
Increased Immunoglobulin and Proteoglycan Synthesis in Resected Hippocampal Tissue Predicts Post-Surgical Seizure Recurrence in Human Temporal Lobe Epilepsy


Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. RNASeq and Statistical Analyses
2.3. Pathway Literature Searches and Feature Selection
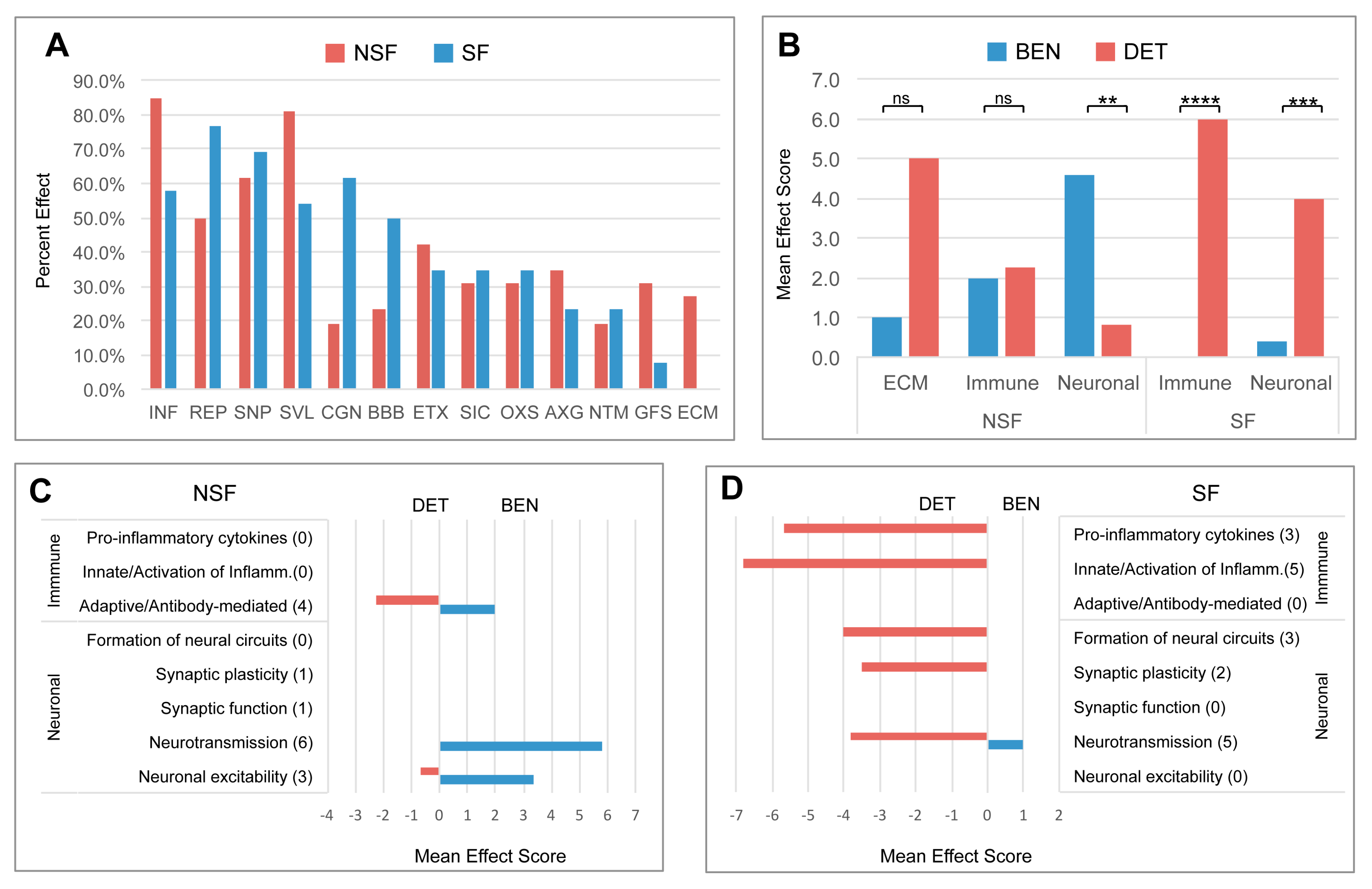
3. Results
3.1. Cohort Characteristics
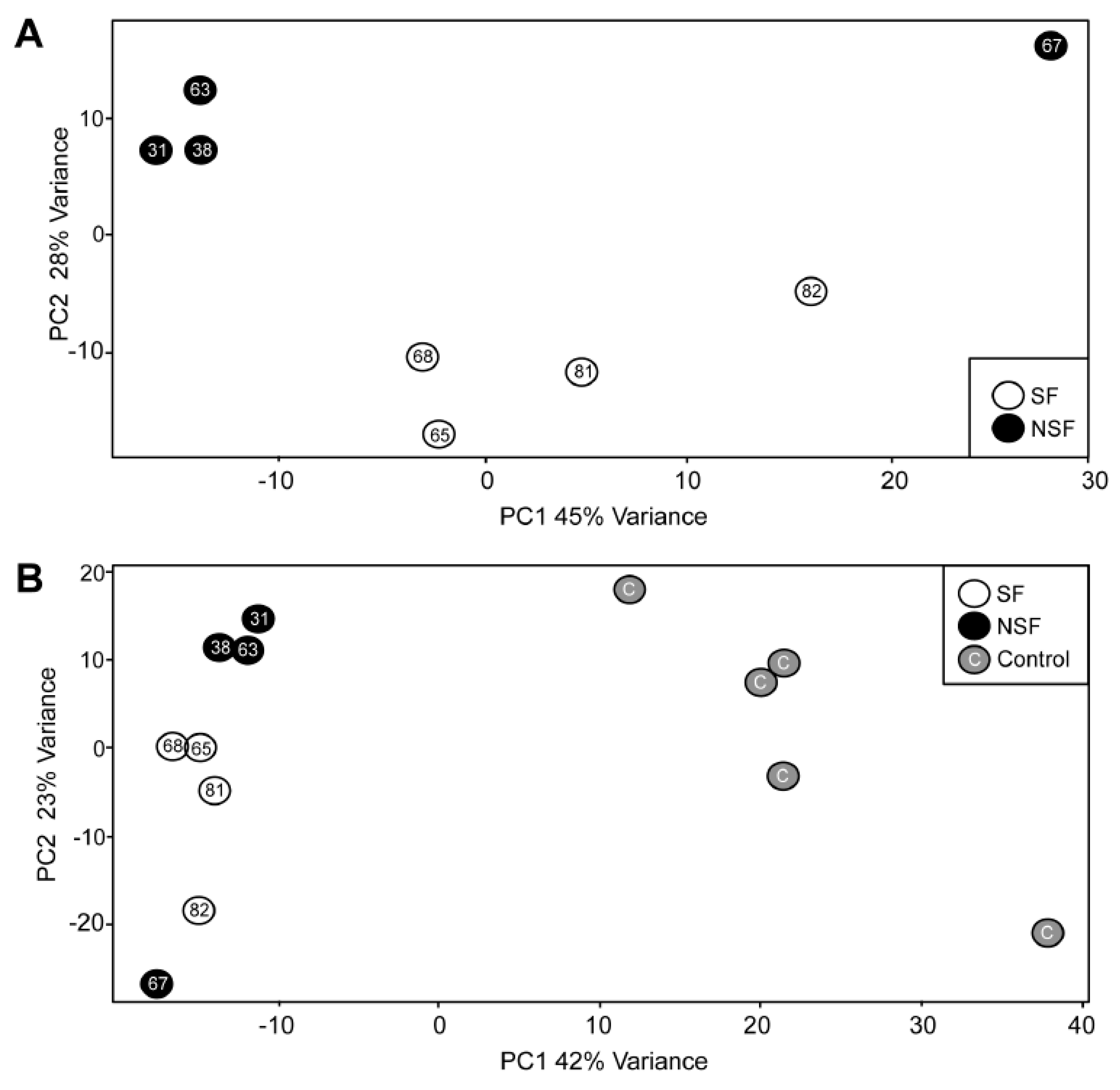
3.2. Principal Component Analysis
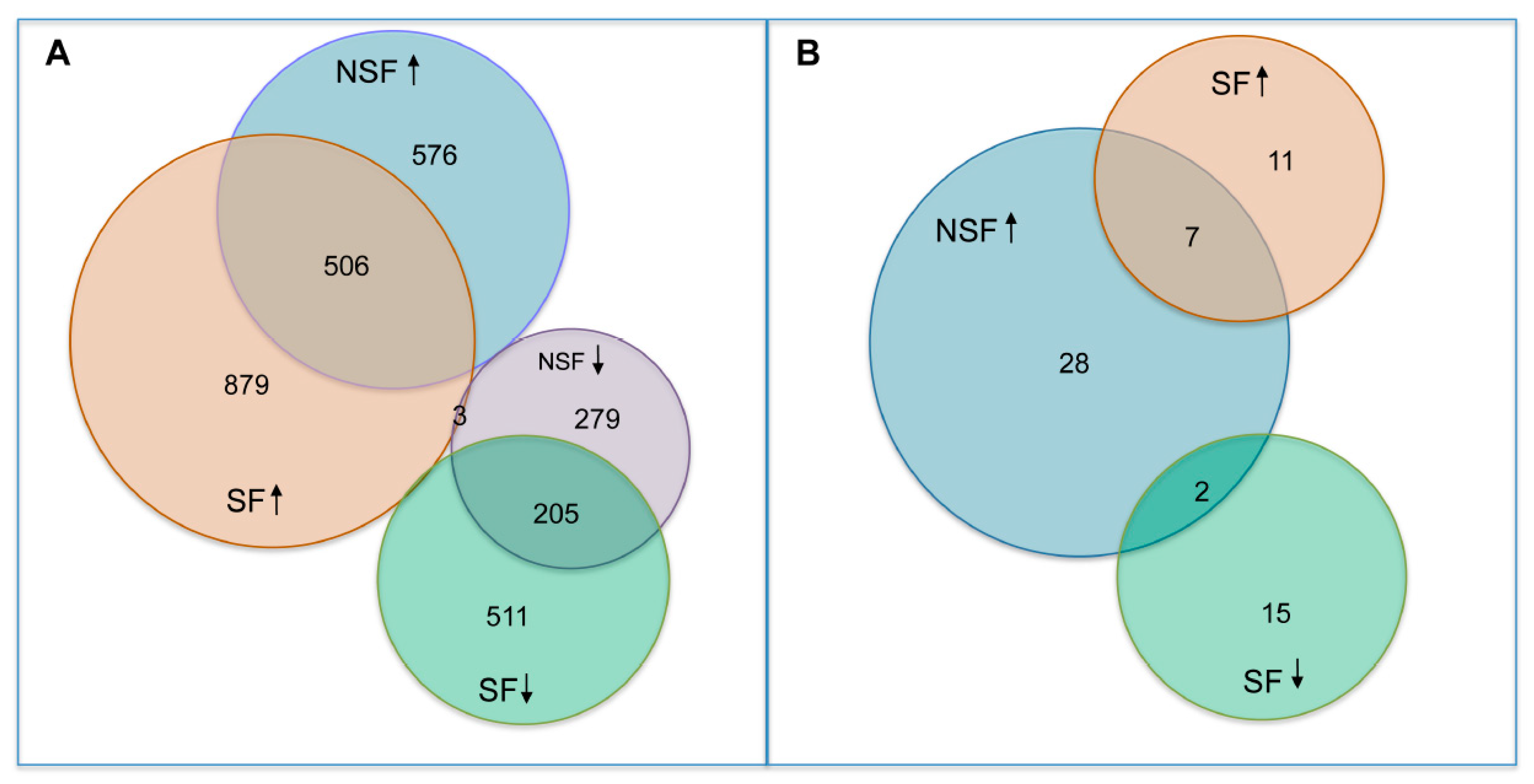
3.3. Differential Expression Analysis
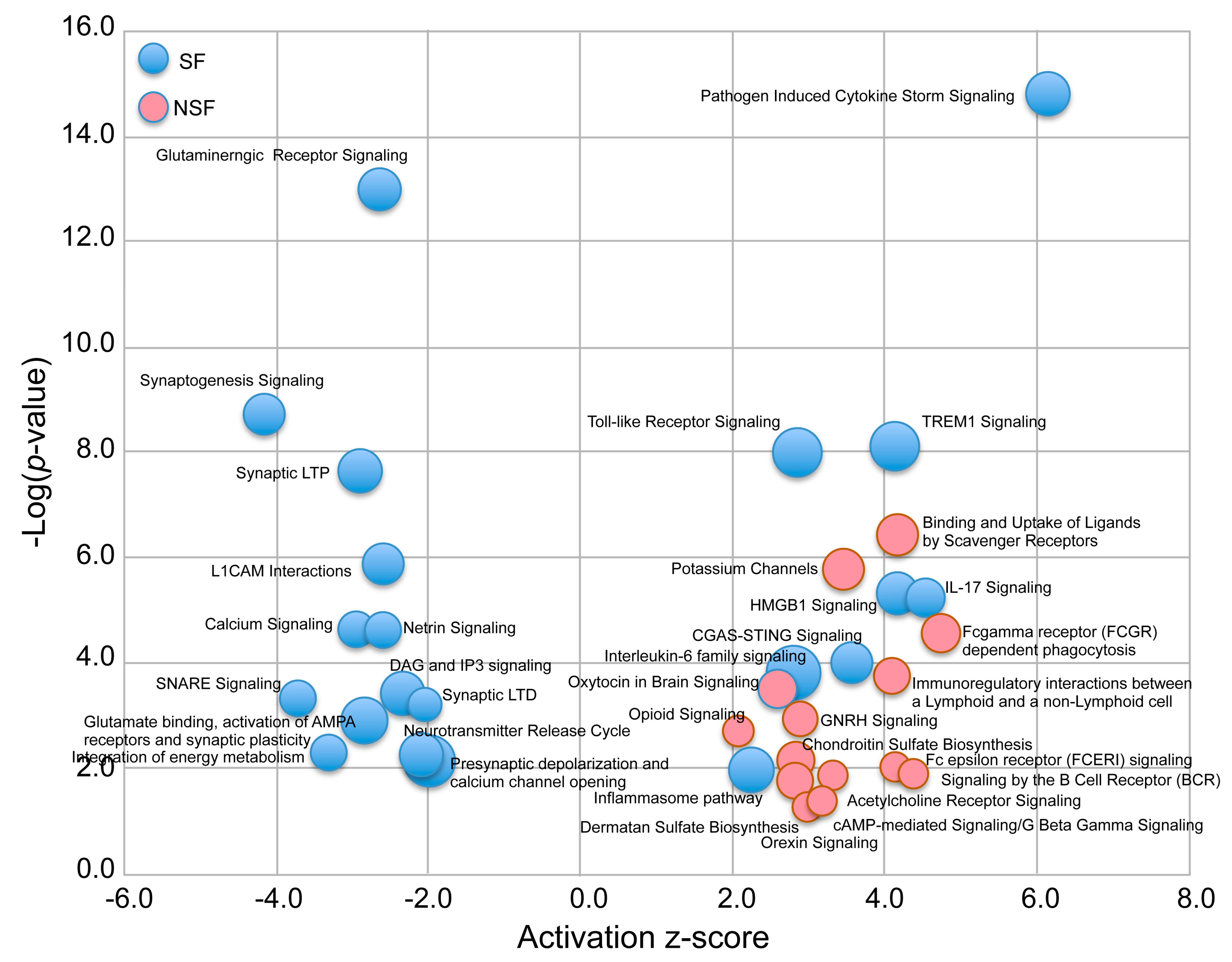
3.4. Canonical Pathways Altered in NSF and SF Cohorts
3.5. Hierarchical Pathway Categories
3.6. Predicted Upstream Regulators
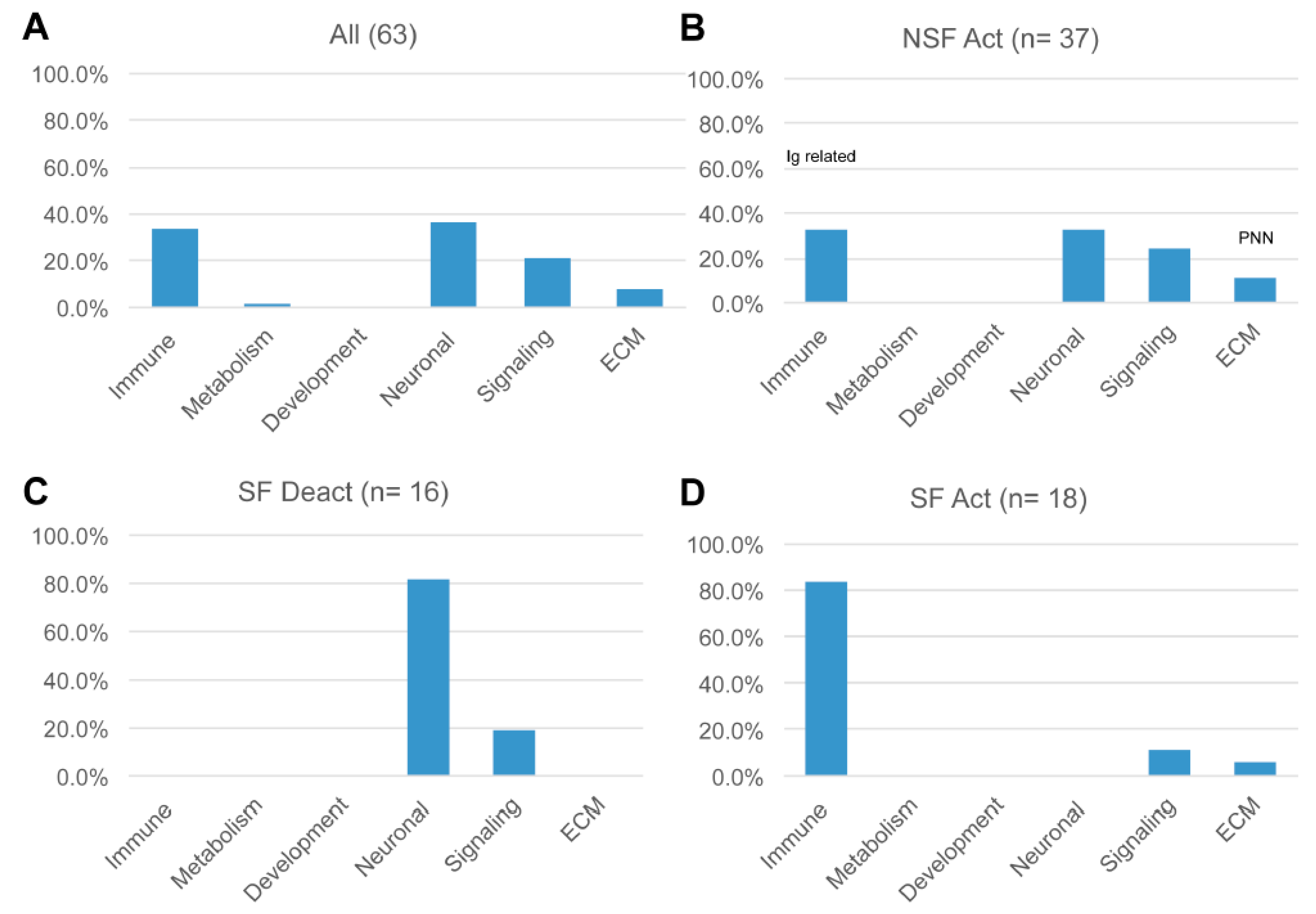
3.7. Pathway Effects
3.8. Divergent Immune System- and Neuronal System-Related Pathway Effects
4. Discussion
4.1. Shared Pathways Indicative of Common Processes in Epileptogenesis
4.2. Pathways Distinguishing NSF from SF
4.3. Increased Expression of Immunoglobulins in NSF
4.4. The Role of Increased Activation of Chondroitin and Dermatan Synthesis in NSF
4.5. What Factors Explain the Association of Increased Pro-Inflammatory Markers and Post-Surgery Seizure Freedom?
4.6. Implications for Pre- and Post-Surgery Surveillance
5. Conclusions and Limitations
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeBarros Lourence, F.H.; Marques, L.H.N.; DeAraujo Filho, G.M. Electroencephalogram alterations associated with psychiatric disorders in temporal lobe epilepsy with mesial sclerosis: A systematic review. Epilepsy Behav. 2020, 108, 107100. [Google Scholar] [CrossRef] [PubMed]
- Cascino, G.D. Temporal lobe epilepsy: More than hippocampal pathology. Epilepsy Curr. 2005, 5, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Panina, Y.S.; Timechko, E.E.; Usoltseva, A.A.; Yakovleva, K.D.; Kantimirova, E.A.; Dmitrenko, D.V. Biomarkers of Drug Resistance in Temporal Lobe Epilepsy in Adults. Metabolites 2023, 13, 83. [Google Scholar] [CrossRef]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathern, G.; Moshe, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Centanaro, M.; Solis, J.; Delgado, F.; Yepez, L. Factors predicting the outcome following medical treatment of mesial temporal epilepsy with hippocampal sclerosis. Seizure 2014, 23, 448–453. [Google Scholar] [CrossRef]
