
The Defensive Interactions of Prominent Infectious Protozoan Parasites: The Host’s Complement System

 , , and
, , and
Abstract
1. Introduction
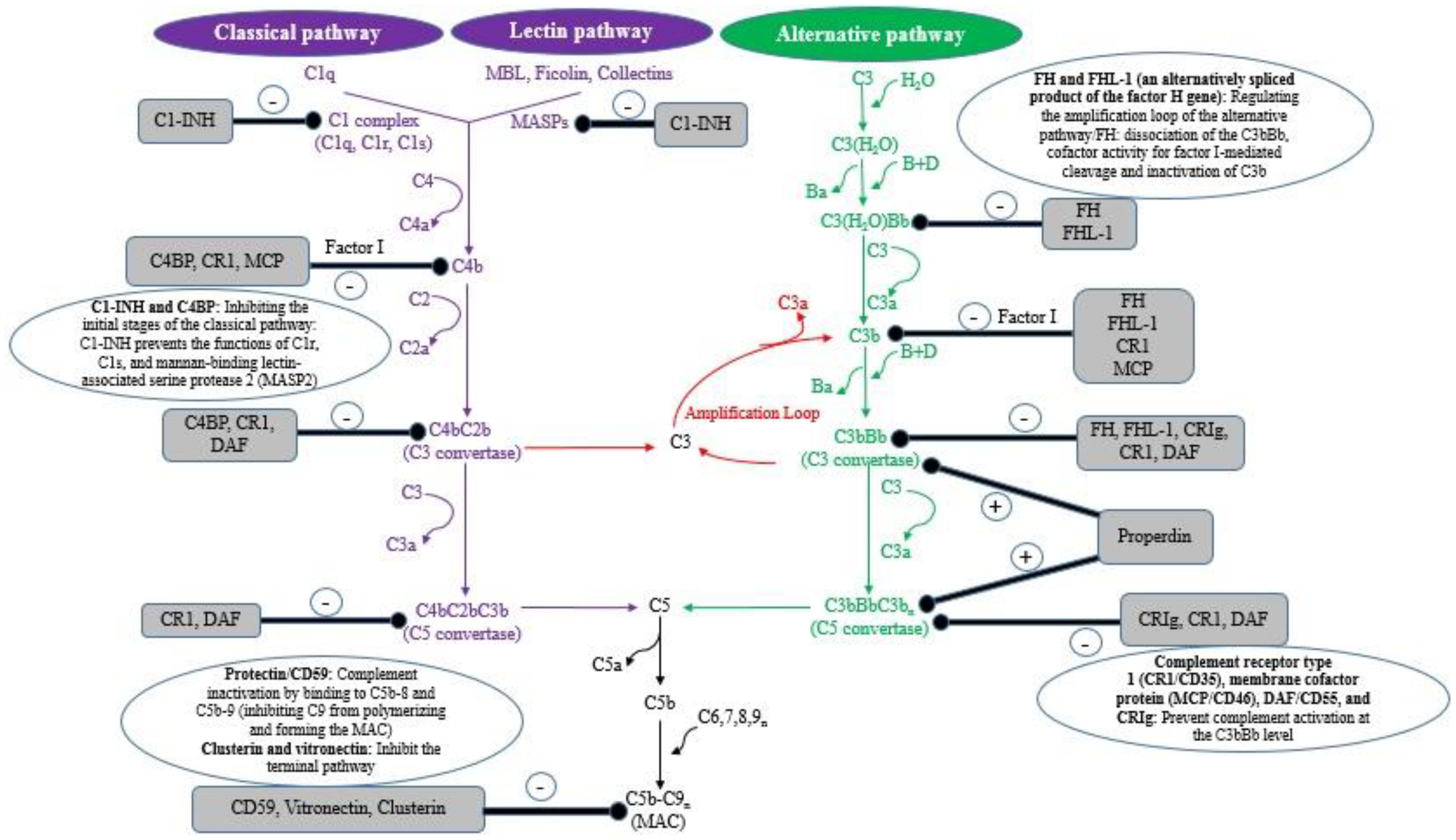
2. Parasite’s Complement Regulatory and Evasion Molecules
2.1. Plasmodium spp.
2.2. Toxoplasma spp.

{kind=link}
{kind=link}
| Interaction Mechanisms with the Tissue (Brain) | References |
|---|---|
| Plasmodium spp. | |
| Upregulation of C1q and C5 in CM patients | [65,66] |
| C5a up-regulation (C5aR deficiency: increasing productivity against CM) C5aR probably mediates persistent neurocognitive deficits | [67,68] |
| Inducing innate immune responses (including the complement system) and associated demyelination (severity of CM) | [65] |
| C5 deficiency and C5aR blockade were protective against CM (protection of C5 deficient mice against CM is mediated via the inhibition of MAC, not through C5a-induced inflammation) | [69,70] |
| C5 plays a role in malaria-induced seizures | [71] |
| T cell-deficiency is protective against CM (which was correlated by decreased complement activation) | [72] |
| Dysregulated C5aR signaling participates in the pathogenesis | [67] |
| C9 deposition throughout the cortex of cerebral malaria (CM progression) | [70] |
| Toxoplasma | |
| C3 and C4b upregulation (especially in the brain with high cyst burden), C5aR and C3aR upregulation in the cerebral cortex and glial cells | [53,63,73] |
| C3, C4, C1q and C1r upregulation in the brain with high cyst burden (complement deposition on the surface of degenerating neurons) | [73] |
| C1q upregulation (especially near parasite cysts and punctate synaptic patterns) | [53,74] |
| C3, C4b, and C1q upregulation and the probable induction of the disruption of tight junctions | [53] |
| Induction or upregulation of the alternative pathway components (FB and FP) and anaphylatoxin receptors (C3aR and C5aR) in the cerebral cortex and glial cells | [63] |
