
Hepatoprotective and Antiatherosclerotic Effects of Oleoylethanolamide-Based Dietary Supplement in Dietary-Induced Obesity in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Production of a Dietary Supplement
2.2. Experimental Model
2.3. Immunohistochemical Staining
2.4. Histological Staining
2.5. Measurement of Serum Cholesterol Levels
2.6. Cell Culture
2.7. Cytotoxicity Assay
2.8. Oxidative Stress Assessment
2.9. Western Blotting
2.10. RT-PCR Assay
2.11. Statistical Analysis
3. Results
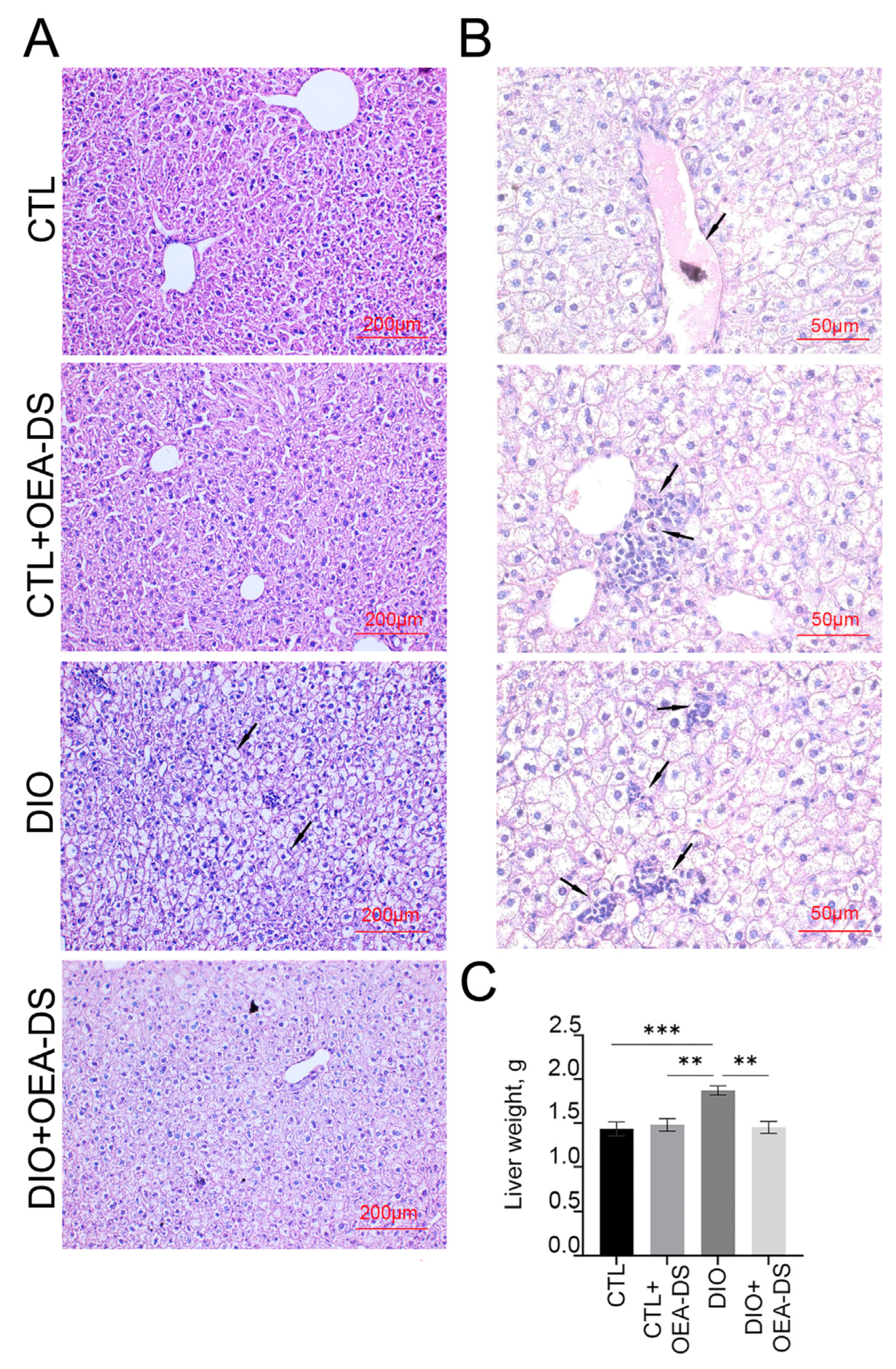
3.1. OEA-DS Administration Alleviates Hepatic Steatosis
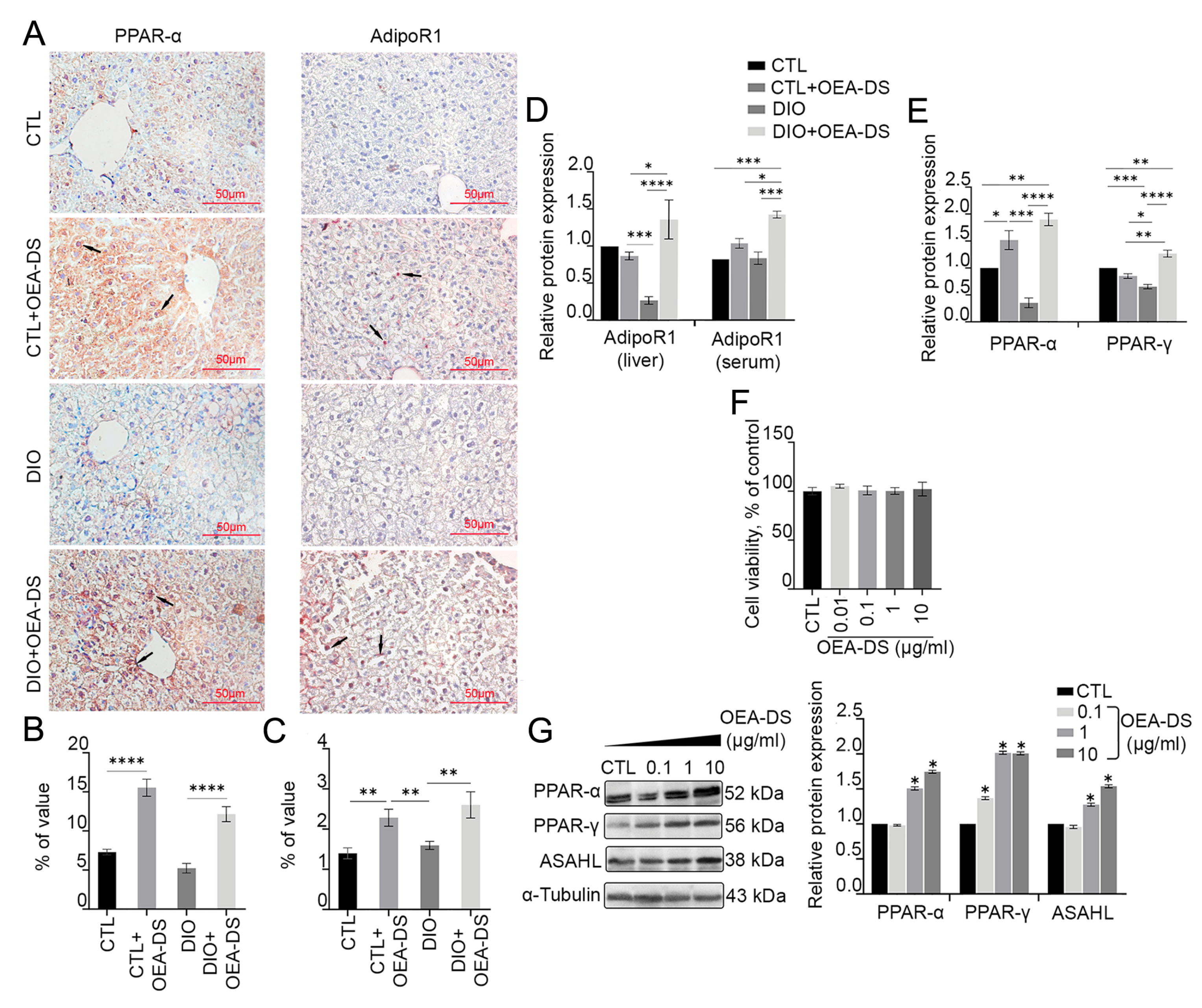
3.2. OEA-DS Administration Causes Activation of PPAR-α and PPAR-γ Receptors and Adiponectin Receptor-1 (AdipoR1) in Liver and Serum
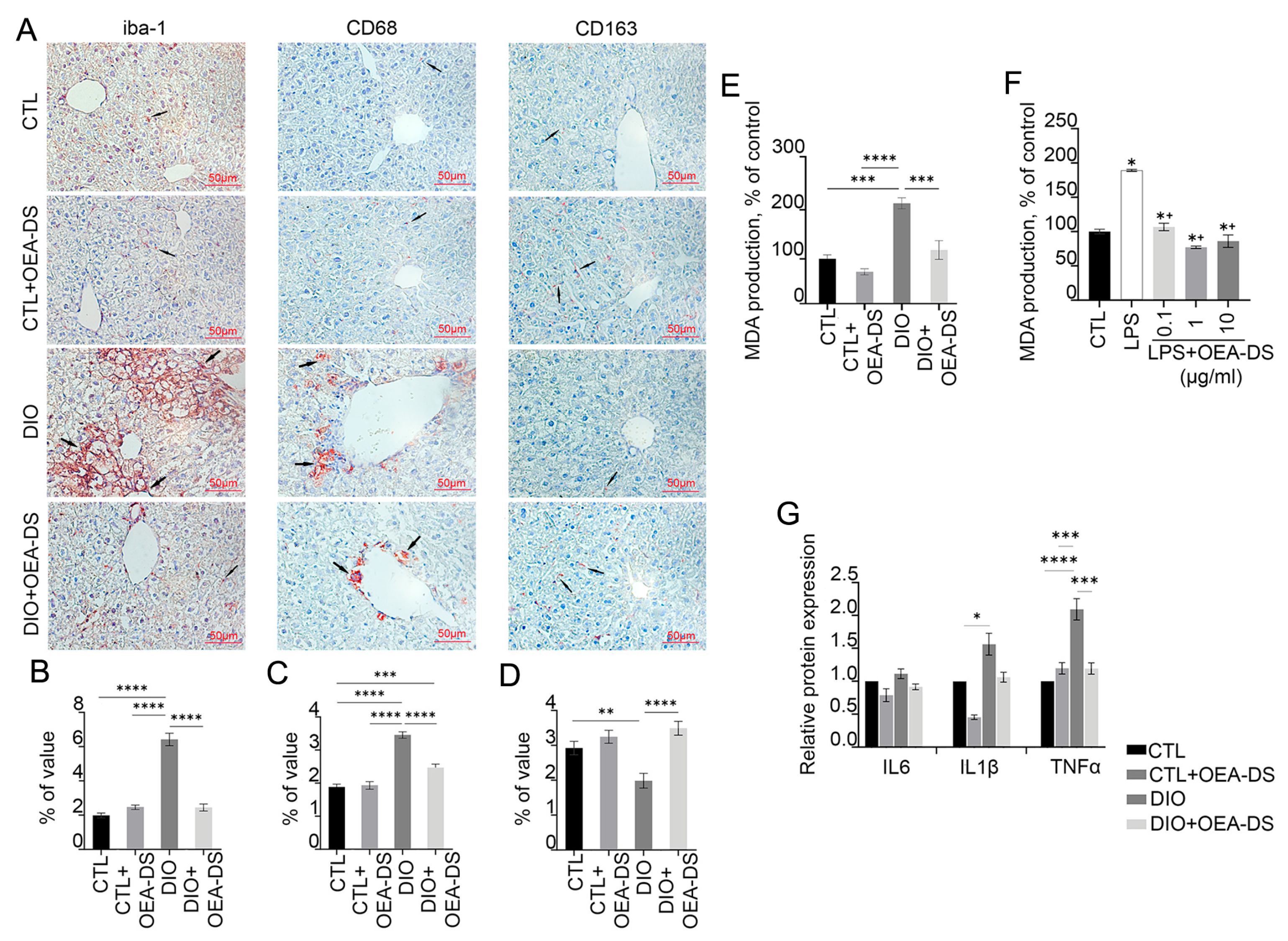
3.3. Administration of OEA-DS Leads to a Decrease in Inflammatory Reactions in the Liver and Oxidative Stress
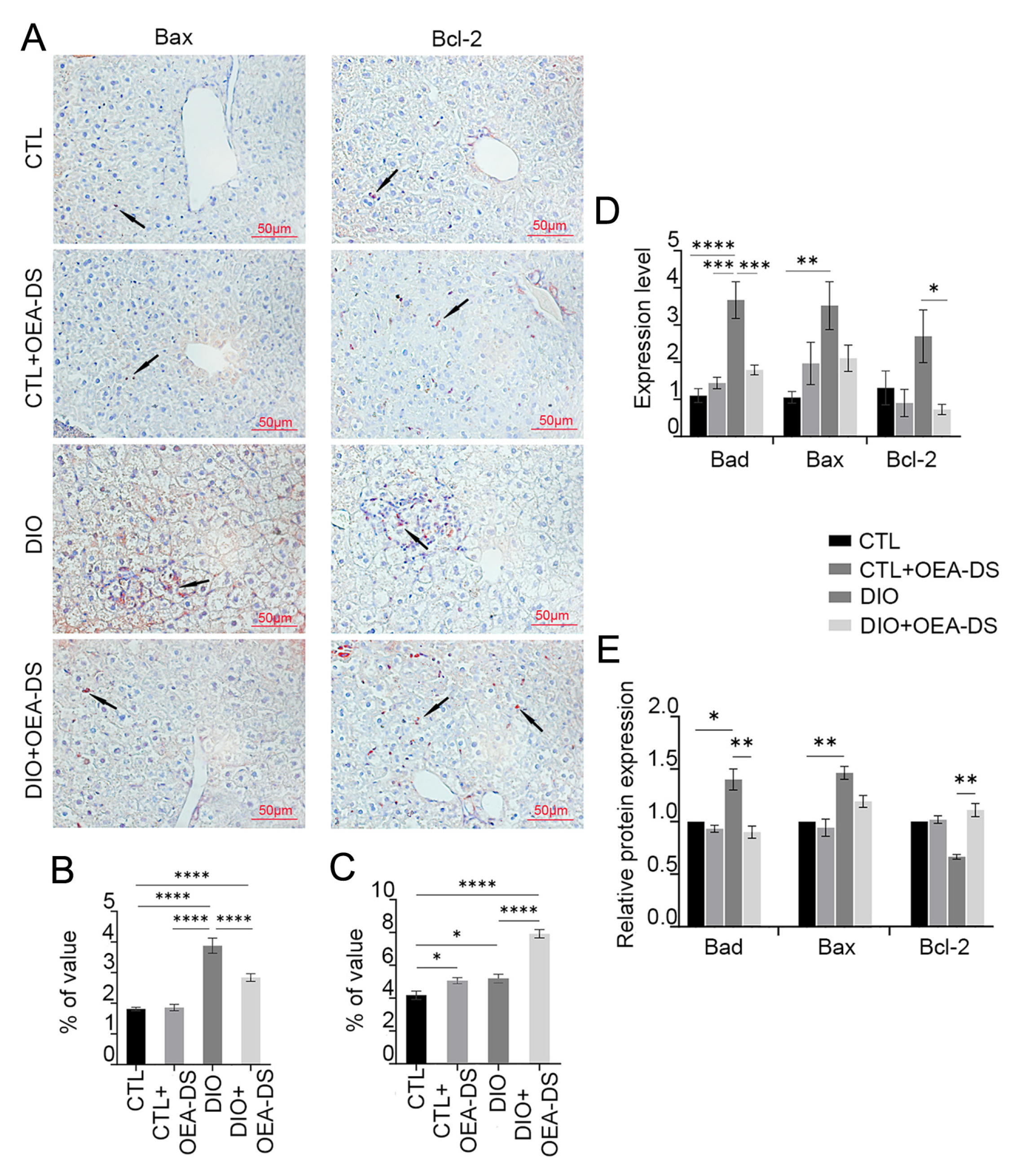
3.4. OEA-DS Reduces the Intensity of Apoptotic Processes in the Liver
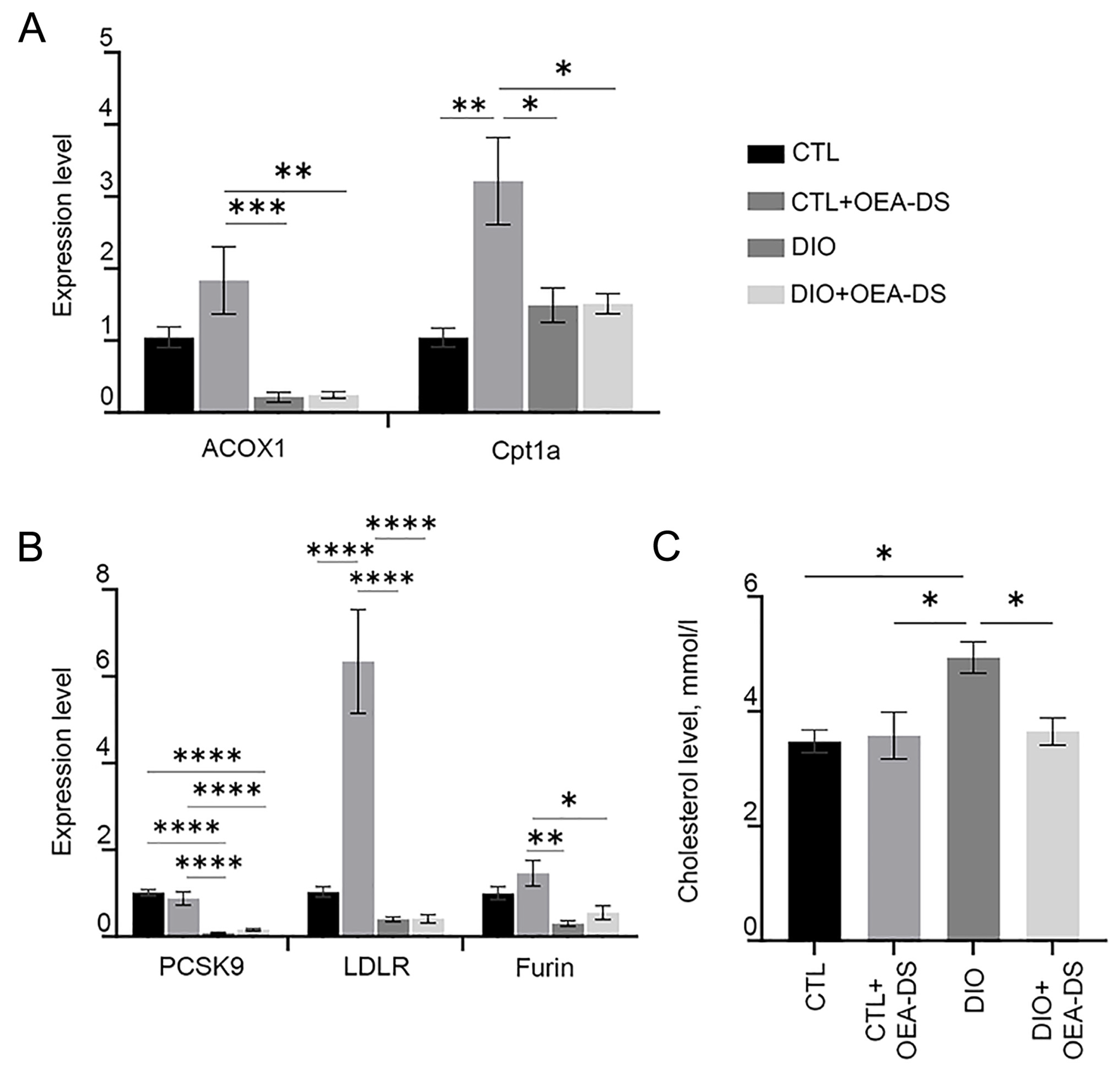
3.5. OEA-DS Affects Indices of Lipid Metabolism in the Liver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, R.J.; Ahmed, A. Obesity and non-alcoholic fatty liver disease: Disparate associations among Asian populations. World J. Hepatol. 2014, 6, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Ng, C.H.; Wong, Z.Y.; Chew, N.W.S.; Chan, K.E.; Xiao, J.; Sayed, N.; Lim, W.H.; Tan, D.J.H.; Loke, R.W.K.; Tay, P.W.L.; et al. Hypertension is prevalent in non-alcoholic fatty liver disease and increases all-cause and cardiovascular mortality. Front. Cardiovasc. Med. 2022, 9, 942753. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kim, B.H.; Seo, H.S.; Lee, Y.J.; Kim, H.H.; Son, H.H.; Choi, M.H. Cholesterol-induced non-alcoholic fatty liver disease and atherosclerosis aggravated by systemic inflammation. PLoS ONE 2014, 9, e97841. [Google Scholar] [CrossRef]
