
Immune Checkpoint Inhibitors and RAS–ERK Pathway-Targeted Drugs as Combined Therapy for the Treatment of Melanoma



Abstract
1. Introduction
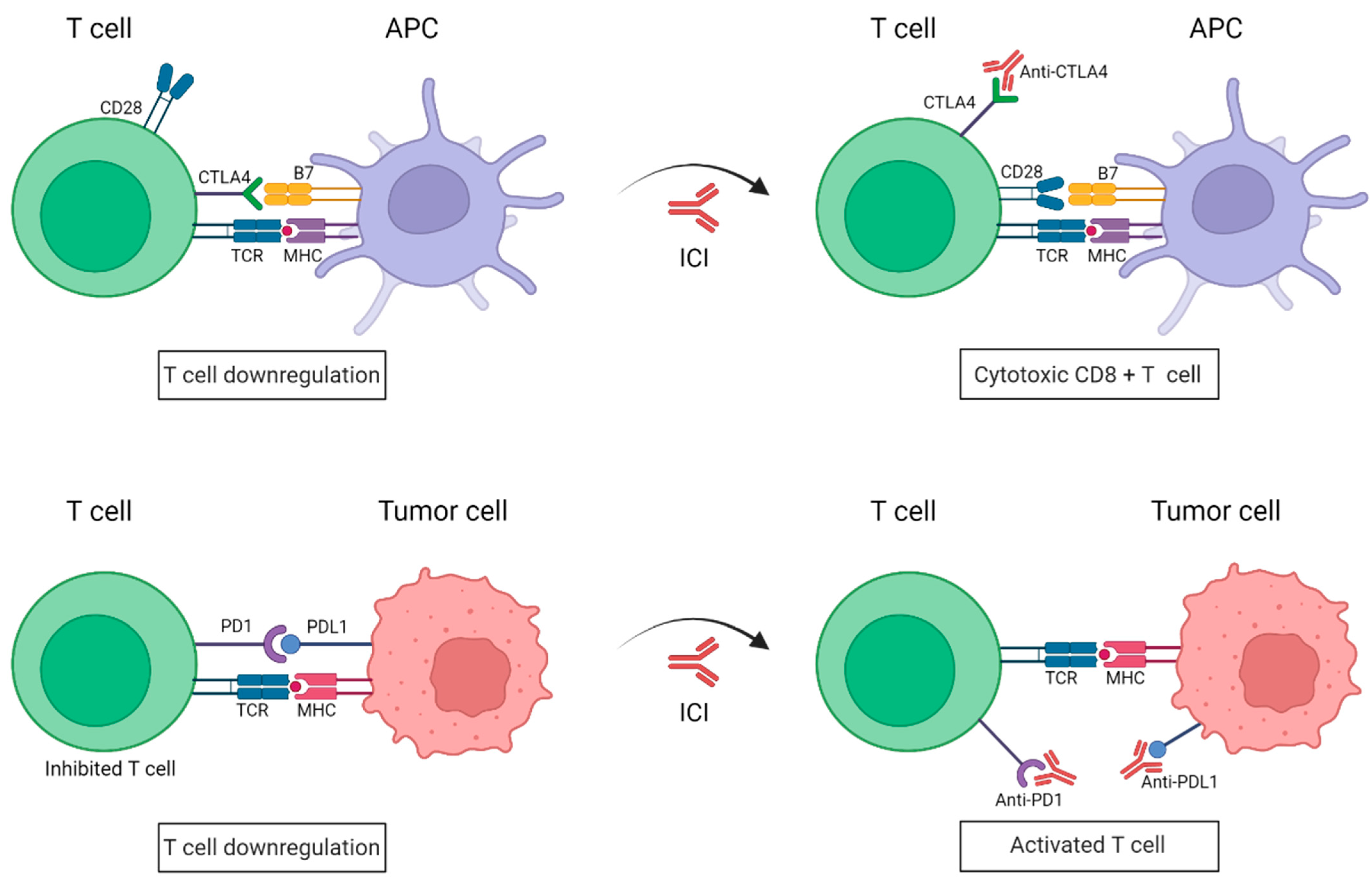
2. Use of Immune Checkpoints Inhibitors in the Treatment of Melanoma
3. The RAS–ERK Pathway
3.1. An Overview
3.2. Immunological Impact of Targeting RAS–ERK Pathway
3.2.1. BRAF Inhibitors
3.2.2. MEK Inhibitors
3.2.3. Combination of BRAF and MEK Inhibitors
3.2.4. Drawbacks in RAS–ERK Pathway-Targeted Therapy
4. Rationale for Combining RAS–ERK Pathway-targeted Therapy and Immunotherapy
Clinical Trials Combining MEK/BRAF Inhibitors with Immunotherapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society Key Statistics for Melanoma Skin Cancer. Available online: https://www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html. (accessed on 21 June 2021).