2.3. Trypanosoma cruzi
2.4. Trypanosoma brucei
2.5. Leishmania spp.
2.6. Growing Evidences Supporting Complement Modulation by Entamoeba, Giardia and Trichomonas spp.
3. Potential Targets Based on Protozoa-Host’s Complement Interactions to Manage Neglected Diseases
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kellermann, M.; Scharte, F.; Hensel, M. Manipulation of Host Cell Organelles by Intracellular Pathogens. Int. J. Mol. Sci. 2021, 22, 6484. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Nakada-Tsukui, K.; Besteiro, S. The Autophagy Machinery in Human-Parasitic Protists; Diverse Functions for Universally Conserved Proteins. Cells 2021, 10, 1258. [Google Scholar] [CrossRef] [PubMed]
- Bosurgi, L.; Rothlin, C.V. Management of cell death in parasitic infections. Semin. Immunopathol. 2021, 43, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, D.L.; Denton, S.L.; Fettel, K.D.; Sondgeroth, K.S.; Munoz Gutierrez, J.; Bangoura, B.; Dunay, I.R.; Gigley, J.P. Innate lymphoid cells in protection, pathology, and adaptive immunity during apicomplexan infection. Front. Immunol. 2019, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Chulanetra, M.; Chaicumpa, W. Revisiting the Mechanisms of Immune Evasion Employed by Human Parasites. Front. Cell. Infect. Microbiol. 2021, 11, 702125. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I–molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Kiyuka, P.K.; Meri, S.; Khattab, A. Complement in malaria: Immune evasion strategies and role in protective immunity. FEBS Lett. 2020, 594, 2502–2517. [Google Scholar] [CrossRef]
- Moore, S.R.; Menon, S.S.; Cortes, C.; Ferreira, V.P. Hijacking factor H for complement immune evasion. Front. Immunol. 2021, 12, 602277. [Google Scholar] [CrossRef]
- Jagatia, H.; Tsolaki, A.G. The Role of Complement System and the Immune Response to Tuberculosis Infection. Medicina 2021, 57, 84. [Google Scholar] [CrossRef]
- Bardhan, M.; Kaushik, R. Physiology, Complement Cascade. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Schmidt, C.Q.; Kennedy, A.T.; Tham, W.-H. More than just immune evasion: Hijacking complement by Plasmodium falciparum. Mol. Immunol. 2015, 67, 71–84. [Google Scholar] [CrossRef]
- Sikorski, P.M.; Commodaro, A.G.; Grigg, M.E. A protective and pathogenic role for complement during acute Toxoplasma gondii infection. Front. Cell. Infect. Microbiol. 2021, 11, 634610. [Google Scholar] [CrossRef] [PubMed]
- Leitner, W.W.; Haraway, M.; Pierson, T.; Bergmann-Leitner, E.S. Role of opsonophagocytosis in immune protection against malaria. Vaccines 2020, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Sun, X.; Chen, Y.; Zhan, B.; Zhu, X. Complement evasion: An effective strategy that parasites utilize to survive in the host. Front. Microbiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Ending the neglect to attain the Sustainable Development Goals: A road map for neglected tropical diseases. Front. Cell. Infect. Microbiol. 2021. [Google Scholar]
- Rathnayake, D.; Aitken, E.H.; Rogerson, S.J. Beyond Binding: The Outcomes of Antibody-Dependent Complement Activation in Human Malaria. Front. Immunol. 2021, 12, 683404. [Google Scholar] [CrossRef] [PubMed]
- Elmahallawy, E.K.; Alkhaldi, A.A.; Saleh, A.A. Host immune response against leishmaniasis and parasite persistence strategies: A review and assessment of recent research. Biomed. Pharmacother. 2021, 139, 111671. [Google Scholar] [CrossRef]
- Yasmin, H.; Adhikary, A.; Al-Ahdal, M.N.; Roy, S.; Kishore, U. Host–Pathogen Interaction in Leishmaniasis: Immune Response and Vaccination Strategies. Immuno 2022, 2, 218–254. [Google Scholar] [CrossRef]
- Okunlola, O.A.; Oyeyemi, O.T. Malaria transmission in Africa: Its relationship with yellow fever and measles. PLoS ONE 2022, 17, e0268080. [Google Scholar] [CrossRef]
- Al-Awadhi, M.; Ahmad, S.; Iqbal, J. Current status and the epidemiology of malaria in the Middle East Region and beyond. Microorganisms 2021, 9, 338. [Google Scholar] [CrossRef]
- Kurtovic, L.; Boyle, M.J.; Opi, D.H.; Kennedy, A.T.; Tham, W.H.; Reiling, L.; Chan, J.A.; Beeson, J.G. Complement in malaria immunity and vaccines. Immunol. Rev. 2020, 293, 38–56. [Google Scholar] [CrossRef]
- Dinko, B.; Pradel, G. Immune evasion by Plasmodium falciparum parasites: Converting a host protection mechanism for the parasite′ s benefit. Adv. Infect. Dis. 2016, 6, 82–95. [Google Scholar]
- Boyle, M.J.; Reiling, L.; Feng, G.; Langer, C.; Osier, F.H.; Aspeling-Jones, H.; Cheng, Y.S.; Stubbs, J.; Tetteh, K.K.; Conway, D.J. Human antibodies fix complement to inhibit Plasmodium falciparum invasion of erythrocytes and are associated with protection against malaria. Immunity 2015, 42, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.; Reiling, L.; Beeson, J. Evaluating Complement-Mediated Humoral Immunity to P. falciparum Blood Stages. EBioMedicine 2016, 14, 9–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Biryukov, S.; Angov, E.; Landmesser, M.E.; Spring, M.D.; Ockenhouse, C.F.; Stoute, J.A. Complement and antibody-mediated enhancement of red blood cell invasion and growth of malaria parasites. EBioMedicine 2016, 9, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Akhouri, R.R.; Goel, S.; Furusho, H.; Skoglund, U.; Wahlgren, M. Architecture of human IgM in complex with P. falciparum erythrocyte membrane protein 1. Cell Rep. 2016, 14, 723–736. [Google Scholar] [CrossRef]
- Kennedy, A.T.; Schmidt, C.Q.; Thompson, J.K.; Weiss, G.E.; Taechalertpaisarn, T.; Gilson, P.R.; Barlow, P.N.; Crabb, B.S.; Cowman, A.F.; Tham, W.-H. Recruitment of factor H as a novel complement evasion strategy for blood-stage Plasmodium falciparum infection. J. Immunol. 2016, 196, 1239–1248. [Google Scholar] [CrossRef]
- Simon, N.; Friedrich, O.; Kappes, B. Quantification of human complement factor H binding to asexual malaria blood stages by an enzyme-linked immunosorbent assay. Vaccine 2018, 36, 1545–1547. [Google Scholar] [CrossRef]
- Rosa, T.F.; Flammersfeld, A.; Ngwa, C.J.; Kiesow, M.; Fischer, R.; Zipfel, P.F.; Skerka, C.; Pradel, G. The Plasmodium falciparum blood stages acquire factor H family proteins to evade destruction by human complement. Cell. Microbiol. 2016, 18, 573–590. [Google Scholar] [CrossRef]