- Gallek, M.J.; Skoch, J.; Ansay, T.; Benbahani, M.; Mount, D.; Manziello, A.; Witte, M.; Bernas, M.; Labiner, D.M.; Weinand, M.E. Cortical gene expression: Prognostic value for seizure outcome following temporal lobectomy and amygdalohippocampectomy. Neurogenetics 2016, 17, 211–218. [Google Scholar] [CrossRef]
- Asadi-Pooya, A.A.; Rostami, C. History of surgery for temporal lobe epilepsy. Epilepsy Behav. 2017, 70, 57–60. [Google Scholar] [CrossRef]
- Donos, C.; Breier, J.; Friedman, E.; Rollo, P.; Johnson, J.; Moss, L.; Thompson, S.; Thomas, M.; Hope, O.; Slater, J.; et al. Laser ablation for mesial temporal lobe epilepsy: Surgical and cognitive outcomes with and without mesial temporal sclerosis. Epilepsia 2018, 59, 1421–1432. [Google Scholar] [CrossRef]
- Bjellvi, J.; Olsson, I.; Malmgren, K.; Wilbe Ramsay, K. Epilepsy duration and seizure outcome in epilepsy surgery: A systematic review and meta-analysis. Neurology 2019, 93, e159–e166. [Google Scholar] [CrossRef]
- Hemb, M.; Palmini, A.; Paglioli, E.; Paglioli, E.B.; Costa da Costa, J.; Azambuja, N.; Portuguez, M.; Viuniski, V.; Booij, L.; Nunes, M.L. An 18-year follow-up of seizure outcome after surgery for temporal lobe epilepsy and hippocampal sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 800–805. [Google Scholar] [CrossRef]
- Ivanovic, J.; Larsson, P.G.; Ostby, Y.; Hald, J.; Krossnes, B.K.; Fjeld, J.G.; Pripp, A.H.; Alfstad, K.A.; Egge, A.; Stanisic, M. Seizure outcomes of temporal lobe epilepsy surgery in patients with normal MRI and without specific histopathology. Acta Neurochir. 2017, 159, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Tomlinson, G.; Snead, C.; Sander, B.; Widjaja, E. Systematic review and network meta-analysis of resective surgery for mesial temporal lobe epilepsy. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1138–1144. [Google Scholar] [CrossRef]
- Widjaja, E.; Jain, P.; Demoe, L.; Guttmann, A.; Tomlinson, G.; Sander, B. Seizure outcome of pediatric epilepsy surgery: Systematic review and meta-analyses. Neurology 2020, 94, 311–321. [Google Scholar] [CrossRef]