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.F., Eds.; Codon Publications: Hong Lim Complex, Singapore, 2017; Chapter 1. [Google Scholar]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Shreiber, R.D. IFNgamma and Lympohcytes Prevent Primary Tomour Development and Shape Tomour Immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The Determinants of Tumour Immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune System and Melanoma Biology: A Balance between Immunosurveillance and Immune Escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef]
- Cormier, J.N.; Hijazi, Y.M.; Abati, A.; Fetsch, P.; Bettinotti, M.; Steinberg, S.M.; Rosenberg, S.A.; Marincola, F.M. Heterogeneous Expression of Melanoma-Associated Antigens and HLA-A2 in Metastatic Melanoma in Vivo. Int J. Cancer 1998, 75, 517–524. [Google Scholar] [CrossRef]
- Maeurer, M.J.; Gollin, S.M.; Martin, D.; Swaney, W.; Bryant, J.; Castelli, C.; Robbins, P.; Parmiani, G.; Storkus, W.J.; Lotze, M.T. Tumor Escape from Immune Recognition: Lethal Recurrent Melanoma in a Patient Associated with Downregulation of the Peptide Transporter Protein TAP-1 and Loss of Expression of the Immunodominant MART-1/Melan-A Antigen. J. Clin. Investig. 1996, 98, 1633–1641. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 Can Function as a Negative Regulator of T Cell Activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Jordan, K.R.; Amaria, R.N.; Ramirez, O.; Callihan, E.B.; Gao, D.; Borakove, M.; Manthey, E.; Borges, V.F.; Mccarter, M.D. Myeloid-Derived Suppressor Cells Are Associated with Disease Progression and Decreased Overall Survival in Advanced-Stage Melanoma Patients. Cancer Immunol. Immunother. 2013, 62, 1711–1722. [Google Scholar]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-Dioxygenase and Tumor-Induced Tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef]
- Pietra, G.; Vitale, M.; Moretta, L.; Mingari, M.C. How Melanoma Cells Inactivate NK Cells. Oncoimmunology 2012, 1, 974–975. [Google Scholar] [CrossRef]
- Jacobs, J.F.M.; Nierkens, S.; Figdor, C.G.; de Vries, I.J.M.; Adema, G.J. Regulatory T Cells in Melanoma: The Final Hurdle towards Effective Immunotherapy? Lancet Oncol. 2012, 13, e32–e42. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; McDermott, D.F. Cytotoxic T-Lymphocyte Antigen-4 Blockade in Melanoma. Clin. Ther. 2015, 37, 755–763. [Google Scholar] [CrossRef]
- Ralli, M.; Botticelli, A.; Visconti, I.C.; Angeletti, D.; Fiore, M.; Marchetti, P.; Lambiase, A.; De Vincentiis, M.; Greco, A. Immunotherapy in the Treatment of Metastatic Melanoma: Current Knowledge and Future Directions. J. Immunol. Res. 2020, 2020, 9235638. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on Both Effector and Regulatory T Cell Compartments Contributes to the Antitumor Activity of Anti-CTLA-4 Antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human Cancer Immunotherapy with Antibodies to the PD-1 and PD-L1 Pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef]
- Herrscher, H.; Robert, C. Immune Checkpoint Inhibitors in Melanoma in the Metastatic, Neoadjuvant, and Adjuvant Setting. Curr. Opin. Oncol. 2020, 32, 106–113. [Google Scholar] [CrossRef]
- Handa, S.; Hans, B.; Goel, S.; Bashorun, H.O.; Dovey, Z.; Tewari, A. Immunotherapy in Prostate Cancer: Current State and Future Perspectives. Ther. Adv. Urol. 2020, 12, 2020. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Approves New Treatment for a Type of Late-Stage Skin Cancer. Available online: www.fda.gov/newsevents/newsroom/pressannouncements/ucm1193237.htm. (accessed on 4 March 2011).
- Raedler, L.A. Opdivo (Nivolumab): Second PD-1 Inhibitor Receives FDA Approval for Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 96–100. [Google Scholar]
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Substudy 02A: Safety and Efficacy of Pembrolizumab in Combination With Investigational Agents in Participants With Programmed Cell-Death 1 (PD-1) Refractory Melanoma (MK-3475-02A/KEYMAKER-U02). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04305041 (accessed on 18 September 2020).
- Bomar, L.; Senithilnathan, A.; Ahn, C. Systemic Therapies for Advanced Melanoma. Dermatol. Clin. 2019, 37, 409–423. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroyakovskiy, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. 3301 Two Year Estimate of Overall Survival in COMBI-v, a Randomized, Open-Label, Phase III Study Comparing the Combination of Dabrafenib (D) and Trametinib (T) with Vemurafenib (Vem) as First-Line Therapy in Patients (Pts) with Unresectable or Metastatic. Eur. J. Cancer 2015, 51, s663. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Stucci, S.; Palmirotta, R.; Passarelli, A.; Silvestris, E.; Argentiero, A.; Lanotte, L.; Acquafredda, S.; Todisco, A.; Silvestris, F. Immune-Related Adverse Events during Anticancer Immunotherapy: Pathogenesis and Management. Oncol. Lett. 2017, 14, 5671–5680. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, S.; Yang, F.; Qi, X.; Wang, X.; Guan, X.; Shen, C.; Duma, N.; Vera Aguilera, J.; Chintakuntlawar, A.; et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-Analysis. JAMA Oncol. 2019, 5, 1008–1019. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.F.; McDermott, D.F.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Combined Nivolumab and Ipilimumab versus Ipilimumab Alone in Patients with Advanced Melanoma: 2-Year Overall Survival Outcomes in a Multicentre, Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2016, 17, 1558–1568. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Stephen Hodi, F.; O, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 8, 711–734. [Google Scholar]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab for Advanced Melanoma: Final Overall Survival Results of a Multicentre, Randomised, Open-Label Phase 3 Study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Sigalotti, L.; Fratta, E.; Coral, S.; Maio, M. Epigenetic Drugs as Immunomodulators for Combination Therapies in Solid Tumors. Pharmacol. Ther. 2014, 142, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; Mckenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.M.; Woan, K.; Cheng, F.; Wang, H.; Perez-Villarroel, P.; Lee, C.; Lienlaf, M.; Atadja, P.; Seto, E.; Weber, J.; et al. The Antimelanoma Activity of the Histone Deacetylase Inhibitor Panobinostat (LBH589) Is Mediated by Direct Tumor Cytotoxicity and Increased Tumor Immunogenicity. Melanoma Res. 2013, 23, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated Regulation of Myeloid Cells by Tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on Tumor Cells Impairs Antitumor T-Cell Responses: A Novel Mechanism of Tumor-Induced Immune Suppression. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef]