- Cserhalmi, M.; Papp, A.; Brandus, B.; Uzonyi, B.; Józsi, M. Regulation of regulators: Role of the complement factor H-related proteins. Semin. Immunol. 2019, 45, 101341. [Google Scholar] [CrossRef]
- Reiss, T.; Thiago, F.d.A.; Blaesius, K.; Bobbert, R.P.; Zipfel, P.F.; Skerka, C.; Pradel, G. Cutting edge: FHR-1 binding impairs factor H–mediated complement evasion by the malaria parasite Plasmodium falciparum. J. Immunol. 2018, 201, 3497–3502. [Google Scholar] [CrossRef]
- Kennedy, A.T.; Wijeyewickrema, L.C.; Huglo, A.; Lin, C.; Pike, R.; Cowman, A.F.; Tham, W.-H. Recruitment of human C1 esterase inhibitor controls complement activation on blood stage Plasmodium falciparum merozoites. J. Immunol. 2017, 198, 4728–4737. [Google Scholar] [CrossRef] [PubMed]
- Mejia, P.; Diez-Silva, M.; Kamena, F.; Lu, F.; Fernandes, S.M.; Seeberger, P.H.; Davis III, A.E.; Mitchell, J.R. Human C1-inhibitor suppresses malaria parasite invasion and cytoadhesion via binding to parasite glycosylphosphatidylinositol and host cell receptors. J. Infect. Dis. 2016, 213, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Oyong, D.A.; Kenangalem, E.; Poespoprodjo, J.R.; Beeson, J.G.; Anstey, N.M.; Price, R.N.; Boyle, M.J. Loss of complement regulatory proteins on uninfected erythrocytes in vivax and falciparum malaria anemia. JCI Insight 2018, 3, e124854. [Google Scholar] [CrossRef] [PubMed]
- Horta, M.F.; Andrade, L.O.; Martins-Duarte, É.S.; Castro-Gomes, T. Cell invasion by intracellular parasites–the many roads to infection. J. Cell Sci. 2020, 133, jcs232488. [Google Scholar] [CrossRef]
- Touray, M.G.; Seeley, D.; Miller, L.H. Plasmodium gallinaceum: Differential lysis of two developmental stages of malaria sporozoites by the alternative pathway of complement. Exp. Parasitol. 1994, 78, 294–301. [Google Scholar] [CrossRef]
- Wiesner, J.; Jomaa, H.; Wilhelm, M.; Tony, H.P.; Kremsner, P.G.; Horrocks, P.; Lanzer, M. Host cell factor CD59 restricts complement lysis of Plasmodium falciparum-infected erythrocytes. Eur. J. Immunol. 1997, 27, 2708–2713. [Google Scholar] [CrossRef]
- Larsen, M.D.; Quintana, M.d.P.; Ditlev, S.B.; Bayarri-Olmos, R.; Ofori, M.F.; Hviid, L.; Garred, P. Evasion of classical complement pathway activation on Plasmodium falciparum-infected erythrocytes opsonized by PfEMP1-specific IgG. Front. Immunol. 2019, 9, 3088. [Google Scholar] [CrossRef]
- Reiss, T.; Theis, H.I.; Gonzalez-Delgado, A.; Vega-Rodriguez, J.; Zipfel, P.F.; Skerka, C.; Pradel, G. Acquisition of human plasminogen facilitates complement evasion by the malaria parasite Plasmodium falciparum. Eur. J. Immunol. 2021, 51, 490–493. [Google Scholar] [CrossRef]
- Ayón-Núñez, D.A.; Fragoso, G.; Bobes, R.J.; Laclette, J.P. Plasminogen-binding proteins as an evasion mechanism of the host’s innate immunity in infectious diseases. Biosci. Rep. 2018, 38, BSR20180705. [Google Scholar] [CrossRef]
- Simon, N.; Lasonder, E.; Scheuermayer, M.; Kuehn, A.; Tews, S.; Fischer, R.; Zipfel, P.F.; Skerka, C.; Pradel, G. Malaria parasites co-opt human factor H to prevent complement-mediated lysis in the mosquito midgut. Cell Host Microbe 2013, 13, 29–41. [Google Scholar] [CrossRef]
- Sologub, L.; Kuehn, A.; Kern, S.; Przyborski, J.; Schillig, R.; Pradel, G. Malaria proteases mediate inside-out egress of gametocytes from red blood cells following parasite transmission to the mosquito. Cell. Microbiol. 2011, 13, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Khattab, A.; Barroso, M.; Miettinen, T.; Meri, S. Anopheles midgut epithelium evades human complement activity by capturing factor H from the blood meal. PLoS Negl. Trop. Dis. 2015, 9, e0003513. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cruz, A.; Garver, L.S.; Alabaster, A.; Bangiolo, L.; Haile, A.; Winikor, J.; Ortega, C.; van Schaijk, B.C.; Sauerwein, R.W.; Taylor-Salmon, E. The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science 2013, 340, 984–987. [Google Scholar] [CrossRef] [PubMed]
- Ramphul, U.N.; Garver, L.S.; Molina-Cruz, A.; Canepa, G.E.; Barillas-Mury, C. Plasmodium falciparum evades mosquito immunity by disrupting JNK-mediated apoptosis of invaded midgut cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cruz, A.; Canepa, G.E.; Kamath, N.; Pavlovic, N.V.; Mu, J.; Ramphul, U.N.; Ramirez, J.L.; Barillas-Mury, C. Plasmodium evasion of mosquito immunity and global malaria transmission: The lock-and-key theory. Proc. Natl. Acad. Sci. USA 2015, 112, 15178–15183. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zheng, H.; Chen, S.; Zhang, K.; Qin, X.; Zhang, J.; Fan, Y.; Wang, L.; Li, X.; Zhang, J. Malaria oocysts require circumsporozoite protein to evade mosquito immunity. Nat. Commun. 2022, 13, 3208. [Google Scholar] [CrossRef]
- Ukegbu, C.V.; Giorgalli, M.; Tapanelli, S.; Rona, L.D.; Jaye, A.; Wyer, C.; Angrisano, F.; Blagborough, A.M.; Christophides, G.K.; Vlachou, D. PIMMS43 is required for malaria parasite immune evasion and sporogonic development in the mosquito vector. Proc. Natl. Acad. Sci. USA 2020, 117, 7363–7373. [Google Scholar] [CrossRef] [PubMed]
- De Barros, R.A.M.; Torrecilhas, A.C.; Marciano, M.A.M.; Mazuz, M.L.; Pereira-Chioccola, V.L.; Fux, B. Toxoplasmosis in Human and Animals Around the World. Diagnosis and Perspectives in the One Health Approach. Acta Trop. 2022, 231, 106432. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, P.M.; Commodaro, A.G.; Grigg, M.E. Toxoplasma gondii recruits factor H and C4b-binding protein to mediate resistance to serum killing and promote parasite persistence in vivo. Front. Immunol. 2020, 10, 3105. [Google Scholar] [CrossRef] [PubMed]
- Shinjyo, N.; Kagaya, W.; Pekna, M. Interaction Between the Complement System and Infectious Agents—A Potential Mechanistic Link to Neurodegeneration and Dementia. Front. Cell. Neurosci. 2021, 15, 710390. [Google Scholar] [CrossRef]