- Jehi, L.; Yehia, L.; Peterson, C.; Niazi, F.; Busch, R.; Prayson, R.; Ying, Z.; Bingaman, W.; Najm, I.; Eng, C. Preliminary report: Late seizure recurrence years after epilepsy surgery may be associated with alterations in brain tissue transcriptome. Epilepsia Open 2018, 3, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.F.; Sprissler, R.; Bina, R.W.; Lau, B.; Johnstone, L.; Walter, C.M.; Labiner, D.M.; Weinand, M.E. Altered expression of signaling pathways regulating neuronal excitability in hippocampal tissue of temporal lobe epilepsy patients with low and high seizure frequency. Epilepsy Res. 2019, 155, 106145. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, C.E.; Louis, S.; Busch, R.M.; Vegh, D.; Najm, I.; Bazeley, P.; Eng, C.; Jehi, L.; Rotroff, D.M. Molecular subtypes of epilepsy associated with post-surgical seizure recurrence. Brain Commun. 2023, 5, fcad251. [Google Scholar] [CrossRef]
- Louis, S.; Busch, R.M.; Lal, D.; Hockings, J.; Hogue, O.; Morita-Sherman, M.; Vegh, D.; Najm, I.; Ghosh, C.; Bazeley, P.; et al. Genetic and molecular features of seizure-freedom following surgical resections for focal epilepsy: A pilot study. Front. Neurol. 2022, 13, 942643. [Google Scholar] [CrossRef]
- Fiala, M.; Avagyan, H.; Merino, J.J.; Bernas, M.; Valdivia, J.; Espinosa-Jeffrey, A.; Witte, M.; Weinand, M. Chemotactic and mitogenic stimuli of neuronal apoptosis in patients with medically intractable temporal lobe epilepsy. Pathophysiology 2013, 20, 59–69. [Google Scholar] [CrossRef]
- Sprissler, R.; Hammer, M.; Labiner, D.; Joshi, N.; Alan, A.; Weinand, M. Leukocyte differential gene expression prognostic value for high versus low seizure frequency in temporal lobe epilepsy. BMC Neurol. 2024, 24, 16. [Google Scholar] [CrossRef]
- Hwang, T.; Park, C.; Leung, A.; Gao, Y.; Hyde, T.; Kleinman, J.; Rajpurohit, A.; Tao, R.; Shin, J.; Weinberger, D. Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 2016, 19, 1093–1099. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Schurch, N.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, C.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Schetinger, M.R.; Morsch, V.M.; Bonan, C.D.; Wyse, A.T. NTPDase and 5′-nucleotidase activities in physiological and disease conditions: New perspectives for human health. Biofactors 2007, 31, 77–98. [Google Scholar] [CrossRef]
