
Pathophysiology of Childhood-Onset Myasthenia: Abnormalities of Neuromuscular Junction and Autoimmunity and Its Background

Abstract
1. Introduction
2. Epidemiological Study
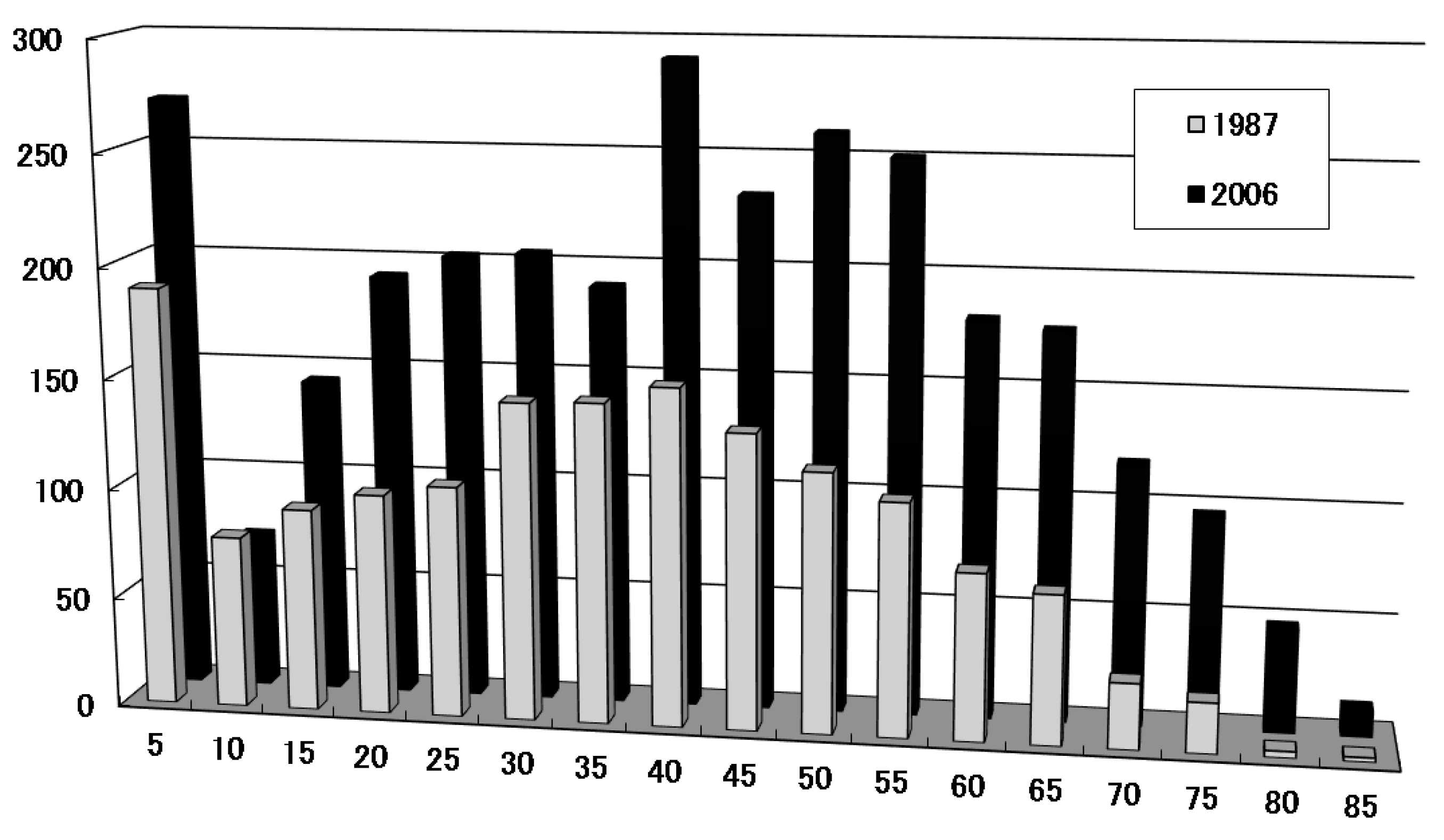
2.1. Characteristics of Childhood Onset; Frequency and Peak Age of Onset in Childhood
2.2. Immunogenetic Studies
2.3. Gender Difference
3. Pathophysiology of Myasthenic State
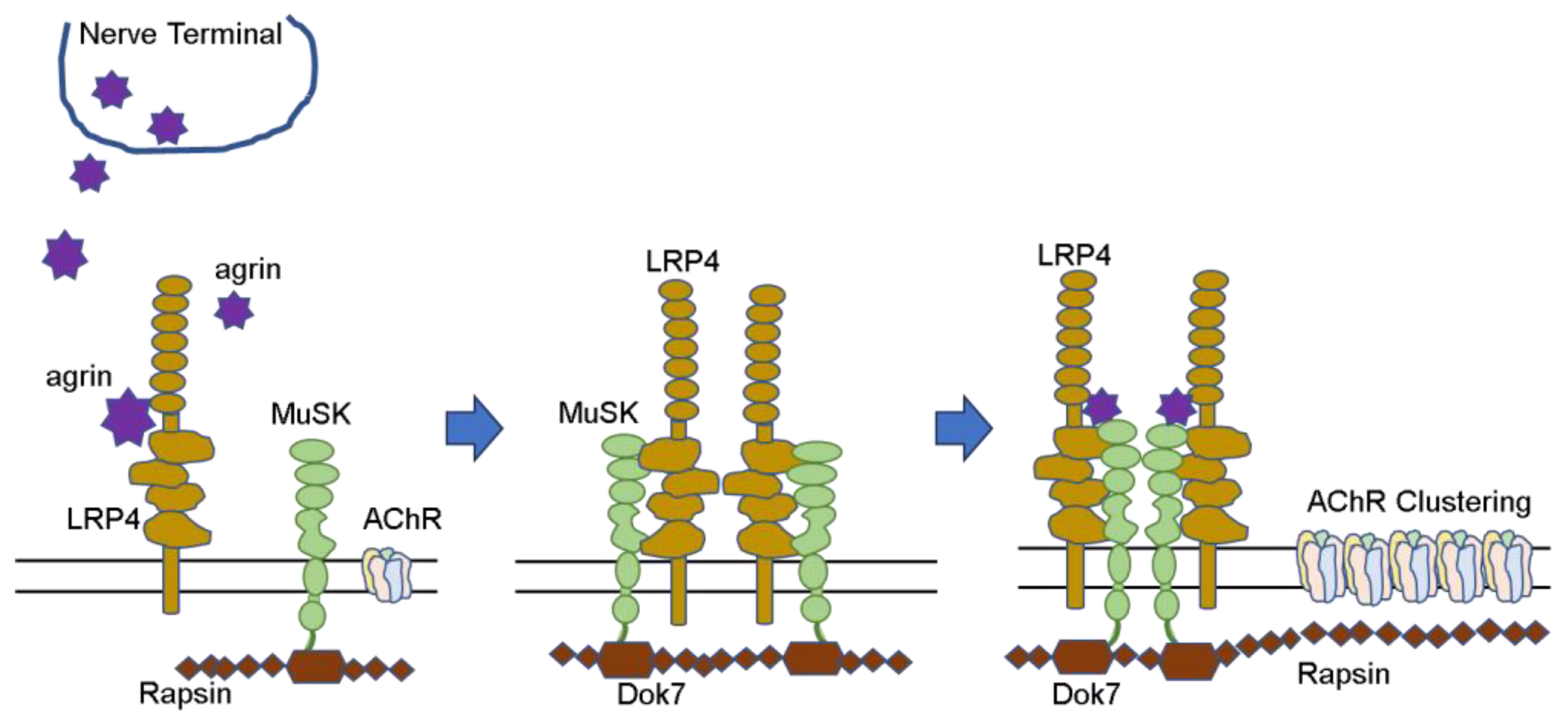
3.1. Formation of Neuromuscular Junction
3.2. Changes at the Neuromuscular Junction
3.2.1. Decreased AChR at the Neuromuscular Junction
3.2.2. Structural Destruction of the Neuromuscular Junction
3.3. Autoantibodies against the Neuromuscular Junction
3.3.1. Involvement of AChR Antibodies
3.3.2. Neonatal Transient Myasthenia Gravis and Fetal Myasthenia
3.3.3. Antibody against Muscle-Specific Tyrosine Kinase (MuSK)
3.3.4. Double or Triple Seronegative MG
3.4. Congenital Myasthenic Syndrome (CMS)
3.5. Disease Classification: Ocular and Generalized MG
3.6. Childhood Thymus and Thymic Selection
3.7. Involvement of Cellular Immunity
3.8. Lymphorrhage
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elmqvist, D.; Hofmann, W.W.; Kugelberg, J.; Quastel, D.M.L. An electrophysiological investigation of neuromuscular transmission in myasthenia gravis. J. Physiol. 1964, 174, 417–434. [Google Scholar] [CrossRef]
- Patrick, J.; Lindstrom, J.M. Autoimmune response to acetylcholine receptor. Science 1973, 180, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.A. Myasthenia gravis: A new hypothesis. Scott. Med. J. 1960, 5, 419. [Google Scholar] [CrossRef]
- Grob, D.; Brunner, N.; Namba, T.; Pagala, M. Lifetime course of myasthenia gravis. Muscle Nerve 2008, 37, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, F.; Long, Z.; Yang, J.; Ren, Y.; Ma, Q.; Li, J.; Wen, X.; Wang, L.; Da, Y.; et al. Mortality of myasthenia gravis: A national population-based study in China. Ann. Clin. Transl. Neurol. 2023, 10, 1095–1105. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Adachi, Y.; Nakamura, Y.; Kuriyama, N.; Murai, H.; Nomura, Y.; Sakai, Y.; Iwasa, K.; Furukawa, Y.; Kuwabara, S.; et al. Two-step nationwide epidemiological survey of myasthenia gravis in Japan 2018. PLoS ONE 2018, 17, e0274161. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, H.J. Observations of the natural history of myasthenia gravis and the effect of thymectomy. Ann. N. Y. Acad. Sci. 1981, 377, 678–690. [Google Scholar] [CrossRef]