- Nanizawa, E.; Otsuka, S.; Hatayama, N.; Tamaki, Y.; Hayashi, Y.; Ishikawa, T.; Hirai, S.; Naito, M. Short-term high-fat and high-carbohydrate diets increase susceptibility to liver injury by inducing hepatic procoagulant and proinflammatory conditions with different balances. Nutrition 2022, 101, 111710. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; Yeung, S.F.; Ke, J.Y.; Antunes, M.M.; Pellizzon, M.A. Considerations When Choosing High-Fat, High-Fructose, and High-Cholesterol Diets to Induce Experimental Nonalcoholic Fatty Liver Disease in Laboratory Animal Models. Curr. Dev. Nutr. 2021, 5, nzab138. [Google Scholar] [CrossRef]
- Byrne, C.D. Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proc. Nutr. Soc. 2013, 72, 412–419. [Google Scholar] [CrossRef]
- Tamura, S.; Shimomura, I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1139–1142. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Paternostro, R.; Trauner, M. Current treatment of non-alcoholic fatty liver disease. J. Intern. Med. 2022, 292, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 36, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Gervois, P.; Mansouri, R.M. PPARα as a therapeutic target in inflammation-associated diseases. Expert. Opin. Ther. Targets 2012, 16, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jia, Y.; Fu, T.; Viswakarma, N.; Bai, L.; Rao, M.S.; Zhu, Y.; Borensztajn, J.; Reddy, J.K. Sustained activation of PPARα by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J. 2012, 26, 628–638. [Google Scholar] [CrossRef]
- Jain, N.; Bhansali, S.; Kurpad, A.V.; Hawkins, M.; Sharma, A.; Kaur, S.; Rastogi, A.; Bhansali, A. Effect of a Dual PPAR α/γ agonist on Insulin Sensitivity in Patients of Type 2 Diabetes with Hypertriglyceridemia- Randomized double-blind placebo-controlled trial. Sci. Rep. 2019, 9, 19017. [Google Scholar] [CrossRef]
- Romano, A.; Coccurello, R.; Giacovazzo, G.; Bedse, G.; Moles, A.; Gaetani, S. Oleoylethanolamide: A novel potential pharmacological alternative to cannabinoid antagonists for the control of appetite. BioMed Res. Int. 2014, 2014, 203425. [Google Scholar] [CrossRef]