- Josephson, C.B.; Dykeman, J.; Fiest, K.M.; Liu, X.; Sadler, R.M.; Jette, N.; Wiebe, S. Systematic review and meta-analysis of standard vs selective temporal lobe epilepsy surgery. Neurology 2013, 80, 1669–1676. [Google Scholar] [CrossRef]
- Grote, A.; Heiland, D.H.; Taube, J.; Helmstaedter, C.; Ravi, V.M.; Will, P.; Hattingen, E.; Schure, J.R.; Witt, J.A.; Reimers, A.; et al. Hippocampal innate inflammatory gliosis only’ in pharmacoresistant temporal lobe epilepsy. Brain 2023, 146, 549–560. [Google Scholar] [CrossRef]
- Athreya, A.; Fasano, R.E.; Drane, D.L.; Millis, S.R.; Willie, J.T.; Gross, R.E.; Karakis, I. Withdrawal of antiepileptic drugs after stereotactic laser amygdalohippocampotomy for mesial temporal lobe epilepsy. Epilepsy Res. 2021, 176, 106721. [Google Scholar] [CrossRef] [PubMed]
- Jehi, L.; Yardi, R.; Chagin, K.; Tassi, L.; Russo, G.L.; Worrell, G.; Hu, W.; Cendes, F.; Morita, M.; Bartolomei, F.; et al. Development and validation of nomograms to provide individualised predictions of seizure outcomes after epilepsy surgery: A retrospective analysis. Lancet Neurol. 2015, 14, 283–290. [Google Scholar] [CrossRef]
- Anyanwu, C.; Motamedi, G.K. Diagnosis and Surgical Treatment of Drug-Resistant Epilepsy. Brain Sci. 2018, 8, 49. [Google Scholar] [CrossRef]
- Pernhorst, K.; Herms, S.; Hoffmann, P.; Cichon, S.; Schulz, H.; Sander, T.; Schoch, S.; Becker, A.J.; Grote, A. TLR4, ATF-3 and IL8 inflammation mediator expression correlates with seizure frequency in human epileptic brain tissue. Seizure 2013, 22, 675–678. [Google Scholar] [CrossRef]
- Strauss, K.I.; Elisevich, K.V. Brain region and epilepsy-associated differences in inflammatory mediator levels in medically refractory mesial temporal lobe epilepsy. J. Neuroinflamm. 2016, 13, 270. [Google Scholar] [CrossRef]
- Teocchi, M.A.; Ferreira, A.E.; Da Luz De Oliveira, E.P.; Tedeschi, H.; D’Souza-Li, L. Hippocampal gene expression dysregulation of Klotho, nuclear factor kappa B and tumor necrosis factor in temporal lobe epilepsy patients. J. Neuroinflamm. 2013, 10, 53. [Google Scholar] [CrossRef]