- Olivera, G.C.; Ross, E.C.; Peuckert, C.; Barragan, A. Blood-brain barrier-restricted translocation of Toxoplasma gondii from cortical capillaries. Elife 2021, 10, e69182. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Wang, Y.P.; Mahmmod, Y.S.; Wang, J.J.; Liu, T.H.; Zheng, Y.X.; Zhou, X.; Zhang, X.X.; Yuan, Z.G. A double-edged sword: Complement component 3 in Toxoplasma gondii infection. Proteomics 2019, 19, 1800271. [Google Scholar] [CrossRef] [PubMed]
- Alsaadawi, M.A.; Alkhuzaie, S.S.; Alasadiy, Y.D.; Alsalih, N.J.; Al-Yasari, A.M.R. Supervision of The Complement System by Toxoplasma During Neural Infections (Areview). IOP Conf. Series Earth Environ. Sci. 2021, 923, 012047. [Google Scholar] [CrossRef]
- Jin, Y.; Yao, Y.; El-Ashram, S.; Tian, J.; Shen, J.; Ji, Y. The neurotropic parasite Toxoplasma gondii induces astrocyte polarization through NFκB pathway. Front. Med. 2019, 6, 267. [Google Scholar] [CrossRef] [PubMed]
- Nasuhidehnavi, A.; Yap, G.S. Microglia and astrocyte responses to neuropathogenic protozoan parasites. Fac. Rev. 2021, 10, 69. [Google Scholar] [CrossRef]
- Carrillo, G.L.; Ballard, V.A.; Glausen, T.; Boone, Z.; Teamer, J.; Hinkson, C.L.; Wohlfert, E.A.; Blader, I.J.; Fox, M.A. Toxoplasma infection induces microglia-neuron contact and the loss of perisomatic inhibitory synapses. Glia 2020, 68, 1968–1986. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.-H.; Suk, K. Microglia-astrocyte crosstalk: An intimate molecular conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Cragnolini, A.B.; Lampitella, G.; Virtuoso, A.; Viscovo, I.; Panetsos, F.; Papa, M.; Cirillo, G. Regional brain susceptibility to neurodegeneration: What is the role of glial cells? Neural. Regen. Res. 2020, 15, 838. [Google Scholar]
- Gray, S.C.; Kinghorn, K.J.; Woodling, N.S. Shifting equilibriums in Alzheimer’s disease: The complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural. Regen. Res. 2020, 15, 1208. [Google Scholar]
- Li, S.-M.; Li, B.; Zhang, L.; Zhang, G.-F.; Sun, J.; Ji, M.-H.; Yang, J.-J. A complement-microglial axis driving inhibitory synapse related protein loss might contribute to systemic inflammation-induced cognitive impairment. Int. Immunopharmacol. 2020, 87, 106814. [Google Scholar] [CrossRef]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.-D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Shinjyo, N.; Hikosaka, K.; Kido, Y.; Yoshida, H.; Norose, K. Toxoplasma infection induces sustained up-regulation of complement factor B and C5a receptor in the mouse brain via microglial activation: Implication for the alternative complement pathway activation and anaphylatoxin signaling in cerebral toxoplasmosis. Front. Immunol. 2021, 11, 603924. [Google Scholar] [CrossRef] [PubMed]
- Briukhovetska, D.; Ohm, B.; Mey, F.T.; Aliberti, J.; Kleingarn, M.; Huber-Lang, M.; Karsten, C.M.; Köhl, J. C5aR1 Activation Drives Early IFN-γ Production to Control Experimental Toxoplasma gondii Infection. Front. Immunol. 2020, 11, 1397. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Varun, C.N.; Dey, G.; Ravikumar, R.; Mahadevan, A.; Shankar, S.K.; Prasad, T.K. Identification of host-response in cerebral malaria patients using quantitative proteomic analysis. Proteom. Clin. Appl. 2018, 12, 1600187. [Google Scholar] [CrossRef] [PubMed]
- Lackner, P.; Hametner, C.; Beer, R.; Burger, C.; Broessner, G.; Helbok, R.; Speth, C.; Schmutzhard, E. Complement factors C1q, C3 and C5 in brain and serum of mice with cerebral malaria. Malar. J. 2008, 7, 207. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Erdman, L.K.; Lu, Z.; Serghides, L.; Zhong, K.; Dhabangi, A.; Musoke, C.; Gerard, C.; Cserti-Gazdewich, C.; Liles, W.C. Functional roles for C5a and C5aR but not C5L2 in the pathogenesis of human and experimental cerebral malaria. Infect. Immun. 2014, 82, 371–379. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.R.; Cahill, L.S.; Ho, K.T.; Yang, J.; Kim, H.; Silver, K.L.; Ward, P.A.; Mount, H.T.; Liles, W.C.; Sled, J.G. Experimental malaria in pregnancy induces neurocognitive injury in uninfected offspring via a C5a-C5a receptor dependent pathway. PLoS Pathog. 2015, 11, e1005140. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.N.; Berghout, J.; Lovegrove, F.E.; Ayi, K.; Conroy, A.; Serghides, L.; Min-Oo, G.; Gowda, D.C.; Sarma, J.V.; Rittirsch, D. C5 deficiency and C5a or C5aR blockade protects against cerebral malaria. J. Exp. Med. 2008, 205, 1133–1143. [Google Scholar] [CrossRef]
- Ramos, T.N.; Darley, M.M.; Hu, X.; Billker, O.; Rayner, J.C.; Ahras, M.; Wohler, J.E.; Barnum, S.R. Cutting edge: The membrane attack complex of complement is required for the development of murine experimental cerebral malaria. J. Immunol. 2011, 186, 6657–6660. [Google Scholar] [CrossRef]
- Buckingham, S.C.; Ramos, T.N.; Barnum, S.R. Complement C5-deficient mice are protected from seizures in experimental cerebral malaria. Epilepsia 2014, 55, e139–e142. [Google Scholar] [CrossRef]
- Finley, R.; Mackey, L.; Lambert, P. Virulent P. berghei malaria: Prolonged survival and decreased cerebral pathology in cell-dependent nude mice. J. Immunol. 1982, 129, 2213–2218. [Google Scholar] [PubMed]
- Li, Y.; Severance, E.G.; Viscidi, R.P.; Yolken, R.H.; Xiao, J. Persistent Toxoplasma infection of the brain induced neurodegeneration associated with activation of complement and microglia. Infect. Immun. 2019, 87, e00139-19. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Li, Y.; Gressitt, K.L.; He, H.; Kannan, G.; Schultz, T.L.; Svezhova, N.; Carruthers, V.B.; Pletnikov, M.V.; Yolken, R.H. Cerebral complement C1q activation in chronic Toxoplasma infection. Brain Behav. Immun. 2016, 58, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Velásquez-Ortiz, N.; Herrera, G.; Hernández, C.; Muñoz, M.; Ramírez, J.D. Discrete typing units of Trypanosoma cruzi: Geographical and biological distribution in the Americas. Sci. Data 2022, 9, 360. [Google Scholar] [CrossRef] [PubMed]
- Caputo, M.B.; Elias, J.; Cesar, G.; Alvarez, M.G.; Laucella, S.A.; Albareda, M.C. Role of the Complement System in the Modulation of T-Cell Responses in Chronic Chagas Disease. Front. Cell. Infect. Microbiol. 2022, 12, 910854. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.S.; Reis-Cunha, J.L.; Bartholomeu, D.C. Evasion of the immune response by Trypanosoma cruzi during acute infection. Front. Immunol. 2016, 6, 659. [Google Scholar] [CrossRef]