- Brown, J.D.; Karimian Azari, E.; Ayala, J.E. Oleoylethanolamide: A fat ally in the fight against obesity. Physiol. Behav. 2017, 176, 50–58. [Google Scholar] [CrossRef]
- Laleh, P.; Yaser, K.; Alireza, O. Oleoylethanolamide: A novel pharmaceutical agent in the management of obesity-an updated review. J. Cell Physiol. 2019, 234, 7893–7902. [Google Scholar] [CrossRef]
- Laleh, P.; Yaser, K.; Abolfazl, B.; Shahriar, A.; Mohammad, A.J.; Nazila, F.; Alireza, O. Oleoylethanolamide increases the expression of PPAR-A and reduces appetite and body weight in obese people: A clinical trial. Appetite 2018, 128, 44–49. [Google Scholar] [CrossRef]
- Ivashkevich, D.; Ponomarenko, A.; Manzhulo, I.; Sultanov, R.; Dyuizen, I. Effect of Oleoylethanolamide-Based Dietary Supplement on Systemic Inflammation in the Development of Alimentary-Induced Obesity in Mice. Nutrients 2023, 15, 4345. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediat. Inflamm. 2010, 2010, 289645. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Oxidative Stress and Acute Hepatic Injury. Curr. Opin. Toxicol. 2018, 7, 17–21. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Kleemann, R.; Gervois, P.P.; Verschuren, L.; Staels, B.; Princen, H.M.; Kooistra, T. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFkappa B-C/EBP-beta complex formation. Blood 2003, 101, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guo, H.; Li, Y.; Meng, X.; Yan, L.; Zhang, D.; Wu, S.; Zhou, H.; Peng, L.; Xie, Q.; et al. Oleoylethanolamide exerts anti-inflammatory effects on LPS-induced THP-1 cells by enhancing PPARα signaling and inhibiting the NF-κB and ERK1/2/AP-1/STAT3 pathways. Sci. Rep. 2016, 6, 34611. [Google Scholar] [CrossRef]
- Giudetti, A.M.; Vergara, D.; Longo, S.; Friuli, M.; Eramo, B.; Tacconi, S.; Fidaleo, M.; Dini, L.; Romano, A.; Gaetani, S. Oleoylethanolamide Reduces Hepatic Oxidative Stress and Endoplasmic Reticulum Stress in High-Fat Diet-Fed Rats. Antioxidants 2021, 10, 1289. [Google Scholar] [CrossRef]
