncertain due to the simplified activity coefficient model used here.
By comparison, end-member Canadian Shield brines from Fritz and Frape [
2] with the highest salt contents of ~240 to 325 g/L have ionic strengths of 4.5 to 6.2. The estimated fluid Ca/Na for granite hydrolysis is 1.5, which is within the range of Ca/Na, 0.7 < Ca/Na < 3 measured by Fritz and Frape [
1]. Our calculations predict relatively low Mg concentrations of 1 to 3 mmol/L, which fall within the wide observed range of Canadian Shield Mg values, 0.5 to 210 mmol/L, measured by Frape and Fritz [
2]. Canadian Shield brines at ~25 °C have a measured Si content of 0.171 to 0.342 mmol/L [
2]. Our calculated 100 °C Si content is substantially higher at 0.909 mmol/L than the Canadian Shield values, which is probably due to our higher temperature. Running the calculation at the lower temperature of Canadian Shield brines results in a Si value of 0.126 mmol/L, similar to the amount measured by Fritz and Frape [
1].
The pH of the fluids remains between 6.6 and 7.1 at 100 °C. Calcium increases in the solution because the calcium supply from biotite dissolution exceeds the amount of calcium taken during the laumontite formation. The brine’s pH level is likely controlled by the K-feldspar/kaolinite equilibria [
13,
14]. Albite/K-feldspar probably sets the Na
+/K
+ ratio [
15]. Increasing the reaction temperature from 100 to 150 °C decreases the calculated Ca and Mg levels but increases the fluid K levels. The ionic strength remains roughly the same as that at 100 °C.
4. Discussion
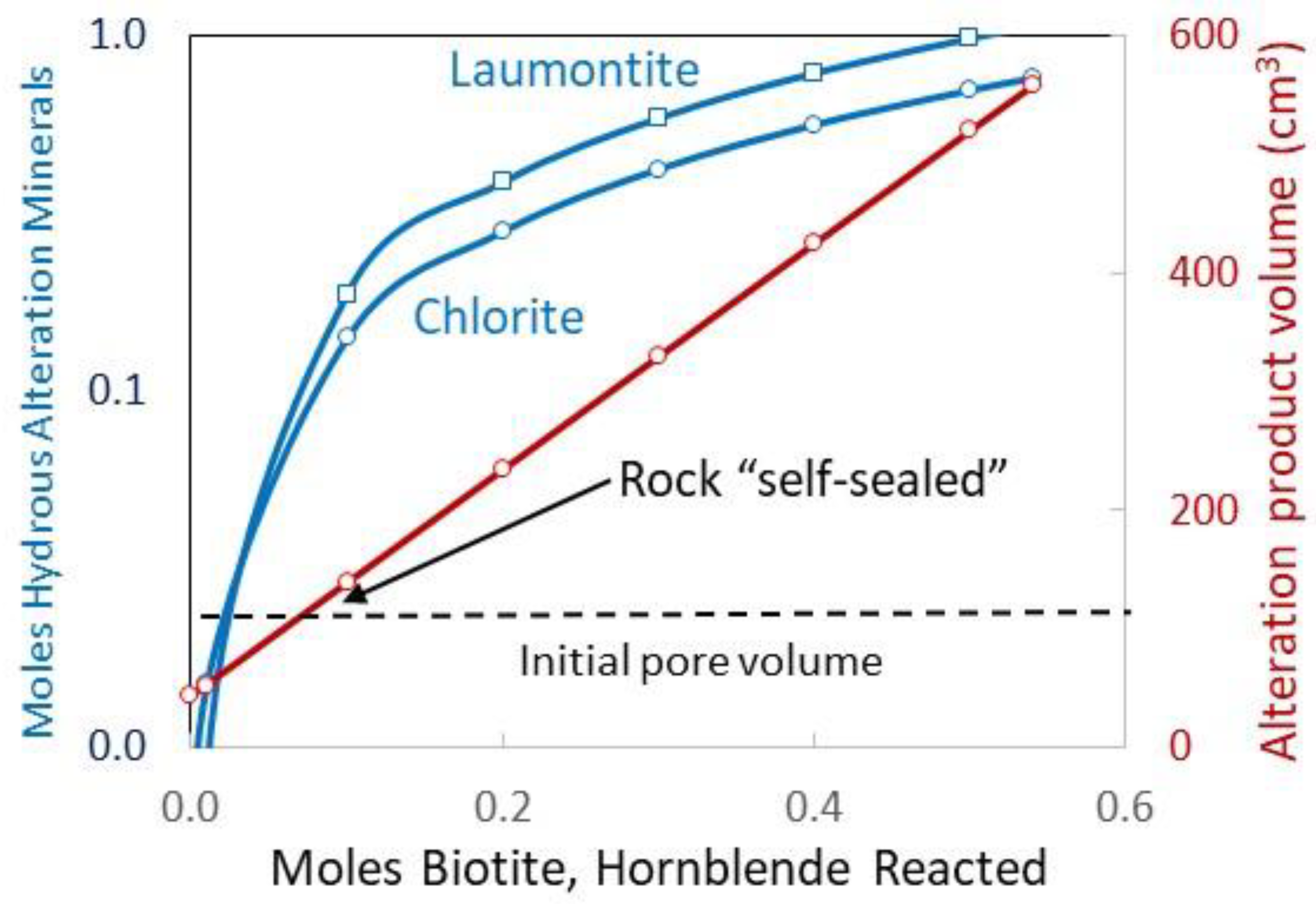
Figure 2 suggests that altering the product accumulation will ultimately limit granite hydrolysis. Recall that the calculation begins with 100 ml of seawater. The change in net rock volume, which is the volume of minerals grown minus the volume of minerals dissolved, exceeds the starting water volume before the 0.1 moles of biotite + hornblende reaction has occurred. Because of this self-sealing characteristic, the fluid–rock reaction is expected to be locally distributed. In flowing wide aperture fractures, where fluids have continual access to granite surfaces, hydrolysis proceeds to the high salinity end-member fluids. Dead-end fractures see less reaction before sealing.