- Arroyo-Olarte, R.D.; Martínez, I.; Cruz-Rivera, M.; Mendlovic, F.; Espinoza, B. Complement system contributes to modulate the infectivity of susceptible TcI strains of Trypanosoma cruzi. Mem. Inst. Oswaldo. Cruz. 2018, 113, e170332. [Google Scholar] [CrossRef]
- Ramírez-Toloza, G.; Ferreira, A. Trypanosoma cruzi evades the complement system as an efficient strategy to survive in the mammalian host: The specific roles of host/parasite molecules and Trypanosoma cruzi calreticulin. Front. Microbiol. 2017, 8, 1667. [Google Scholar] [CrossRef]
- Evans-Osses, I.; Mojoli, A.; Beltrame, M.H.; Da Costa, D.E.; DaRocha, W.D.; Velavan, T.P.; de Messias-Reason, I.; Ramirez, M.I. Differential ability to resist to complement lysis and invade host cells mediated by MBL in R4 and 860 strains of Trypanosoma cruzi. FEBS Lett. 2014, 588, 956–961. [Google Scholar] [CrossRef]
- Gavinho, B.; Rossi, I.V.; Evans-Osses, I.; Inal, J.; Ramirez, M.I. A new landscape of host–protozoa interactions involving the extracellular vesicles world. Parasitology 2018, 145, 1521–1530. [Google Scholar] [CrossRef]
- Torrecilhas, A.C.; Soares, R.P.; Schenkman, S.; Fernández-Prada, C.; Olivier, M. Extracellular vesicles in trypanosomatids: Host cell communication. Front. Cell. Infect. Microbiol. 2020, 10, 602502. [Google Scholar] [CrossRef] [PubMed]
- Deolindo, P.; Evans-Osses, I.; Ramirez, M.I. Microvesicles and exosomes as vehicles between protozoan and host cell communication. Biochem. Soc. Trans. 2013, 41, 252–257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wyllie, M.; Ramirez, M. Microvesicles released during the interaction between Trypanosoma cruzi TcI and TcII strains and host blood cells inhibit complement system and increase the infectivity of metacyclic forms of host cells in a strain-independent process. Pathog. Dis. 2017, 75, ftx077. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.; Valck, C.; Sánchez, G.; Gingras, A.; Tzima, S.; Molina, M.C.; Sim, R.; Schwaeble, W.; Ferreira, A. The classical activation pathway of the human complement system is specifically inhibited by calreticulin from Trypanosoma cruzi. J. Immunol. 2004, 172, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.R.; Cecchi, G.; Paone, M.; Diarra, A.; Grout, L.; Kadima Ebeja, A.; Simarro, P.P.; Zhao, W.; Argaw, D. The elimination of human African trypanosomiasis: Achievements in relation to WHO road map targets for 2020. PLoS Negl. Trop. Dis. 2022, 16, e0010047. [Google Scholar] [CrossRef]
- Márquez-Contreras, M.E. Mechanisms of immune evasion by Trypanosoma brucei. Microbiol. Curr. Res. 2018, 2, 39–44. [Google Scholar] [CrossRef]
- Onyilagha, C.; Uzonna, J.E. Host immune responses and immune evasion strategies in African trypanosomiasis. Front. Immunol. 2019, 10, 2738. [Google Scholar] [CrossRef]
- Russo, D.; Williams, D.; Grab, D. Mechanisms for the elimination of potentially lytic complement-fixing variable surface glycoprotein antibody-complexes in Trypanosoma brucei. Parasitol. Res. 1994, 80, 487–492. [Google Scholar] [CrossRef]
- Stijlemans, B.; Caljon, G.; Van Den Abbeele, J.; Van Ginderachter, J.A.; Magez, S.; De Trez, C. Immune evasion strategies of Trypanosoma brucei within the mammalian host: Progression to pathogenicity. Front. Immunol. 2016, 7, 233. [Google Scholar] [CrossRef]
- Samanta, D.; Mulye, M.; Clemente, T.M.; Justis, A.V.; Gilk, S.D. Manipulation of host cholesterol by obligate intracellular bacteria. Front. Cell. Infect. Microbiol. 2017, 7, 165. [Google Scholar] [CrossRef]
- O’Neal, A.J.; Butler, L.R.; Rolandelli, A.; Gilk, S.D.; Pedra, J.H. Lipid hijacking: A unifying theme in vector-borne diseases. Elife 2020, 9, e61675. [Google Scholar] [CrossRef] [PubMed]
- Machado, H.; Bizarra-Rebelo, T.; Costa-Sequeira, M.; Trindade, S.; Carvalho, T.; Rijo-Ferreira, F.; Rentroia-Pacheco, B.; Serre, K.; Figueiredo, L.M. Trypanosoma brucei triggers a broad immune response in the adipose tissue. PLoS Pathog. 2021, 17, e1009933. [Google Scholar] [CrossRef] [PubMed]
- Mans, B.J.; Ribeiro, J.M. A novel clade of cysteinyl leukotriene scavengers in soft ticks. Insect. Biochem. Mol. Biol. 2008, 38, 862–870. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rios-Barros, L.V.; Silva-Moreira, A.L.; Horta, M.F.; Gontijo, N.F.; Castro-Gomes, T. How to get away with murder: The multiple strategies employed by pathogenic protozoa to avoid complement killing. Mol. Immunol. 2022, 149, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Oghumu, S.; Satoskar, A.R. Mechanisms of immune evasion in leishmaniasis. Adv. Appl. Microbiol. 2013, 82, 155–184. [Google Scholar]
- Hermoso, T.; Fishelson, Z.; Becker, S.; Hirschberg, K.; Jaffe, C. Leishmanial protein kinases phosphorylate components of the complement system. EMBO J. 1991, 10, 4061–4067. [Google Scholar] [CrossRef]
- Ouaissi, A.; Ouaissi, M. Molecular basis of Trypanosoma cruzi and Leishmania interaction with their host (s): Exploitation of immune and defense mechanisms by the parasite leading to persistence and chronicity, features reminiscent of immune system evasion strategies in cancer diseases. Arch. Immunol. Ther. Exp. 2005, 53, 102–114. [Google Scholar]
- Kedzierski, L.; Montgomery, J.; Bullen, D.; Curtis, J.; Gardiner, E.; Jimenez-Ruiz, A.; Handman, E. A leucine-rich repeat motif of Leishmania parasite surface antigen 2 binds to macrophages through the complement receptor 3. J. Immunol. 2004, 172, 4902–4906. [Google Scholar] [CrossRef]
- Pereira Filho, A.A.; de Sousa Nascimento, A.A.; Saab, N.A.A.; Fugiwara, R.T.; Pessoa, G.C.D.Á.; Koerich, L.B.; Pereira, M.H.; Araújo, R.N.; Sant’Anna, M.R.V.; Gontijo, N.F. Evasion of the complement system by Leishmania through the uptake of factor H, a complement regulatory protein. Acta Trop. 2021, 224, 106152. [Google Scholar] [CrossRef]
- Kim, Y.U.; Hong, Y. Functional analysis of the first mannosyltransferase (PIG-M) involved in glycosylphosphatidylinositol synthesis in Plasmodium falciparum. Mol. Cells 2007, 24, 294–300. [Google Scholar]