- Pandey, Μ.R.; Ernstoff, M.S.; Pandey, M.R. Resistance Mechanism of Resistance to Immune Checkpoint Inhibitors. Cancer Drug Resist. 2019, 2, 178–188. [Google Scholar] [CrossRef]
- Zhu, J.; Powis De Tenbossche, C.G.; Cané, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.-M.; Liljeström, P.; Uyttenhove, C.; van den Eynde, B.J. Resistance to Cancer Immunotherapy Mediated by Apoptosis of Tumor-Infiltrating Lymphocytes. Nat. Commun. 2017, 8, 1404. [Google Scholar] [CrossRef]
- Liu, D.; Lin, J.R.; Robitschek, E.J.; Kasumova, G.G.; Heyde, A.; Shi, A.; Kraya, A.; Zhang, G.; Moll, T.; Frederick, D.T.; et al. Evolution of Delayed Resistance to Immunotherapy in a Melanoma Responder. Nat. Med. 2021, 27, 985–992. [Google Scholar] [CrossRef]
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic b-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Luke, J.J.; Zha, Y.; Segal, J.P.; Ritterhouse, L.L.; Spranger, S.; Matijevich, K.; Gajewski, T.F. Secondary Resistance to Immunotherapy Associated with β-Catenin Pathway Activation or PTEN Loss in Metastatic Melanoma. J. Immunother Cancer 2019, 7, 295. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to Checkpoint Blockade Therapy through Inactivation of Antigen Presentation. Nat. Commun. 2017, 8, 1–11. [Google Scholar]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor Aneuploidy Correlates with Markers of Immune Evasion and with Reduced Response to Immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated Molecular Analysis of Tumor Biopsies on Sequential CTLA-4 and PD-1 Blockade Reveals Markers of Response and Resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Fonsatti, E.; Nicolay, H.J.M.; Sigalotti, L.; Calabro’, L.; Pezzani, L.; Colizzi, F.; Altomonte, M.; Guidoboni, M.; Marincola, F.M.; Maio, M. Functional Up-Regulation of Human Leukocyte Antigen Class I Antigens Expression by 5-Aza-2 ¶-Deoxycytidine in Cutaneous Melanoma: Immunotherapeutic Implications Cancer Therapy: Preclinical. Clin. Cancer Res. 2007, 13, 3333–3338. [Google Scholar] [CrossRef]
- Herrero, A.; Crespo, P. Ras Dimers: The Novice Couple at the Ras-Erk Pathway Ball. Genes 2021, 12, 1556. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. ERK1/2 MAP Kinases: Structure, Function, and Regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Reszka, A.A.; Seger, R.; Diltz, C.D.; Krebs, E.G.; Fischer, E.H. Association of Mitogen-Activated Protein Kinase with the Microtubule Cytoskeleton. Proc. Natl. Acad. Sci. USA 1995, 92, 8881–8885. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, C.; Torres, J.; Pulido, R. A Novel Regulatory Mechanism of MAP Kinases Activation and Nuclear Translocation Mediated by PKA and the PTP-SL Tyrosine Phosphatase. J. Cell Biol. 1999, 147, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Gotoh, I.; Adachi, M.; Gotoh, Y.; Nishida, E. A Novel Regulatory Mechanism in the Mitogen-Activated Protein (MAP) Kinase Cascade. Role of Nuclear Export Signal of MAP Kinase Kinase. J. Biol. Chem. 1997, 272, 32642–32648. [Google Scholar] [CrossRef]
- Lenormand, P.; Sardet, C.; Pages, G.; L’Allemain, G.; Brunet, A.; Pouyssegur, J. Growth Factors Induce Nuclear Translocation of MAP Kinases (P42(Mapk) and P44(Mapk)) but Not Their Activator MAP Kinase Kinase (P45(Mapkk)) in Fibroblasts. J. Cell Biol. 1993, 122, 1079–1088. [Google Scholar] [CrossRef]
- Yoon, S.; Seger, R. The Extracellular Signal-Regulated Kinase: Multiple Substrates Regulate Diverse Cellular Functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Ünal, E.B.; Uhlitz, F.; Blüthgen, N. A Compendium of ERK Targets. FEBS Lett. 2017, 591, 2607–2615. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-Independent PARP-1 Activation by Phosphorylated ERK2 Increases Elk1 Activity: A Link to Histone Acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Steelman, L.S.; Shelton, J.G.; Lee, J.T.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.; McCubrey, J.A. Regulation of Cell Cycle Progression and Apoptosis by the Ras/Raf/MEK/ERK Pathway (Review). Int. J. Oncol. 2003, 22, 469–480. [Google Scholar] [PubMed]
- Lichtenstein, M.P.; Carriba, P.; Baltrons, M.A.; Wojciak-Stothard, B.; Peterson, J.R.; García, A.; Galea, E. Secretase-Independent and RhoGTPase/PAK/ERK-Dependent Regulation of Cytoskeleton Dynamics in Astrocytes by NSAIDs and Derivatives. J. Alzheimer’s Dis. 2010, 22, 1135–1155. [Google Scholar] [CrossRef] [PubMed]
- Fincham, V.J.; James, M.; Frame, M.C.; Winder, S.J. Active ERK/MAP Kinase Is Targeted to Newly Forming Cell-Matrix Adhesions by Integrin Engagement and v-Src. EMBO J. 2000, 19, 2911–2923. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.; Chou, F.L.; Glading, A.; Schaefer, E.; Ginsberg, M.H. Phosphorylation of Phosphoprotein Enriched in Astrocytes (PEA-15) Regulates Extracellular Signal-Regulated Kinase-Dependent Transcription and Cell Proliferation. Mol. Biol. Cell 2005, 16, 3552–3561. [Google Scholar] [CrossRef]
- Glading, A.; Überall, F.; Keyse, S.M.; Lauffenburger, D.A.; Wells, A. Membrane Proximal ERK Signaling Is Required for M-Calpain Activation Downstream of Epidermal Growth Factor Receptor Signaling. J. Biol. Chem. 2001, 276, 23341–23348. [Google Scholar] [CrossRef]