- Bien, C.G.; Vincent, A.; Barnett, M.H.; Becker, A.J.; Blumcke, I.; Graus, F.; Jellinger, K.A.; Reuss, D.E.; Ribalta, T.; Schlegel, J.; et al. Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain 2012, 135, 1622–1638. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, D.P.; Guan, Z.; Popovich, P.G. B cells produce pathogenic antibodies and impair recovery after spinal cord injury in mice. J. Clin. Investig. 2009, 119, 2990–2999. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, D.P.; Lucin, K.M.; Sanders, V.M.; McGaughy, V.M.; Popovich, P.G. Spinal cord injury triggers systemic autoimmunity: Evidence for chronic B lymphocyte activation and lupus-like autoantibody synthesis. J. Neurochem. 2006, 99, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Popovich, P.G.; Stokes, B.T.; Whitacre, C.C. Concept of autoimmunity following spinal cord injury: Possible roles for T lymphocytes in the traumatized central nervous system. J. Neurosci. Res. 1996, 45, 349–363. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, Z.; Juan, Z.; Zhang, R.; Zhang, C. The high-affinity IgG receptor FcgammaRI modulates peripheral nerve injury-induced neuropathic pain in rats. Mol. Brain 2019, 12, 83. [Google Scholar] [CrossRef]
- Flammer, J.; Neziraj, T.; Ruegg, S.; Probstel, A.K. Immune Mechanisms in Epileptogenesis: Update on Diagnosis and Treatment of Autoimmune Epilepsy Syndromes. Drugs 2023, 83, 135–158. [Google Scholar] [CrossRef]
- Ryding, M.; Mikkelsen, A.W.; Nissen, M.S.; Nilsson, A.C.; Blaabjerg, M. Pathophysiological Effects of Autoantibodies in Autoimmune Encephalitides. Cells 2023, 13, 15. [Google Scholar] [CrossRef]
- Chen, B.; Lopez Chiriboga, A.S.; Sirven, J.I.; Feyissa, A.M. Autoimmune Encephalitis-Related Seizures and Epilepsy: Diagnostic and Therapeutic Approaches. Mayo Clin. Proc. 2021, 96, 2029–2039. [Google Scholar] [CrossRef]
- Geis, C.; Planaguma, J.; Carreno, M.; Graus, F.; Dalmau, J. Autoimmune seizures and epilepsy. J. Clin. Investig. 2019, 129, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Husari, K.S.; Dubey, D. Autoimmune Epilepsy. Neurotherapeutics 2019, 16, 685–702. [Google Scholar] [CrossRef] [PubMed]
- Dyck, S.M.; Karimi-Abdolrezaee, S. Chondroitin sulfate proteoglycans: Key modulators in the developing and pathologic central nervous system. Exp. Neurol. 2015, 269, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Kwok, J.C.F. Proteoglycan Sulphation in the Function of the Mature Central Nervous System. Front. Integr. Neurosci. 2022, 16, 895493. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Moon, L.D.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef]