- Carr, A.S.; Cardwell, C.R.; McCarron, P.O.; McConville, J. A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol. 2010, 10, 46. [Google Scholar] [CrossRef]
- Dresser, L.; Wlodarski, R.; Rezania, K.; Soliven, B. Myasthenia gravis: Epidemiology, pathophysiology and clinical manifestations. J. Clin. Med. 2021, 10, 2235. [Google Scholar] [CrossRef]
- Bettini, M.; Chaves, M.; Cristiano, E.; Pagotto, V.; Perez, L.; Giunta, D.; Rugiero, M. Incidence of autoimmune myasthenia gravis in a health maintenance organization in Buenos Aires, Argentina. Neuroepidemiology 2017, 48, 119–123. [Google Scholar] [CrossRef]
- Park, J.-S.; Eah, K.Y.; Park, J.-M. Epidemiological profile of myasthenia gravis in South Korea using the national health insurance database. Acta Neurol. Scand. 2022, 145, 633–640. [Google Scholar] [CrossRef]
- Casetta, I.; Groppo, E.; Gennaro, R.D.; Cesnik, E.; Piccolo, L.; Volpato, S.; Granieri, E. Myasthenia gravis: A changing pattern of incidence. J. Neurol. 2010, 257, 2015–2019. [Google Scholar] [CrossRef]
- Osserman, K.E.; Genkins, G. Studies in myasthenia gravis: Review of a twenty-year experience in over 1200 patients. Mt. Sinai J. Med. 1971, 38, 497–537. [Google Scholar]
- Chiu, H.-C.; Vincent, A.; Newsom-Davis, J.; Hsieh, K.-H.; Hung, T.-P. Myasthenia gravis: Population differences in disease expression and acetylcholine receptor antibody titers between Chinese and Caucasians. Neurology 1987, 37, 1854–1857. [Google Scholar] [CrossRef] [PubMed]
- Murai, H.; Yamashita, N.; Watanabe, M.; Nomura, Y.; Motomura, M.; Yoshikawa, H.; Nakamura, Y.; Kawaguchi, N.; Onodera, H.; Araga, S.; et al. Characteristics of myasthenia gravis according to onset-age: Japanese Nationwide survey. J. Neurol. Sci. 2011, 305, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, W.B.; Men, L.N.; Feng, H.Y.; Li, Y.; Luo, C.M.; Qiu, L. Clinical