- Peng, J.; Li, J.; Huang, J.; Xu, P.; Huang, H.; Liu, Y.; Yu, L.; Yang, Y.; Zhou, B.; Jiang, H.; et al. p300/CBP inhibitor A-485 alleviates acute liver injury by regulating macrophage activation and polarization. Theranostics 2019, 9, 8344–8361. [Google Scholar] [CrossRef]
- Park, S.J.; Garcia Diaz, J.; Um, E.; Hahn, Y.S. Major roles of kupffer cells and macrophages in NAFLD development. Front. Endocrinol. 2023, 14, 1150118. [Google Scholar] [CrossRef] [PubMed]
- Penas, F.; Mirkin, G.A.; Vera, M.; Cevey, Á.; González, C.D.; Gómez, M.I.; Sales, M.E.; Goren, N.B. Treatment in vitro with PPARα and PPARγ ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim. Biophys. Acta 2015, 1852, 893–904. [Google Scholar] [CrossRef]
- Cao, H.; Wen, G.; Li, H. Role of peroxisome proliferator-activated receptor α in atherosclerosis. Mol. Med. Rep. 2014, 9, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Wang, Q.; Wu, C.; Du, G.; Yang, L. Oleoylethanolamide Protects against Acute Ischemic Stroke by Promoting PPARα-Mediated Microglia/Macrophage M2 Polarization. Pharmaceuticals 2023, 16, 621. [Google Scholar] [CrossRef]
- Zuo, Y.; Xiao, T.; Qiu, X.; Liu, Z.; Zhang, S.; Zhou, N. Adiponectin reduces apoptosis of diabetic cardiomyocytes by regulating miR-711/TLR4 axis. Diabetol. Metab. Syndr. 2022, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gan, L.; Wu, T.; Feng, F.; Luo, D.; Gu, H.; Liu, S.; Sun, C. Adiponectin reduces ER stress-induced apoptosis through PPARα transcriptional regulation of ATF2 in mouse adipose. Cell Death Dis. 2016, 7, e2487. [Google Scholar] [CrossRef]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef]
- Steckley, D.; Karajgikar, M.; Dale, L.B.; Fuerth, B.; Swan, P.; Drummond-Main, C.; Poulter, M.O.; Ferguson, S.S.; Strasser, A.; Cregan, S.P. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J. Neurosci. 2007, 27, 12989–12999. [Google Scholar] [CrossRef]
- Li, L.; Li, L.; Chen, L.; Lin, X.; Xu, Y.; Ren, J.; Fu, J.; Qiu, Y. Effect of oleoylethanolamide on diet-induced nonalcoholic fatty liver in rats. J. Pharmacol. Sci. 2015, 127, 244–250. [Google Scholar] [CrossRef]