- Reyes, A.C.; Encina, J.L.R. Trypanosoma cruzi infection: Mechanisms of evasion of immune response. In Biology of Trypanosoma cruzi; IntechOpen: London, UK, 2019. [Google Scholar]
- Cestari, I.; Ansa-Addo, E.; Deolindo, P.; Inal, J.M.; Ramirez, M.I. Trypanosoma cruzi immune evasion mediated by host cell-derived microvesicles. J. Immunol. 2012, 188, 1942–1952. [Google Scholar] [CrossRef] [PubMed]
- De Castro Neto, A.L.; Da Silveira, J.F.; Mortara, R.A. Comparative analysis of virulence mechanisms of trypanosomatids pathogenic to humans. Front. Cell. Infect. Microbiol. 2021, 11, 669079. [Google Scholar] [CrossRef] [PubMed]
- Sosoniuk, E.; Vallejos, G.; Kenawy, H.; Gaboriaud, C.; Thielens, N.; Fujita, T.; Schwaeble, W.; Ferreira, A.; Valck, C. Trypanosoma cruzi calreticulin inhibits the complement lectin pathway activation by direct interaction with L-Ficolin. Mol. Immunol. 2014, 60, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Brittingham, A.; Morrison, C.J.; McMaster, W.R.; McGwire, B.S.; Chang, K.-P.; Mosser, D.M. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J. Immunol. 1995, 155, 3102–3111. [Google Scholar] [CrossRef]
- Chaudhuri, G.; Chang, K.-P. Acid protease activity of a major surface membrane glycoprotein (gp63) from Leishmania mexicana promastigotes. Mol. Biochem. Parasitol. 1988, 27, 43–52. [Google Scholar] [CrossRef]
- Nunes, A.; Almeida-Campos, F.; Horta, M.; Ramalho-Pinto, F. Leishmania amazonensis promastigotes evade complement killing by interfering with the late steps of the cascade. Parasitology 1997, 115, 601–609. [Google Scholar] [CrossRef]
- Verma, S.; Mandal, A.; Ansari, M.Y.; Kumar, A.; Abhishek, K.; Ghosh, A.K.; Kumar, A.; Kumar, V.; Das, S.; Das, P. Leishmania donovani inhibitor of serine peptidases 2 mediated inhibition of lectin pathway and upregulation of C5aR signaling promote parasite survival inside host. Front. Immunol. 2018, 9, 63. [Google Scholar] [CrossRef]
- Lima, P.C.; Mottram, J.C. Trypanosomatid-encoded inhibitors of peptidases: Unique structural features and possible roles as virulence factors. Open Parasitol. J. 2010, 4, 132–138. [Google Scholar] [CrossRef]
- Lamotte, S.; Späth, G.F.; Rachidi, N.; Prina, E. The enemy within: Targeting host–parasite interaction for antileishmanial drug discovery. PLoS Negl. Trop. Dis. 2017, 11, e0005480. [Google Scholar] [CrossRef]
- Miller, H.W.; Tam, T.S.; Ralston, K.S. Entamoeba histolytica Develops Resistance to Complement Deposition and Lysis after Acquisition of Human Complement-Regulatory Proteins through Trogocytosis. Mbio 2022, 13, e03163-21. [Google Scholar] [CrossRef]
- Faria, C.P.; Neves, B.M.; Lourenço, Á.; Cruz, M.T.; Martins, J.D.; Silva, A.; Pereira, S.; Sousa, M.d.C. Giardia lamblia decreases NF-κB p65RelA protein levels and modulates LPS-induced pro-inflammatory response in macrophages. Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Escribano, A.; Nogal-Ruiz, J.J.; Pérez-Serrano, J.; Gómez-Barrio, A.; Escario, J.A.; Alderete, J. Sequestration of host-CD59 as potential immune evasion strategy of Trichomonas vaginalis. Acta Trop. 2015, 149, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kurtovic, L.; Behet, M.C.; Feng, G.; Reiling, L.; Chelimo, K.; Dent, A.E.; Mueller, I.; Kazura, J.W.; Sauerwein, R.W.; Fowkes, F.J. Human antibodies activate complement against Plasmodium falciparum sporozoites, and are associated with protection against malaria in children. BMC Med. 2018, 16, 61. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.; Chan, J.; Handayuni, I.; Reiling, L.; Feng, G.; Hilton, A.; Kurtovic, L.; Oyong, D.; Piera, K.; Barber, B. IgM in human immunity to Plasmodium falciparum malaria. Sci. Adv. 2019, 5, eaax4489. [Google Scholar] [CrossRef] [PubMed]
- Behet, M.C.; Kurtovic, L.; van Gemert, G.-J.; Haukes, C.M.; Siebelink-Stoter, R.; Graumans, W.; van de Vegte-Bolmer, M.G.; Scholzen, A.; Langereis, J.D.; Diavatopoulos, D.A. The complement system contributes to functional antibody-mediated responses induced by immunization with Plasmodium falciparum malaria sporozoites. Infect. Immun. 2018, 86, e00920-17. [Google Scholar] [CrossRef]
- Reiling, L.; Boyle, M.J.; White, M.T.; Wilson, D.W.; Feng, G.; Weaver, R.; Opi, D.H.; Persson, K.E.; Richards, J.S.; Siba, P.M. Targets of complement-fixing antibodies in protective immunity against malaria in children. Nat. Commun. 2019, 10, 610. [Google Scholar] [CrossRef]
- Ubillos, I.; Ayestaran, A.; Nhabomba, A.J.; Dosoo, D.; Vidal, M.; Jiménez, A.; Jairoce, C.; Sanz, H.; Aguilar, R.; Williams, N.A. Baseline exposure, antibody subclass, and hepatitis B response differentially affect malaria protective immunity following RTS, S/AS01E vaccination in African children. BMC Med. 2018, 16, 197. [Google Scholar] [CrossRef]
- Zenklusen, I.; Jongo, S.; Abdulla, S.; Ramadhani, K.; Lee Sim, B.K.; Cardamone, H.; Flannery, E.L.; Nguyen, T.; Fishbaugher, M.; Steel, R.W. Immunization of malaria-preexposed volunteers with PfSPZ vaccine elicits long-lived IgM invasion-inhibitory and complement-fixing antibodies. J. Infect. Dis. 2018, 217, 1569–1578. [Google Scholar] [CrossRef]
- Autheman, D.; Crosnier, C.; Clare, S.; Goulding, D.A.; Brandt, C.; Harcourt, K.; Tolley, C.; Galaway, F.; Khushu, M.; Ong, H. An invariant Trypanosoma vivax vaccine antigen induces protective immunity. Nature 2021, 595, 96–100. [Google Scholar] [CrossRef]
- Gruszynski, A.E.; van Deursen, F.J.; Albareda, M.C.; Best, A.; Chaudhary, K.; Cliffe, L.J.; del Rio, L.; Dunn, J.D.; Ellis, L.; Evans, K.J. Regulation of surface coat exchange by differentiating African trypanosomes. Mol. Biochem. Parasitol. 2006, 147, 211–223. [Google Scholar] [CrossRef]
- Moreno, C.J.G.; Temporão, A.; Torres, T.; Sousa Silva, M. Trypanosoma brucei interaction with host: Mechanism of VSG release as target for drug discovery for african trypanosomiasis. Int. J. Mol. Sci. 2019, 20, 1484. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.P.; Pangburn, M.K.; Cortés, C. Complement control protein factor H: The good, the bad, and the inadequate. Mol. Immunol. 2010, 47, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, A.E.; Sarr, I.; Correa, S.; Nwakanma, D.; Brouwer, M.C.; Wouters, D.; Secka, F.; Anderson, S.T.; Conway, D.J.; Walther, M. Complement factor H levels associate with Plasmodium falciparum malaria susceptibility and severity. Open Forum Infect. Dis. 2018, 20, ofy166. [Google Scholar] [CrossRef] [PubMed]