- Teis, D.; Taub, N.; Kurzbauer, R.; Hilber, D.; De Araujo, M.E.; Erlacher, M.; Offterdinger, M.; Villunger, A.; Geley, S.; Bohn, G.; et al. P14-MP1-MEK1 Signaling Regulates Endosomal Traffic and Cellular Proliferation during Tissue Homeostasis. J. Cell Biol. 2006, 175, 861–868. [Google Scholar] [CrossRef]
- Acharya, U.; Mallabiabarrena, A.; Acharya, J.K.; Malhotra, V. Signaling via Mitogen-Activated Protein Kinase Kinase (MEK1) Is Required for Golgi Fragmentation during Mitosis. Cell 1998, 92, 183–192. [Google Scholar] [CrossRef]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK Signaling Promotes Site-Specific Ribosomal Protein S6 Phosphorylation via RSK and Stimulates Cap-Dependent Translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef]
- Stefanovsky, V.Y.; Moss, T. The Splice Variants of UBF Differentially Regulate RNA Polymerase I Transcription Elongation in Response to ERK Phosphorylation. Nucleic Acids Res. 2008, 36, 5093–5101. [Google Scholar] [CrossRef]
- Ray, L.B.; Sturgill, T.W. Characterization of Insulin-Stimulated Microtubule-Associated Protein Kinase. Rapid Isolation and Stabilization of a Novel Serine/Threonine Kinase from 3T3-L1 Cells. J. Biol. Chem. 1988, 263, 12721–12727. [Google Scholar] [CrossRef]
- Mukhopadhyay, N.K.; Price, D.J.; Kyriakis, J.M.; Pelech, S.; Sanghera, J.; Avruch, J. An Array of Insulin-Activated, Proline-Directed Serine/Threonine Protein Kinases Phosphorylate the P70 S6 Kinase. J. Biol. Chem. 1992, 267, 3325–3335. [Google Scholar] [CrossRef]
- Lin, L.L.; Wartmann, M.; Lin, A.Y.; Knopf, J.L.; Seth, A.; Davis, R.J. CPLA2 Is Phosphorylated and Activated by MAP Kinase. Cell 1993, 72, 269–278. [Google Scholar] [CrossRef]
- Hoffmann, R.; Baillie, G.S.; MacKenzie, S.J.; Yarwood, S.J.; Houslay, M.D. The MAP Kinase ERK2 Inhibits the Cyclic AMP-Specific Phosphodiesterase HSPDE4D3 by Phosphorylating It at Ser579. EMBO J. 1999, 18, 893–903. [Google Scholar] [CrossRef]
- Der, C.J.; Krontiris, T.G.; Cooper, G.M. Transforming Genes of Human Bladder and Lung Carcinoma Cell Lines Are Homologous to the Ras Genes of Harvey and Kirsten Sarcoma Viruses. Proc. Natl. Acad. Sci. USA 1982, 79, 3637–3640. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.J. An Unidentified Virus Which Causes the Rapid Production of Tumours in Mice. Nature 1964, 204, 1104–1105. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef]
- Fernández-Medarde, A.; Santos, E. Ras in Cancer and Developmental Diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef]
- Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 5 September 2021).
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The Role of BRAF V600 Mutation in Melanoma. J. Transl. Med. 2012, 10. [Google Scholar] [CrossRef]
- Bedognetti, D.; Roelands, J.; Decock, J.; Wang, E.; Hendrickx, W. The MAPK Hypothesis: Immune-Regulatory Effects of MAPK-Pathway Genetic Dysregulations and Implications for Breast Cancer Immunotherapy. Emerg. Top. Life Sci. 2017, 1, 429–445. [Google Scholar]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF Inhibition Is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, B.; Hill, C.E.; Pollack, B.P. Vemurafenib Enhances MHC Induction in BRAFV600E Homozygous Melanoma Cells. Oncoimmunology 2013, 2, e22890. [Google Scholar] [CrossRef] [PubMed]
- Wilmott, J.S.; Long, G.V.; Howle, J.R.; Haydu, L.E.; Sharma, R.N.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Scolyer, R.A. Selective BRAF Inhibitors Induce Marked T-Cell Infiltration into Human Metastatic Melanoma. Clin. Cancer Res. 2012, 18, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF Inhibition Increases Tumor Infiltration by T Cells and Enhances the Antitumor Activity of Adoptive Immunotherapy in Mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK Signaling Pathway Is Essential for Cancer-Immune Evasion in Human Melanoma Cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved Antitumor Activity of Immunotherapy with BRAF and MEK Inhibitors in BRAFV600E Melanoma. Sci. Transl. Med. 2015, 7, 279. [Google Scholar] [CrossRef]
- Knight, D.A.; Ngiow, S.F.; Li, M.; Parmenter, T.; Mok, S.; Cass, A.; Haynes, N.M.; Kinross, K.; Yagita, H.; Koya, R.C.; et al. Host Immunity Contributes to the Anti-Melanoma Activity of BRAF Inhibitors. J. Clin. Investig. 2015, 126, 402–403. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Frederick, D.T.; Juneja, V.R.; Sullivan, R.J.; Lawrence, D.P.; Piris, A.; Sharpe, A.H.; Fisher, D.E.; Flaherty, K.T.; Wargo, J.A. BRAF Inhibition Is Associated with Increased Clonality in Tumor-Infiltrating Lymphocytes. Oncoimmunology 2013, 2, e26615. [Google Scholar] [CrossRef]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E Inhibition Enhances T-Cell Recognition of Melanoma without Affecting Lymphocyte Function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Pasam, A.; Dimopoulos, N.; Andrews, M.; Knights, A.; Puaux, A.L.; Louahed, J.; Chen, W.; Woods, K.; Cebon, J.S. MEK Inhibition, Alone or in Combination with BRAF Inhibition, Affects Multiple Functions of Isolated Normal Human Lymphocytes and Dendritic Cells. Cancer Immunol. Res. 2014, 2, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Dushyanthen, S.; Teo, Z.L.; Caramia, F.; Savas, P.; Mintoff, C.P.; Virassamy, B.; Henderson, M.A.; Luen, S.J.; Mansour, M.; Kershaw, M.H.; et al. Agonist Immunotherapy Restores T Cell Function Following MEK Inhibition Improving Efficacy in Breast Cancer. Nat. Commun. 2017, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lieskovan, S.; Robert, L.; Moreno, B.H.; Ribas, A. Combining Targeted Therapy with Immunotherapy in BRAF-Mutant Melanoma: Promise and Challenges. J. Clin. Oncol. 2014, 32, 2248–2254. [Google Scholar] [CrossRef]