A much more diluted groundwater (taken from
Table 3 of [
16]) was also tested as a starting fluid to determine whether the starting water composition has a significant effect on the hydrolysis. At 100 °C, the net granite hydrolysis reaction into diluted groundwater is:
This produces a brine with a pH of ~6.7, similar to the reaction calculated using seawater. The same hydrous alteration minerals and ionic strengths were observed in the calculation, suggesting that the granite hydrolysis reaction is relatively unaffected by the starting water composition.
Including Fe-rich end-members for chlorite and biotite resulted in a granite hydrolysis reaction close to the Mg-rich end-member reaction calculated initially. The effect of plagioclase calcium on granite hydrolysis was also examined. Plagioclase in granites is typically low in Ca, An0–An10 [
3], but is potentially important because the anorthite component of plagioclase dissolves non-stoichiometrically [
17] and far faster than the albitic end-member. In near-surface environments, anorthite appears to dissolve at least 5–10 times faster than the albite component of plagioclase [
18,
19,
20]. The non-stoichiometry is observed in both short-term lab experiments (<2000 hours) and long-term watershed studies and is proposed to occur in brines in contact with deep granites [
21]. The accelerated leaching of Ca likely comes from the exsolution of intergrowths, dislocations, and/or Ca-rich zones. Preliminary granite hydrolysis calculations indicated that anorthite never dissolved to saturation (i.e., remained far from equilibrium), unlike albite. When 0.5 moles of anorthite was included in the groundwater–granite hydrolysis calculation, the overall reaction was:
The overall reactions for An-included and An-free granite are similar.
5. Conclusions
Brine formation appears to be the inevitable result of a prolonged contact between groundwater and deep granites. Yet, because of the alteration—the increase in product volume—the reaction between the two should be locally self-limiting. The picture of granite hydrolysis presented here might be improved with a Pitzer approach and solid solution models for the major phases. The calculation might also be improved by accounting for salt addition to brines from fracture surfaces [
3], fluid inclusions (e.g., [
5,
22]), and chloride exchange for hydroxyls in the secondary chlorite and zeolite [
23].
Author Contributions
Conceptualization, D.S.; formal analysis, P.V.B. and C.L.; writing—P.V.B. and D.S.
Funding
This research was funded by Sandia National Laboratories.
Acknowledgments
We greatly appreciate the technical advice of Carlos Jové-Colon (SNL) and Kristopher Kuhlman (SNL). Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA-0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Frape, S.; Fritz, P.; McNutt, R. Water-rock interaction and chemistry of groundwaters from the Canadian Shield. Geochim. Cosmochim. Acta 1984, 48, 1617–1627. [Google Scholar] [CrossRef]
- Frape, S.; Fritz, P. Geochemical Trends for Groundwaters from the Canadian Shield. In Saline Water and Gases in Crystalline Rocks; Geological Association of Canada: St. John’s, NL, Canda, 1987; pp. 19–38. [Google Scholar]
- Bucher, K.; Stober, I. Fluids in the upper continental crust. Geofluids 2010, 10, 241–253. [Google Scholar]
- Bottomley, D.J.; Gregoire, D.C.; Raven, G.K. Saline ground waters and brines in the Canadian Shield: Geochemical and isotopic evidence for a residual evaporite brine component. Geochim. Cosmochim. Acta 1994, 58, 1483–1498. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; Olsson, T. Fluid inclusions as a source of dissolved salts in deep granitic groundwaters, in Saline water and gases in crystalline rocks. Geol. Assn. Can. Spec. Pap. 1987, 33, 111–119. [Google Scholar]
- Vovk, I. Radiolytic salt enrichment and brines in the crystalline basement of the East European Platform, in Saline water and gases in crystalline rocks. Geol. Assn. Can. Spec. Pap. 1987, 33, 197–210. [Google Scholar]
- Helgeson, H.C.; Brown, T.H.; Nigrini, A.; Jones, T.A. Calculation of mass transfer in geochemical processes involving aqueous solutions. Geochim. Cosmochim. Acta 1970, 34, 569–592. [Google Scholar] [CrossRef]
- Wolery, T.J. A Computer Program for Geochemical Aqueous Speciation-Solubility Calculations: User’s Guide and Documentation; UCRL-53414; Lawence Livermore National Labratory: Livermore, CA, USA, 1983.