- Macleod, O.J.; Bart, J.-M.; MacGregor, P.; Peacock, L.; Savill, N.J.; Hester, S.; Ravel, S.; Sunter, J.D.; Trevor, C.; Rust, S. A receptor for the complement regulator factor H increases transmission of trypanosomes to tsetse flies. Nat. Commun. 2020, 11, 1326. [Google Scholar] [CrossRef]
- Mulamba, C.; Williams, C.; Kreppel, K.; Ouedraogo, J.B.; Olotu, A.I. Evaluation of the Pfs25-IMX313/Matrix-M malaria transmission-blocking candidate vaccine in endemic settings. Malar. J. 2022, 21, 159. [Google Scholar] [CrossRef]
- Kipnis, T.L.; David, J.R.; Alper, C.A.; Sher, A.; Da Silva, W.D. Enzymatic treatment transforms trypomastigotes of Trypanosoma cruzi into activators of alternative complement pathway and potentiates their uptake by macrophages. Proc. Natl. Acad. Sci. USA 1981, 78, 602–605. [Google Scholar] [CrossRef]
- Giuliani, M.M.; Adu-Bobie, J.; Comanducci, M.; Aricò, B.; Savino, S.; Santini, L.; Brunelli, B.; Bambini, S.; Biolchi, A.; Capecchi, B. A universal vaccine for serogroup B meningococcus. Proc. Natl. Acad. Sci. USA 2006, 103, 10834–10839. [Google Scholar] [CrossRef]
- Orsini, F.; De Blasio, D.; Zangari, R.; Zanier, E.R.; De Simoni, M.-G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell. Neurosci. 2014, 8, 380. [Google Scholar] [CrossRef]
- Sultan, B.A.; AL-Fatlawi, S.N. Assessment of C3 and C4 component of complement system in aborted women infected with Toxoplasma gondii. Al-Qadisiyah Med. J. 2016, 12, 110–114. [Google Scholar]
- Leite, P.E.C.; de Araujo Portes, J.; Pereira, M.R.; Russo, F.B.; Martins-Duarte, E.S.; Dos Santos, N.A.; Attias, M.; Barrantes, F.J.; Beltrão-Braga, P.C.B.; de Souza, W. Morphological and biochemical repercussions of Toxoplasma gondii infection in a 3D human brain neurospheres model. Brain Behav. Immun. Health 2021, 11, 100190. [Google Scholar] [CrossRef]
- Yang, H.-Y.; Chien, W.-C.; Chung, C.-H.; Su, R.-Y.; Lai, C.-Y.; Yang, C.-C.; Tzeng, N.-S. Risk of dementia in patients with toxoplasmosis: A nationwide, population-based cohort study in Taiwan. Parasit. Vectors 2021, 14, 435. [Google Scholar] [CrossRef] [PubMed]
- Nayeri, T.; Sarvi, S.; Sharif, M.; Daryani, A. Toxoplasma gondii: A possible etiologic agent for Alzheimer’s disease. Heliyon 2021, 7, e07151. [Google Scholar] [CrossRef] [PubMed]
- de Bles, N.J.; van der Does, J.E.; Kortbeek, L.M.; Hofhuis, A.; van Grootheest, G.; Vollaard, A.M.; Schoevers, R.A.; van Hemert, A.M.; Penninx, B.W.; Rius-Ottenheim, N. Toxoplasma gondii seropositivity in patients with depressive and anxiety disorders. Brain Behav. Immun. Health 2021, 11, 100197. [Google Scholar] [CrossRef] [PubMed]
- Roe, K. The link between Toxoplasma gondii infections and higher mortality in COVID-19 patients having schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 272, 167–168. [Google Scholar] [CrossRef]
- Lv, L.; Wang, Y.; Feng, W.; Hernandez, J.A.; Huang, W.; Zheng, Y.; Zhou, X.; Lv, S.; Chen, Y.; Yuan, Z.-G. iTRAQ-based differential proteomic analysis in Mongolian gerbil brains chronically infected with Toxoplasma gondii. J. Proteom. 2017, 160, 74–83. [Google Scholar] [CrossRef]
- Ziabska, K.; Ziemka-Nalecz, M.; Pawelec, P.; Sypecka, J.; Zalewska, T. Aberrant Complement System Activation in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4675. [Google Scholar] [CrossRef]
- Sekar, A.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V. Author Correction: Schizophrenia risk from complex variation of complement component 4. Nature 2022, 601, E4–E5. [Google Scholar] [CrossRef]
- Rachidi, N.; Taly, J.F.; Durieu, E.; Leclercq, O.; Aulner, N.; Prina, E.; Pescher, P.; Notredame, C.; Meijer, L.; Späth, G.F. Pharmacological assessment defines Leishmania donovani casein kinase 1 as a drug target and reveals important functions in parasite viability and intracellular infection. Antimicrob. Agents Chemother. 2014, 58, 1501–1515. [Google Scholar] [CrossRef]
- Ishino, T.; Yano, K.; Chinzei, Y.; Yuda, M.; Ward, G. Cell-passage activity is required for the malarial parasite to cross the liver sinusoidal cell layer. PLoS Biol. 2004, 2, e4. [Google Scholar] [CrossRef]
- Ishino, T.; Chinzei, Y.; Yuda, M. A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cell. Microbiol. 2005, 7, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hamaoka, B.Y.; Ghosh, P. Structure of the essential Plasmodium host cell traversal protein SPECT1. PLoS ONE 2014, 9, e114685. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kumar, G.; Vashishta, M.; Pandey, R.; Rathore, S.; Chourasia, B.K.; Singhal, J.; Deshmukh, A.; Kalamuddin, M.; Paul, G. Biochemical characterization of Plasmodium complement factors binding protein for its role in immune modulation. Biochem. J. 2018, 475, 2877–2891. [Google Scholar] [CrossRef] [PubMed]
- Sijwali, P.S. Interaction with complement proteins and dendritic cells implicates LCCL domain-containing proteins (CCps) of malaria parasites in immunomodulation. Biochem. J. 2018, 475, 3311–3314. [Google Scholar] [CrossRef] [PubMed]
- Torrecilhas, A.C.; Schumacher, R.I.; Alves, M.J.M.; Colli, W. Vesicles as carriers of virulence factors in parasitic protozoan diseases. Microbes Infect. 2012, 14, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Murphy, E.E.; Wong, W.W.; Klickstein, L.; Weis, J.H.; Fearon, D.T. Identification of a restriction fragment length polymorphism by a CR1 cDNA that correlates with the number of CR1 on erythrocytes. J. Exp. Med. 1986, 164, 50–59. [Google Scholar] [CrossRef]
- Opi, D.H.; Swann, O.; Macharia, A.; Uyoga, S.; Band, G.; Ndila, C.M.; Harrison, E.M.; Thera, M.A.; Kone, A.K.; Diallo, D.A. Two complement receptor one alleles have opposing associations with cerebral malaria and interact with α+thalassaemia. Elife 2018, 7, e31579. [Google Scholar] [CrossRef]