- Homet Moreno, B.; Mok, S.; Comin-Anduix, B.; Hu-Lieskovan, S.; Ribas, A. Combined Treatment with Dabrafenib and Trametinib with Immune-Stimulating Antibodies for BRAF Mutant Melanoma. Oncoimmunology 2016, 5, e1052212. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Liu, Y.; Gray, N.S. Rational Design of Inhibitors That Bind to Inactive Kinase Conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Whittaker, S.; Kirk, R.; Hayward, R.; Zambon, A.; Viros, A.; Cantarino, N.; Affolter, A.; Nourry, A.; Niculescu-Duvaz, D.; Springer, C.; et al. Gatekeeper Mutations Mediate Resistance to BRAF-Targeted Therapies. Sci. Transl. Med. 2010, 2, 35–41. [Google Scholar] [CrossRef]
- Tseng, J.R.; Stuart, D.; Aardalen, K.; Kaplan, A.; Aziz, N.; Hughes, N.P.; Gambhir, S.S. Use of DNA Microarray and Small Animal Positron Emission Tomography in Preclinical Drug Evaluation of RAF265, a Novel B-Raf/VEGFR-2 Inhibitor. Neoplasia 2011, 13, 266–275. [Google Scholar] [CrossRef]
- Sharfman, W.H.; Hodi, F.S.; Lawrence, D.P.; Flaherty, K.T.; Amaravadi, R.K.; Kim, K.B.; Dummer, R.; Gobbi, S.; Puzanov, I.; Sosman, J.A.; et al. Results from the First-in-Human (FIH) Phase I Study of the Oral RAF Inhibitor RAF265 Administered Daily to Patients with Advanced Cutaneous Melanoma. J. Clin. Oncol. 2011, 29, 8508. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical Efficacy of a RAF Inhibitor Needs Broad Target Blockade in BRAF-Mutant Melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Cseh, B.; Doma, E.; Baccarini, M. “rAF” Neighborhood: Protein-Protein Interaction in the Raf/Mek/Erk Pathway. FEBS Lett. 2014, 588, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a Selective Inhibitor of Oncogenic B-Raf Kinase with Potent Antimelanoma Activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Shah, S.R.; Illum, H.; Dowell, J. Vemurafenib: Targeted Inhibition of Mutated BRAF for Treatment of Advanced Melanoma and Its Potential in Other Malignancies. Drugs 2012, 72, 2207–2222. [Google Scholar] [CrossRef]
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Go, Z.; Iyer, R.; Kolis, S.; Zhao, S.; Lee, R.; Grippo, J.F.; et al. RG7204 (PLX4032), a Selective BRAFV600E Inhibitor, Displays Potent Antitumor Activity in Preclinical Melanoma Models. Cancer Res. 2010, 70, 5518–5527. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and Efficacy of Vemurafenib in BRAFV600E and BRAFV600K Mutation-Positive Melanoma (BRIM-3): Extended Follow-up of a Phase 3, Randomised, Open-Label Study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Kim, G.; McKee, A.E.; Ning, Y.M.; Hazarika, M.; Theoret, M.; Johnson, J.R.; Xu, Q.C.; Tang, S.; Sridhara, R.; Jiang, X.; et al. FDA Approval Summary: Vemurafenib for Treatment of Unresectable or Metastatic Melanoma with the BRAFV600E Mutation Mutation. Clin. Cancer Res. 2014, 20, 4994–5000. [Google Scholar] [CrossRef] [PubMed]
- Roche Roche’s Zelboraf Receives EU Approval for the Treatment of People with Deadly Form of Skin Cancer. 2012. Available online: https://www.roche.com/media/releases/med-cor-2012-02-20.htm (accessed on 2 February 2012).
- McClure, E.; Carr, M.J.; Zager, J.S. The MAP Kinase Signal Transduction Pathway: Promising Therapeutic Targets Used in the Treatment of Melanoma. Expert Rev. Anticancer Ther. 2020, 20, 687–701. [Google Scholar] [CrossRef] [PubMed]
- GlaxoSmithKline Two New GSK Oral Oncology Treatments, BRAF-Inhibitor Tafinlar®(Dabrafenib) Capsules and the First MEK-Inhibitor MekinistTM (Trametinib) Tablets, Approved by FDA as Single-Agent Therapies. 2013. Available online: https://www.gsk.com/en-gb/media/press-releases/two-new-gsk-oral-oncology-treatments-braf-inhibitor-tafinlar-dabrafenib-capsules-and-the-first-mek-inhibitor-mekinist-trametinib-tablets-approved-by-fda-as-single-agent-therapies/ (accessed on 15 May 2013).
- Gibney, G.T.; Zager, J.S. Clinical Development of Dabrafenib in BRAF Mutant Melanoma and Other Malignancies. Expert Opin. Drug Metab. Toxicol. 2013, 9, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Laquerre, S.; Arnone, M.; Moss, K.; Yang, J.; Fisher, K.; Kane-Carson, L.S.; Smitheman, K.; Ward, J.; Heidrich, B.; Rheault, T.; et al. Abstract B88: A Selective Raf Kinase Inhibitor Induces Cell Death and Tumor Regression of Human Cancer Cell Lines Encoding B-RafV600E Mutation. Mol. Cancer Ther. 2009, 8, b88. [Google Scholar] [CrossRef]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in Patients with Melanoma, Untreated Brain Metastases, and Other Solid Tumours: A Phase 1 Dose-Escalation Trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.J.; et al. Phase II Trial (BREAK-2) of the BRAF Inhibitor Dabrafenib (GSK2118436) in Patients with Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Long, G.V.; Trefzer, U.; Davies, M.A.; Kefford, R.F.; Ascierto, P.A.; Chapman, P.B.; Puzanov, I.; Hauschild, A.; Robert, C.; Algazi, A.; et al. Dabrafenib in Patients with Val600Glu or Val600Lys BRAF-Mutant Melanoma Metastatic to the Brain (BREAK-MB): A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2012, 13, 1087–1095. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Mirakhur, B.; Guckert, M.E.; et al. Phase III, Randomized, Open-Label, Multicenter Trial (BREAK-3) Comparing the BRAF Kinase Inhibitor Dabrafenib (GSK2118436) with Dacarbazine (DTIC) in Patients with BRAFV600E-Mutated Melanoma. J. Clin. Oncol. 2012, 30, LBA8500. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-Mutated Metastatic Melanoma: A Multicentre, Open-Label, Phase 3 Randomised Controlled Trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus Binimetinib versus Vemurafenib or Encorafenib in Patients with BRAF-Mutant Melanoma (COLUMBUS): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [PubMed]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF Mutation Predicts Sensitivity to MEK Inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A Randomised, Double-Blind, Placebo-Controlled Trial of Trametinib, an Oral MEK Inhibitor, in Combination with Gemcitabine for Patients with Untreated Metastatic Adenocarcinoma of the Pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. 2013. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000TOC.cfm (accessed on 23 July 2013).