- Burnside, E.R.; Bradbury, E.J. Manipulating the extracellular matrix and its role in brain and spinal cord plasticity and repair. Neuropathol. Appl. Neurobiol. 2014, 40, 26–59. [Google Scholar] [CrossRef]
- Sorg, B.A.; Berretta, S.; Blacktop, J.M.; Fawcett, J.W.; Kitagawa, H.; Kwok, J.C.; Miquel, M. Casting a Wide Net: Role of Perineuronal Nets in Neural Plasticity. J. Neurosci. 2016, 36, 11459–11468. [Google Scholar] [CrossRef]
- Bartus, K.; James, N.D.; Didangelos, A.; Bosch, K.D.; Verhaagen, J.; Yanez-Munoz, R.J.; Rogers, J.H.; Schneider, B.L.; Muir, E.M.; Bradbury, E.J. Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. J. Neurosci. 2014, 34, 4822–4836. [Google Scholar] [CrossRef]
- Yutsudo, N.; Kitagawa, H. Involvement of chondroitin 6-sulfation in temporal lobe epilepsy. Exp. Neurol. 2015, 274, 126–133. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Burnside, E.R. Moving beyond the glial scar for spinal cord repair. Nat. Commun. 2019, 10, 3879. [Google Scholar] [CrossRef]
- Quirico-Santos, T.; Meira, I.D.; Gomes, A.C.; Pereira, V.C.; Pinto, M.; Monteiro, M.; Souza, J.M.; Alves-Leon, S.V. Resection of the epileptogenic lesion abolishes seizures and reduces inflammatory cytokines of patients with temporal lobe epilepsy. J. Neuroimmunol. 2013, 254, 125–130. [Google Scholar] [CrossRef]
- Ravizza, T.; Gagliardi, B.; Noe, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef]
- Sprissler, R.; Bina, R.; Kasoff, W.; Witte, M.H.; Bernas, M.; Walter, C.; Labiner, D.M.; Lau, B.; Hammer, M.F.; Weinand, M.E. Leukocyte expression profiles reveal gene sets with prognostic value for seizure-free outcome following stereotactic laser amygdalohippocampotomy. Sci. Rep. 2019, 9, 1086. [Google Scholar] [CrossRef] [PubMed]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A multivalent proteoglycan providing structure and signals. J. Histochem. Cytochem. 2012, 60, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Toledano, M.; Britton, J.W.; McKeon, A.; Shin, C.; Lennon, V.A.; Quek, A.M.; So, E.; Worrell, G.A.; Cascino, G.D.; Klein, C.J.; et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology 2014, 82, 1578–1586. [Google Scholar] [CrossRef]