- Drover, V.A.; Abumrad, N.A. CD36-dependent fatty acid uptake regulates expression of peroxisome proliferator activated receptors. Biochem. Soc. Trans. 2005, 33 Pt 1, 311–315. [Google Scholar] [CrossRef]
- Tutunchi, H.; Ostadrahimi, A.; Saghafi-Asl, M.; Maleki, V. The effects of oleoylethanolamide, an endogenous PPAR-α agonist, on risk factors for NAFLD: A systematic review. Obes. Rev. 2019, 20, 1057–1069. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Tsuchida, A.; Yamauchi, T.; Takekawa, S.; Hada, Y.; Ito, Y.; Maki, T.; Kadowaki, T. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: Comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes 2005, 54, 3358–3370. [Google Scholar] [CrossRef]
- Kourimate, S.; Le May, C.; Langhi, C.; Jarnoux, A.L.; Ouguerram, K.; Zaïr, Y.; Nguyen, P.; Krempf, M.; Cariou, B.; Costet, P. Dual mechanisms for the fibrate-mediated repression of proprotein convertase subtilisin/kexin type 9. J. Biol. Chem. 2008, 283, 9666–9673. [Google Scholar] [CrossRef]
- Costet, P.; Cariou, B.; Lambert, G.; Lalanne, F.; Lardeux, B.; Jarnoux, A.L.; Grefhorst, A.; Staels, B.; Krempf, M. Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J. Biol. Chem. 2006, 281, 6211–6218. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The proprotein convertases are potential targets in the treatment of dyslipidemia. J. Mol. Med. 2007, 85, 685–696. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Walsh, K. Obesity, adiponectin and vascular inflammatory disease. Curr. Opin. Lipidol. 2003, 14, 561–566. [Google Scholar] [CrossRef]
- Ouedraogo, R.; Gong, Y.; Berzins, B.; Wu, X.; Mahadev, K.; Hough, K.; Chan, L.; Goldstein, B.J.; Scalia, R. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J. Clin. Investig. 2007, 117, 1718–1726. [Google Scholar] [CrossRef]
- Recasens, M.; Ricart, W.; Fernández-Real, J.M. Obesity and inflammation. Rev. De Med. De La Univ. De Navar. 2004, 48, 49–54. [Google Scholar]