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained in Vivo Pathway Inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, B.; Liu, D.; Shen, R.; Yan, Y.; Yang, L.; Zhang, M.; Zhang, L.; Cao, G.; Cao, H.; et al. EBI-907, a Novel BRAFV600E Inhibitor, Has Potent Oral Anti-Tumor Activity and a Broad Kinase Selectivity Profile. Cancer Biol. Ther. 2016, 17, 199–207. [Google Scholar] [CrossRef]
- Bedogni, B.; Welford, S.M.; Kwan, A.C.; Ranger-Moore, J.; Saboda, K.; Powell, M.B. Inhibition of Phosphatidylinositol-3-Kinase and Mitogen-Activated Protein Kinase Kinase 1/2 Prevents Melanoma Development and Promotes Melanoma Regression in the Transgenic TPRas Mouse Model. Mol. Cancer Ther. 2006, 5, 3071–3077. [Google Scholar] [CrossRef]
- Menzies, A.M.; Long, G. V Dabrafenib and Trametinib, Alone and in Combination for BRAF-Mutant Metastatic Melanoma. Clin. Cancer Res. 2014, 20, 2035–2043. [Google Scholar]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Approves COTELLICTM (Cobimetinib) for Use in Combination with Vemurafenib to Treat Advanced Melanoma. 2015. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206192Orig1s000PharmR.pdf (accessed on 1 May 2015).
- U.S. Food and Drug Administration. FDA Approves Encorafenib and Binimetinib in Combination for Unresectable or Metastatic Melanoma with BRAF Mutations. 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-encorafenib-and-binimetinib-combination-unresectable-or-metastatic-melanoma-braf (accessed on 21 June 2018).
- Atefi, M.; Titz, B.; Avramis, E.; Ng, C.; Wong, D.J.L.; Lassen, A.; Cerniglia, M.; Escuin-Ordinas, H.; Foulad, D.; Comin-Anduix, B.; et al. Combination of Pan-RAF and MEK Inhibitors in NRAS Mutant Melanoma. Mol. Cancer 2015, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Trojaniello, C.; Vitale, M.G.; Ascierto, P.A. Triplet Combination of BRAF, MEK and PD-1/PD-L1 Blockade in Melanoma: The More the Better? Curr. Opin. Oncol. 2021, 33, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Ahronian, L.G.; Sennott, E.M.; Van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Guo, W. A Review of the Molecular Pathways Involved in Resistance to BRAF Inhibitors in Patients with Advanced-Stage Melanoma. Med. Sci. Monit. 2020, 26, e920957. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad Targeting of Resistance to Apoptosis in Cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef] [PubMed]
- Borst, A.; Haferkamp, S.; Grimm, J.; Rösch, M.; Zhu, G.; Guo, S.; Li, C.; Gao, T.; Meierjohann, S.; Schrama, D.; et al. BIK Is Involved in BRAF/MEK Inhibitor Induced Apoptosis in Melanoma Cell Lines. Cancer Lett. 2017, 404, 70–78. [Google Scholar] [CrossRef]
- Grossman, D.; Altieri, D.C. Drug Resistance in Melanoma: Mechanisms, Apoptosis, and New Potential Therapeutic Targets. Cancer Metastasis Rev. 2001, 20, 3–11. [Google Scholar] [CrossRef]
- Lai, F.; Jin, L.; Gallagher, S.; Mijatov, B.; Zhang, X.D.; Hersey, P. Histone Deacetylases (HDACs) as Mediators of Resistance to Apoptosis in Melanoma and as Targets for Combination Therapy with Selective BRAF Inhibitors. Adv. Pharmacol. 2012, 65, 27–43. [Google Scholar]
- Han, Y.; Fang, J.; Xiao, Z.; Deng, J.; Zhang, M.; Gu, L. Downregulation of LncRNA TSLNC8 Promotes Melanoma Resistance to BRAF Inhibitor PLX4720 through Binding with PP1α to Re-Activate MAPK Signaling. J. Cancer Reh. Clin. Oncol. 2021, 147, 767–777. [Google Scholar] [CrossRef]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of CRAF-Mediated MEK Activation Is Required for Effective Mek Inhibition in KRAS Mutant Tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef]
- Little, A.S.; Smith, P.D.; Cook, S.J. Mechanisms of Acquired Resistance to ERK1/2 Pathway Inhibitors. Oncogene 2013, 32, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Goetz, E.M.; Ghandi, M.; Treacy, D.J.; Wagle, N.; Garraway, L.A. ERK Mutations Confer Resistance to Mitogen-Activated Protein Kinase Pathway Inhibitors. Cancer Res. 2014, 74, 7079–7089. [Google Scholar] [CrossRef] [PubMed]
- Babagana, M.; Johnson, S.; Slabodkin, H.; Bshara, W.; Morrison, C.; Kandel, E.S. P21-Activated Kinase 1 Regulates Resistance to BRAF Inhibition in Human Cancer Cells. Mol. Carcinog. 2017, 56, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Pelster, M.S.; Amaria, R.N. Combined Targeted Therapy and Immunotherapy in Melanoma: A Review of the Impact on the Tumor Microenvironment and Outcomes of Early Clinical Trials. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Dummer, R. Immunological Effects of BRAF+MEK Inhibition. Oncoimmunology 2018, 7, e1468955. [Google Scholar] [CrossRef]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of Patients with Advanced Metastatic Melanoma: The Impact of Novel Therapies–Update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Juneja, V.R.; Sage, P.T.; Frederick, D.T.; Piris, A.; Mitra, D.; Lo, J.A.; Hodi, F.S.; Freeman, G.J.; Bosenberg, M.W.; et al. Responseto BRAF Inhibitionin Melanoma Is Enhanced When Combined with Immune Checkpoint Blockade. Cancer Immunol. Res. 2014, 2, 643–654. [Google Scholar] [CrossRef]
- Deken, M.A.; Gadiot, J.; Jordanova, E.S.; Lacroix, R.; van Gool, M.; Kroon, P.; Pineda, C.; Geukes Foppen, M.H.; Scolyer, R.; Song, J.Y.; et al. Targeting the MAPK and PI3K Pathways in Combination with PD1 Blockade in Melanoma. Oncoimmunology 2016, 5, e1238557. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Database of Privately and Publicly Funded Clinical Studies Conducted around the World. Available online: https://clinicaltrials.gov/ (accessed on 1 September 2021).