- Beaudoin, C.A.; Jamasb, A.R.; Alsulami, A.F.; Copoiu, L.; van Tonder, A.J.; Hala, S.; Bannerman, B.P.; Thomas, S.E.; Vedithi, S.C.; Torres, P.H. Predicted structural mimicry of spike receptor-binding motifs from highly pathogenic human coronaviruses. Comput. Struct. Biotechnol. J. 2021, 19, 3938–3953. [Google Scholar] [CrossRef]
- Wong, S.S.W.; Daniel, I.; Gangneux, J.-P.; Jayapal, J.M.; Guegan, H.; Dellière, S.; Lalitha, P.; Shende, R.; Madan, T.; Bayry, J. Differential interactions of serum and bronchoalveolar lavage fluid complement proteins with conidia of airborne fungal pathogen Aspergillus fumigatus. Infect. Immun. 2020, 88, e00212-20. [Google Scholar] [CrossRef]
- Bayer-Santos, E.; Aguilar-Bonavides, C.; Rodrigues, S.P.; Cordero, E.M.; Marques, A.F.; Varela-Ramirez, A.; Choi, H.; Yoshida, N.; Da Silveira, J.F.; Almeida, I.C. Proteomic analysis of Trypanosoma cruzi secretome: Characterization of two populations of extracellular vesicles and soluble proteins. J. Proteome Res. 2013, 12, 883–897. [Google Scholar] [CrossRef]
- Rashidi, S.; Mojtahedi, Z.; Shahriari, B.; Kalantar, K.; Ghalamfarsa, G.; Mohebali, M.; Hatam, G. An immunoproteomic approach to identifying immunoreactive proteins in Leishmania infantum amastigotes using sera of dogs infected with canine visceral leishmaniasis. Pathog. Glob. Health 2019, 113, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, S.; Torelli, F.; Lockyer, E.J.; Wagener, J.; Song, O.-R.; Broncel, M.; Russell, M.; Young, J.C.; Treeck, M. Toxoplasma gondii ROP1 subverts murine and human innate immune restriction. bioRxiv 2022. [Google Scholar]
- Nguyen, V.A.; Riddell, N.; Crewther, S.G.; Faou, P.; Rajapaksha, H.; Howells, D.W.; Hankey, G.J.; Wijeratne, T.; Ma, H.; Davis, S. Longitudinal Stroke Recovery Associated With Dysregulation of Complement System—A Proteomics Pathway Analysis. Front. Neurol. 2020, 11, 692. [Google Scholar] [CrossRef] [PubMed]


| Biomolecules, Proteins, Receptors | Mechanism of Action to Inhibit Complement | References |
|---|---|---|
| Plasmodium spp. | ||
| MSP3.1 | The inactivation of C1s and mucin-associated surface protein-2 (MASP-2) through C1-INH | [32] |
| Mannosyltransferase (PIG-M) | Employing CD59 to decrease C9 polymerization on the cell surface by binding to C8a and C9 | [101] |
| T. cruzi | ||
| Calreticulin (TcCRT) | Interacts with L-ficolin and preventing C4 conversion to C4b, interacting with C1q collagen-like domain (inhibiting both the classical and lectin pathways) | [5,14,77,79,95,102,103,104,105] |
| T. cruzi complement regulatory protein (TcCRP) | Binding to C3b and C4b and inhibiting the formation of the classical and alternative complement C3 convertase | |
| T. cruzi complement C2 receptor inhibition trispanning (TcCRIT) | Blocking C2 cleavage by C1s or MASP-2 into C2a and inhibiting C3 convertase formation, hijacking C2 (modulating the activation of the lectin and classical complement pathways) | |
| Trypomastigote decay-accelerating factor (T-DAF) | Mimicking the activity of the complement regulatory protein DAF, blocking C3 and C4, accelerating the dissociation or assembly of C3 convertases (modulating/inhibiting the activation of the alternative, classical, and probably the lectins pathways) | |
| Glycoprotein 58/68 | Preventing the formation of cell-bound C3 convertase (decay-accelerating activity) by inhibiting the initial association of factor B (FB) to surface fixed C3b, attaching to human complements C3b and C4b to prevent the activation of the complement) | |
| Membrane-derived vesicles (microvesicles) | Inhibiting the classical and lectin pathways by binding to C3 convertase C4b2a on the parasite surface and decreasing its catalytic activity | |
| N- and O-glycosylated biomolecules | To inhibit activation of the lectin complement pathway through L-ficolin, H-ficolins, and mannose-binding lectin (MBL) (resulting in the failure of MASP-2-induced C2 and C4 cleavage) | |
| GP72 | Inhibiting the formation of the C3 convertase in the alternative pathway | |
| GP160 | As a member of the C3/C4 binding family of complement regulators: Inhibits the formation of the alternative and classical C3 convertase (preventing the activation of the complement cascade) | |
| Leishmania spp. | ||
| GP63 | Cleaving parasite-bound C3b into inactive form iC3b, (prevent the formation of C3 and C5 convertase and the MAC-mediated lysis of the parasite) | [5,95,106,107] |
| GP46 (as a membrane-associated protein inhibitors expressed on L. amazonensis complement-resistant promastigotes) | Inhibiting the lytic activity of AP, impairing C9, but not C3, attaching to complement-activating complex (probably block the complement activation after C3b deposition and at the stage of C9 deposition) | [95,108] |
| Inhibitors of serine proteinase (ISP) (L. donovani) | Interacting with host C1r, C1s, MASP-1 and MASP-2, preventing the formation of CP and LP initiators (preventing the formation of C3 convertase, decreasing the production of the anaphylatoxins C3a and C5a) | [5,95,109,110] |
| Casein kinase 1 isoform 2 (CK1.2) | Interacting with C3a and modulating complement system and inducing immune evasion | [97,111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashidi, S.; Mansouri, R.; Ali-Hassanzadeh, M.; Muro, A.; Nguewa, P.; Manzano-Román, R. The Defensive Interactions of Prominent Infectious Protozoan Parasites: The Host’s Complement System. Biomolecules 2022, 12, 1564. https://doi.org/10.3390/biom12111564
Rashidi S, Mansouri R, Ali-Hassanzadeh M, Muro A, Nguewa P, Manzano-Román R. The Defensive Interactions of Prominent Infectious Protozoan Parasites: The Host’s Complement System. Biomolecules. 2022; 12(11):1564. https://doi.org/10.3390/biom12111564
Chicago/Turabian StyleRashidi, Sajad, Reza Mansouri, Mohammad Ali-Hassanzadeh, Antonio Muro, Paul Nguewa, and Raúl Manzano-Román. 2022. "The Defensive Interactions of Prominent Infectious Protozoan Parasites: The Host’s Complement System" Biomolecules 12, no. 11: 1564. https://doi.org/10.3390/biom12111564
APA StyleRashidi, S., Mansouri, R., Ali-Hassanzadeh, M., Muro, A., Nguewa, P., & Manzano-Román, R. (2022). The Defensive Interactions of Prominent Infectious Protozoan Parasites: The Host’s Complement System. Biomolecules, 12(11), 1564. https://doi.org/10.3390/biom12111564