- Ouchi, N.; Walsh, K. Adiponectin as an anti-inflammatory factor. Clin. Chim. Acta 2007, 380, 24–30. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Compounds, Compositions and Treatment of Oleoylethanolamide-Like Modulators of PPARalpha. PubChem Patent Summary for US-2005054730-A1, 10 March 2025.
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.M.; Fu, N.; Wu, W.J.; Liang, B.L.; Wang, R.Q.; Zhao, S.X.; Zhao, J.M.; Yu, J. Rosiglitazone prevents nutritional fibrosis and steatohepatitis in mice. Scand. J. Gastroenterol. 2009, 44, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Lutchman, G.; Modi, A.; Kleiner, D.E.; Promrat, K.; Heller, T.; Ghany, M.; Borg, B.; Loomba, R.; Liang, T.J.; Premkumar, A.; et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology 2007, 46, 424–429. [Google Scholar] [CrossRef]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.; Bandyopadyhay, G.; Leung, H.Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.K.; et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef]
- Peraldi, P.; Xu, M.; Spiegelman, B.M. Thiazolidinediones block tumor necrosis factor-alpha-induced inhibition of insulin signaling. J. Clin. Investig. 1997, 100, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Rajala, M.W.; Obici, S.; Scherer, P.E.; Rossetti, L. Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J. Clin. Investig. 2003, 111, 225–230. [Google Scholar] [CrossRef]
- Combs, T.P.; Berg, A.H.; Obici, S.; Scherer, P.E.; Rossetti, L. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J. Clin. Investig. 2001, 108, 1875–1881. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Sabahi, M.; Ahmadi, S.A.; Kazemi, A.; Mehrpooya, M.; Khazaei, M.; Ranjbar, A.; Mowla, A. The Effect of Oleoylethanolamide (OEA) Add-On Treatment on Inflammatory, Oxidative Stress, Lipid, and Biochemical Parameters in the Acute Ischemic Stroke Patients: Randomized Double-Blind Placebo-Controlled Study. Oxid. Med. Cell Longev. 2022, 2022, 5721167. [Google Scholar] [CrossRef]
- Eng, J.M.; Estall, J.L. Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol. Cells 2022, 10, 1805. [Google Scholar] [CrossRef]