- Shafqat, A.; Albalkhi, I.; Magableh, H.M.; Saleh, T.; Alkattan, K.; Yaqinuddin, A. Tackling the glial scar in spinal cord regeneration: New discoveries and future directions. Front. Cell. Neurosci. 2023, 17, 1180825. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject # | Surgery Outcome | Sex | Age (yr) | Duration (yr) | Etiology | Onset Age (yr) | Seizures/Mo | H.S. | #ASMs |
|---|---|---|---|---|---|---|---|---|---|
| 1 | NSF | M | 32 | 17 | Unk | 15 | 4 | No (HA) | 8 |
| 2 | NSF | M | 32 | 29 | Unk | 3 | 4 | no | 3 |
| 3 | NSF | M | 21 | Unk | Unk | Unk | 8 | yes | 3 |
| 4 | NSF | F | 31 | 16 | Unk | 15 | 8 | yes | 3 |
| 5 | SF | F | 32 | 8 | CVA | 24 | 0.33 | No (HA) | 1 |
| 6 | SF | M | 16 | 10 | Unk | 6 | 4 | yes | 6 |
| 7 | SF | M | 30 | 8 | OD | 22 | 4 | yes | 4 |
| 8 | SF | M | 38 | 37 | Feb | 1 | 10 | No (HA) | 2 |
| Category | Gene Name | NSF vs. SF | NSF vs. Control | SF vs. Control | Role in Autoantibody Function |
|---|---|---|---|---|---|
| IgH | IGHV5-51 | 159.0 | 527.6 | Antigen specificity, can target self | |
| IGHV4-39 | 131.8 | 103.6 | Antigen specificity, can target self | ||
| IGHG3 | 127.6 | 156.9 | IgG component; autoimmunity | ||
| IGHV1-18 | 118.1 | 202.6 | Antigen specificity, can target self | ||
| IGHG1 | 107.9 | 449.0 | 4.2 | IgG component; autoimmunity | |
| IGHV2-5 | 96.4 | 216.9 | Antigen specificity, can target self | ||
| IGHV1-69 | 47.8 | 2117.3 | 44.6 | Antigen specificity, can target self | |
| IGHG4 | 38.3 | 206.0 | IgG component; autoimmunity | ||
| IGHGP | 31.1 | 45.7 | Pseudogene, no autoantibody role | ||
| IGHV3-7 | 20.0 | 102.1 | Antigen specificity, can target self | ||
| IGHM | 6.6 | 4.7 | IgMcomponent; form autoantibodies | ||
| IGHV1-46 | 5.5 | 64.5 | Antigen specificity, can target self | ||
| IGHA1 | 8.8 | IgAcomponent; form autoantibodies | |||
| IGHV1-67 | 46.4 | Antigen specificity, can target self | |||
| IGHV3-33 | 32.4 | Antigen specificity, can target self | |||
| IgL | IGLC3 | 195.9 | 58.3 | Lambda light chains, autoantibodies | |
| IGLV2-14 | 139.6 | 52.0 | Antigen specificity, can target self | ||