- Ribas, A.; Hodi, F.S.; Callahan, M.; Konto, C.; Wolchok, J. Hepatotoxicity with Combination of Vemurafenib and Ipilimumab. N. Engl. J. Mede 2013, 368, 1365–1366. [Google Scholar] [CrossRef]
- Ribas, A.; Hodi, F.S.; Kurland, J.F.; Shahabi, V.; Francis, S.; Konto, C.K.; Joe, A.; Lainas, I.; Wolchok, J. CA184-161: A Phase I/II Trial of Vemurafenib and Ipilimumab in Patients with BRAF V600 Mutation-Positive Metastatic Melanoma. J. Clin. Oncol. 2012, 30. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Gonzalez, R.; Lewis, K.D.; Hamid, O.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Wallin, J.; Pitcher, B.; Cha, E.; et al. Atezolizumab (A) + Cobimetinib (C) + Vemurafenib (V) in BRAF V600 -Mutant Metastatic Melanoma (Mel): Updated Safety and Clinical Activity. J. Clin. Oncol. 2017, 35, 3063. [Google Scholar] [CrossRef]
- Ribas, A.; Hodi, F.S.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Carlino, M.S.; Fisher, R.; Long, G.V.; Miller, W.H.; Huang, Y.; et al. KEYNOTE-022 Update: Phase 1 Study of First-Line Pembrolizumab (Pembro) plus Dabrafenib (D) and Trametinib (T) for BRAF-Mutant Advanced Melanoma. Ann. Oncol. 2017, 28, v430. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Stephens, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. KEYNOTE-022 Part 3: Phase II Randomized Study of 1L Dabrafenib (D) and Trametinib (T) plus Pembrolizumab (Pembro) or Placebo (PBO) for BRAF-Mutant Advanced Melanoma. Ann. Oncol. 2018, 29, viii442. [Google Scholar] [CrossRef]
- Ribas, A.; Butler, M.; Lutzky, J.; Lawrence, D.P.; Robert, C.; Miller, W.; Linette, G.P.; Ascierto, P.A.; Kuzel, T.; Algazi, A.P.; et al. Phase I Study Combining Anti-PD-L1 (MEDI4736) with BRAF (Dabrafenib) and/or MEK (Trametinib) Inhibitors in Advanced Melanoma. J. Clin. Oncol 2015, 33, 3003. [Google Scholar] [CrossRef]
- Burton, E.M.; Amaria, R.N.; Glitza, I.C.; Shephard, M.; Diab, A.; Milton, D.; Patel, S.; Mcquade, J.; Wong, M.; Hwu, P.; et al. Safety and Efficacy of TRIplet Combination of Nivolumab (N) with Dabrafenib (D) and Trametinib (T) [TRIDeNT] in Patients (Pts) with BRAF-Mutated Metastatic Melanoma (MM): A Single Center Phase II Study. Ann. Oncol. 2019, 30, v534–v535. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, Vemurafenib, and Cobimetinib as First-Line Treatment for Unresectable Advanced BRAFV600 Mutation-Positive Melanoma (IMspire150): Primary Analysis of the Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Approves Atezolizumab for BRAF V600 Unresectable or Metastatic Melanoma. 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-braf-v600-unresectable-or-metastatic-melanoma (accessed on 1 July 2020).
- Novartis Novartis Reports Late-Breaking Data from Phase III COMBI-i Trial of Spartalizumab (PDR001) with Tafinlar®and Mekinist®in Advanced Melanoma. 2020. Available online: https://www.novartis.com/news/media-releases/novartis-reports-late-breaking-data-from-phase-iii-combi-i-trial-spartalizumab-pdr001-tafinlar-and-mekinist-advanced-melanoma (accessed on 16 September 2020).