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Water-Resources Investigations Report 99-4259; US Geological Survey: Reston, VA, USA, 1999.
- Juhlin, C.; Wallroth, T.; Smellie, J.; Leijon, B.; Eliasson, T.; Ljunggren, C.; Beswick, J. The Very Deep Hole Concept-Geoscientific Appraisal of Conditions at Great Depth; Swedish Nuclear Fuel and Waste Management Co.: Solna, Sweden, 1998. [Google Scholar]
- Johnson, J.; Anderson Gand Parkhurst, D. Thermo. Com. V8. R6, 230; Lawence Livermore National Labratory: Livermore, CA, USA, 2000.
- Robie, R.A.; Hemingway, B.S.; Fisher, R.J. Thermodynamic Properties of Minerals and Related Substances at 298.15K and 1 Bar (105 Pascals) Pressure and at Higher Temperatures; US Government Printing Office: Washington, DC, USA, 1978.
- Baccar, M.B.; Fritz, B. Geochemical modeling of sandstone diagenesis and its consequence on the evolution of porosity. Appl. Geochem. 1993, 8, 285–298. [Google Scholar] [CrossRef]
- Ehrenberg, S.N.; Aagaard, P.; Wilson, M.J.; Fraser, A.R.; Duthie, D.M.L. Depth-dependent transformation of Kaoline to Dickite in sandstones of the Norwegian Continental Shelf. Clay Miner. 1993, 28, 325–352. [Google Scholar] [CrossRef]
- Pearson, F., Jr. Models of Mineral Controls on the Composition of Saline Groundwaters of the Canadian Shield. Saline Water and Gases in Crystalline Rocks; Fritz, P., Frape, S.K., Eds.; Geological Association of Canada Special Paper: St. John’s, NL, Canda, 1987; Volume 33, pp. 39–51. [Google Scholar]
- Plummer, L.N.; Bexfield, L.M.; Anderholm, S.K.; Sanford, W.E.; Busenberg, E. Geochemical Characterization of Ground Water Flow in the Santa Fe Group Aquifer System, Middle Rio Grande Basin, New Mexico. Water-Res. Investig. Rep. 2004, 3, 4131. [Google Scholar]
- Gardner, L.R. Mechanics and kinetics of incongruent feldspar dissolution. Geology 1983, 11, 418–421. [Google Scholar] [CrossRef]
- Clayton, J.L. Some observations on the stoichiometry of feldspar hydrolysis in granitic soil. J. Environ. Qual. 1988, 17, 153–157. [Google Scholar] [CrossRef]
- Mast, M.A.; Drever, J.I. The effect of oxalate on the dissolution rates of oligoclase and tremolite. Geochim. Cosmochim. Acta 1987, 51, 2559–2568. [Google Scholar] [CrossRef]
- Oxburgh, R.; Drever, J.L.; Sun, Y.-T. Mechanism of plagioclase dissolution in acid solution at 25 °C. Geochim. Cosmochim. Acta 1994, 58, 661–669. [Google Scholar] [CrossRef]
- Edmunds, W.; Kay, R.; Miles, D.; Cook, J. The origin of saline groundwaters in the Carnmenellis granite, Cornwall (UK): Further evidence from minor and trace elements. Saline Water Gases Cryst. Rocks 1987, 33, 127–143. [Google Scholar]
- Nordstrom, D.K.; Lindblom, S.; Donahoe RJand Barton, C.C. Fluid inclusions in the Stripa granite and their possible influence on the groundwater chemistry. Geochim. Cosmochim. Acta 1989, 53, 1741–1755. [Google Scholar] [CrossRef]
- Bucher, K.; Stober, I. Water-rock reaction experiments with Black Forest gneiss and granite. In Water-Rock Interaction; Springer: Berlin, Germany, 2002; pp. 61–95. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).


{kind=link}
{kind=link}