| IGLV1-44 | 96.4 | 65.2 | Antigen specificity, can target self | ||
| IGLC2 | 39.4 | 195.9 | Lambda light chains, autoantibodies | ||
| IGLV1-40 | 28.4 | 55.7 | Antigen specificity, can target self | ||
| IGLV4-69 | 42.7 | Antigen specificity, can target self | |||
| IgK | IGKV1-39 | 443.2 | Antigen specificity, can target self | ||
| IGK1-5 | 456.9 | 169.4 | Antigen specificity, can target self | ||
| IGK1-9 | 23.7 | Antigen specificity, can target self | |||
| IGKV1D-33 | 32.4 | Antigen specificity, can target self | |||
| IGKV1D-39 | 17.7 | 269.8 | Antigen specificity, can target self | ||
| IGKV3-11 | 65.1 | 13.2 | Antigen specificity, can target self | ||
| IGKV3-15 | 12.6 | Antigen specificity, can target self | |||
| IGKV3-20 | 251.4 | 570.8 | Antigen specificity, can target self | ||
| IGKV3D-15 | 131.6 | Antigen specificity, can target self | |||
| IGKV4-1 | 37.3 | 7.3 | Antigen specificity, can target self | ||
| Others | IGSF9B | 2.5 | 2.8 | Not directly in autoantibodies | |
| ISLR | 2.2 | Not directly in autoantibodies | |||
| ISLR2 | 2.2 | Not directly in autoantibodies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammer, M.F.; Weinand, M.E. Increased Immunoglobulin and Proteoglycan Synthesis in Resected Hippocampal Tissue Predicts Post-Surgical Seizure Recurrence in Human Temporal Lobe Epilepsy. Pathophysiology 2025, 32, 15. https://doi.org/10.3390/pathophysiology32020015
Hammer MF, Weinand ME. Increased Immunoglobulin and Proteoglycan Synthesis in Resected Hippocampal Tissue Predicts Post-Surgical Seizure Recurrence in Human Temporal Lobe Epilepsy. Pathophysiology. 2025; 32(2):15. https://doi.org/10.3390/pathophysiology32020015
Chicago/Turabian StyleHammer, Michael F., and Martin E. Weinand. 2025. "Increased Immunoglobulin and Proteoglycan Synthesis in Resected Hippocampal Tissue Predicts Post-Surgical Seizure Recurrence in Human Temporal Lobe Epilepsy" Pathophysiology 32, no. 2: 15. https://doi.org/10.3390/pathophysiology32020015
APA StyleHammer, M. F., & Weinand, M. E. (2025). Increased Immunoglobulin and Proteoglycan Synthesis in Resected Hippocampal Tissue Predicts Post-Surgical Seizure Recurrence in Human Temporal Lobe Epilepsy. Pathophysiology, 32(2), 15. https://doi.org/10.3390/pathophysiology32020015