- Fang, T.; Wang, H.; Pan, X.; Little, P.J.; Xu, S.; Weng, J. Mouse models of nonalcoholic fatty liver disease (NAFLD): Pathomechanisms and pharmacotherapies. Int. J. Biol. Sci. 2022, 18, 5681–5697. [Google Scholar] [CrossRef]
- Geidl-Flueck, B.; Gerber, P.A. Fructose drives de novo lipogenesis affecting metabolic health. J. Endocrinol. 2023, 257, e220270. [Google Scholar] [CrossRef]
- Chu, D.T.; Malinowska, E.; Jura, M.; Kozak, L.P.L. C57BL/6J mice as a polygenic developmental model of diet-induced obesity. Physiol. Rep. 2017, 5, e13093. [Google Scholar] [CrossRef]
- Vercalsteren, E.; Vranckx, C.; Frederix, L.; Lox, M.; Lijnen, H.R.; Scroyen, I.; Hemmeryckx, B. Advanced-age C57BL/6JRj mice do not develop obesity upon western-type diet exposure. Adipocyte 2019, 8, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Kim, H.S.; Choi, Y.W.; Kim, Y.M.; Kang, K.W. Therapeutic application of GPR119 ligands in metabolic disorders. Diabetes Obes. Metab. 2018, 20, 257–269. [Google Scholar] [CrossRef]
- Jung, I.; Koo, D.J.; Lee, W.Y. Insulin Resistance, Non-Alcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: Clinical and Experimental Perspective. Diabetes Metab. J. 2024, 48, 327–339. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, M.; Zakharia, A.; Werstuck, G.H. The Role of Estrogen in Insulin Resistance: A Review of Clinical and Preclinical Data. Am. J. Pathol. 2021, 191, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.M.; Clegg, D.J. Central effects of estradiol in the regulation of food intake, body weight, and adiposity. J. Steroid Biochem. Mol. Biol. 2010, 122, 65–73. [Google Scholar] [CrossRef]
- Ariadi, A.; Jamsari, J.; Yanwirasti, Y.; Siregar, M.F.G.; Yusrawati, Y. Correlation between Estrogen Levels with Lipid Profile in Menopause Women in West Sumatera. Open Access Maced. J. Med. Sci. 2019, 7, 2084–2087. [Google Scholar] [CrossRef]
- Stapleton, S.; Welch, G.; DiBerardo, L.; Freeman, L.R. Sex differences in a mouse model of diet-induced obesity: The role of the gut microbiome. Biol. Sex Differ. 2024, 15, 5. [Google Scholar] [CrossRef]