- Gogas, H.; Dréno, B.; Larkin, J.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Francesco Ferrucci, P.; Pigozzo, J.; Rutkowski, P.; Mackiewicz, J.; et al. Cobimetinib plus Atezolizumab in BRAFV600 Wild-Type Melanoma: Primary Results from the Randomized Phase III IMspire170 Study. Ann. Oncol. 2021, 32, 384–394. [Google Scholar] [CrossRef]
- Simeone, E.; Grimaldi, A.M.; Festino, L.; Giannarelli, D.; Vanella, V.; Palla, M.; Curvietto, M.; Esposito, A.; Palmieri, G.; Mozzillo, N.; et al. Correlation between Previous Treatment with BRAF Inhibitors and Clinical Response to Pembrolizumab in Patients with Advanced Melanoma. Oncoimmunology 2017, 6, e1283462. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Munhoz, R.R.; Kuk, D.; Ott, P.A.; Johnson, D.B.; Tsai, K.K.; Rapisuwon, S.; Eroglu, Z.; Sullivan, R.J.; Luke, J.J.; et al. The Efficacy of Anti-PD-1 Agents in Acral and Mucosal Melanoma. Cancer 2016, 122, 3354–3362. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Mandala, M.; Ferrucci, P.F.; Rutkowski, P.; Guidoboni, M.; Fernandez, A.M.A.; Ferraresi, V.; Maiello, E.; Guida, M.; Del Vecchio, M.; et al. LBA45 First Report of Efficacy and Safety from the Phase II Study SECOMBIT (SEquential COMBo Immuno and Targeted Therapy Study). Ann. Oncol. 2020, 31, s1173–s1174. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| NTC Number | Phase | Targeted Therapy | Immunotherapy | Conditions | Status |
|---|---|---|---|---|---|
| NCT04835805 | I | Belvarafenib, Cobimetinib | Atezolizumab | Advanced Melanoma | Recruiting |
| NCT04722575 | II | Cobimetinib, Vemurafenib | Atezolizumab | Resectable Melanoma | Recruiting |
| NCT01400451 | I | Vemurafenib | Ipilimumab | Advanced melanoma | Terminated |
| NCT01673854 | II | Vemurafenib | Ipilimumab | Advanced melanoma | Completed |
| NCT02200562 | I | Dabrafenib | Ipilimumab | Stage III/IV Melanoma | Terminated |
| NCT02130466 | I/II | Dabrafenib, Trametinib | Pembrolizumab | Advanced Melanoma | Completed |
| NCT03149029 | II | Dabrafenib, Trametinib | Pembrolizumab | Metastatic Melanoma | Active, not recruiting |
| NCT02858921 | II | Dabrafenib, Trametinib | Pembrolizumab | Stage III Melanoma | Active, not recruiting |
| NCT02625337 | II | Dabrafenib, Trametinib | Pembrolizumab | Metastatic Melanoma | Unknown status |
| NCT02027961 | I | Dabrafenib, Trametinib | Durvalumab | Advanced melanoma | Completed |
| NCT02910700 | II | Dabrafenib, Trametinib | Nivolumab | Stage III/IV Metastatic Melanoma | Recruiting |
| NCT02908672 | III | Cobimetinib, Vemurafenib | Atezolizumab | Metastatic or Unresectable Melanoma | Active, not recruiting |
| NCT03273153 | III | Cobimetinib | Atezolizumab, Pembrolizumab | Advanced Melanoma | Completed |
| NCT02967692 | III | Dabrafenib, Trametinib | Spartalizumab | Metastatic or Unresectable Melanoma | Active, not recruiting |
| NCT04657991 | III | Encorafenib, Binimetinib | Pembrolizumab | Melanoma | Recruiting |
| NCT04655157 | I/II | Encorafenib, Binimetinib | Nivolumab, Ipilimumab | Metastatic Melanoma | Recruiting |
| NCT04511013 | II | Encorafenib, Binimetinib | Nivolumab, Ipilimumab | Melanoma | Recruiting |
| NCT03554083 | II | Cobimetinib, Vemurafenib | Atezolizumab | Stage III Melanoma | Recruiting |
| NCT03235245 | II | Encorafenib, Binimetinib | Nivolumab, Ipilimumab | Unresectable Stage III/IV Melanoma | Recruiting |
| NCT03175432 | II | Cobimetinib | Atezolizumab, Bevacizumab | Untreated Melanoma Brain Metastases | Recruiting |
| NCT02902029 | II | Cobimetinib, Vemurafenib | Atezolizumab | Metastatic or Unresectable Melanoma | Active, not recruiting |
| NCT02818023 | I | Cobimetinib, Vemurafenib | Pembrolizumab | Advanced Melanoma | Terminated |
| NCT02303951 | II | Cobimetinib, Vemurafenib | Atezolizumab | Malignant Melanoma | Terminated |
| NCT01940809 | I | Dabrafenib, Trametinib | Nivolumab, Ipilimumab | Stage III/IV Metastatic Melanoma | Terminated |
| NCT01767454 | I | Dabrafenib, Trametinib | Ipilimumab | Metastatic or Unresectable Melanoma | Completed |
| NCT01656642 | I | Cobimetinib, Vemurafenib | Atezolizumab | Metastatic Melanoma | Completed |
| NCT03178851 | I | Cobimetinib | Atezolizumab | Malignant Melanoma | Completed |
| NCT02902042 | I/II | Encorafenib, Binimetinib | Pembrolizumab | Malignant Melanoma | Completed |
| NCT02224781 | III | Dabrafenib, Trametinib | Ipilimumab, Nivolumab | Metastatic Melanoma | Active, not recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morante, M.; Pandiella, A.; Crespo, P.; Herrero, A. Immune Checkpoint Inhibitors and RAS–ERK Pathway-Targeted Drugs as Combined Therapy for the Treatment of Melanoma. Biomolecules 2022, 12, 1562. https://doi.org/10.3390/biom12111562
Morante M, Pandiella A, Crespo P, Herrero A. Immune Checkpoint Inhibitors and RAS–ERK Pathway-Targeted Drugs as Combined Therapy for the Treatment of Melanoma. Biomolecules. 2022; 12(11):1562. https://doi.org/10.3390/biom12111562
Chicago/Turabian StyleMorante, Marta, Atanasio Pandiella, Piero Crespo, and Ana Herrero. 2022. "Immune Checkpoint Inhibitors and RAS–ERK Pathway-Targeted Drugs as Combined Therapy for the Treatment of Melanoma" Biomolecules 12, no. 11: 1562. https://doi.org/10.3390/biom12111562
APA StyleMorante, M., Pandiella, A., Crespo, P., & Herrero, A. (2022). Immune Checkpoint Inhibitors and RAS–ERK Pathway-Targeted Drugs as Combined Therapy for the Treatment of Melanoma. Biomolecules, 12(11), 1562. https://doi.org/10.3390/biom12111562