- Beery, A.K. Inclusion of females does not increase variability in rodent research studies. Curr. Opin. Behav. Sci. 2018, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Aubry, A.V.; Joseph Burnett, C.; Goodwin, N.L.; Li, L.; Navarrete, J.; Zhang, Y.; Tsai, V.; Durand-de Cuttoli, R.; Golden, S.A.; Russo, S.J. Sex differences in appetitive and reactive aggression. Neuropsychopharmacology 2022, 47, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xue, R.; Ji, L.; Zhang, X.; Wu, J.; Gu, J.; Zhou, M.; Chen, S. Activation of farnesoid X receptor (FXR) protects against fructose-induced liver steatosis via inflammatory inhibition and ADRP reduction. Biochem. Biophys. Res. 2014, 450, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11, 584303. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivashkevich, D.; Ponomarenko, A.; Manzhulo, I.; Egoraeva, A.; Dyuizen, I. Hepatoprotective and Antiatherosclerotic Effects of Oleoylethanolamide-Based Dietary Supplement in Dietary-Induced Obesity in Mice. Pathophysiology 2025, 32, 16. https://doi.org/10.3390/pathophysiology32020016
Ivashkevich D, Ponomarenko A, Manzhulo I, Egoraeva A, Dyuizen I. Hepatoprotective and Antiatherosclerotic Effects of Oleoylethanolamide-Based Dietary Supplement in Dietary-Induced Obesity in Mice. Pathophysiology. 2025; 32(2):16. https://doi.org/10.3390/pathophysiology32020016
Chicago/Turabian StyleIvashkevich, Darya, Arina Ponomarenko, Igor Manzhulo, Anastasia Egoraeva, and Inessa Dyuizen. 2025. "Hepatoprotective and Antiatherosclerotic Effects of Oleoylethanolamide-Based Dietary Supplement in Dietary-Induced Obesity in Mice" Pathophysiology 32, no. 2: 16. https://doi.org/10.3390/pathophysiology32020016
APA StyleIvashkevich, D., Ponomarenko, A., Manzhulo, I., Egoraeva, A., & Dyuizen, I. (2025). Hepatoprotective and Antiatherosclerotic Effects of Oleoylethanolamide-Based Dietary Supplement in Dietary-Induced Obesity in Mice. Pathophysiology, 32(2), 16. https://doi.org/10.3390/pathophysiology32020016