features of myasthenia gravis in southern China: A retrospective review of 2154 cases over 22 years. Neurol. Sci. 2013, 34, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Luo, X.; Lin, J.; Li, Y.; Zhang, M.; Zhang, X.; Yang, M.; Wang, W.; Bu, B. Long-term outcome of 424 childhood-onset myasthenia gravis patients. J. Neurol. 2015, 262, 823–830. [Google Scholar] [CrossRef]
- Chou, C.-C.; CHEESE study group; Su, I.-C.; Chou, I.-J.; Lin, J.-J.; Lan, S.-Y.; Wang, Y.-S.; Kong, S.-S.; Chen, Y.-J.; Hsieh, M.-Y.; et al. Correlation of anti-acetylcholine receptor antibody levels and long-term outcomes of juvenile myasthenia gravis in Taiwan: A case control study. BMC Neurol. 2019, 19, 170. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.N.; Kang, H.-C.; Lee, J.S.; Kim, H.D.; Shin, H.Y.; Kim, S.M.; Sunwoo, I.N.; Lee, Y.-M. Juvenile myasthenia gravis in Korea: Subgroup analysis according to sex and onset age. J. Chil. Neurol. 2016, 31, 1561–1568. [Google Scholar] [CrossRef]
- Finnis, M.F.; Jayawant, S. Juvenile myasthenia gravis: A pediatric perspective. Autoimmune Dis. 2011, 2011, 404101. [Google Scholar] [CrossRef]
- Popperud, T.H.; Boldingh, M.I.; Rasmussen, M.; Kerty, E. Juvenile myasthenia gravis in Norway: Clinical characteristics, treatment, and long-term outcome in a nationwide population-based cohort. Eur. J. Pediatr. Neurol. 2017, 21, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M. Childhood-onset myasthenia gravis. No Hattatsu 2022, 54, 235–242. (In Japanese) [Google Scholar]
- Mansukhani, S.A.; Bothun, E.; Diehl, N.N.; Mohney, B.G. Incidence and ocular features of pediatric myasthenias. Am. J. Ophthalmol. 2019, 200, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, D.; Ramdas, S.; Munot, P.; Pitt, M.; Beeson, D.; Knight, R.; Cruz, P.R.; Vincent, A.; Jayawant, S.; DeVile, C.; et al. Pediatric myasthenia gravis: Prognostic factors for drug free remission. Neuromuscul. Disord. 2020, 30, 120–127. [Google Scholar] [CrossRef]
- Yang, L.; Tang, Y.; He, F.; Zhang, C.; Kessi, M.; Peng, J.; Yin, F. Clinical characteristics and outcome predictors of a Chinese childhood-onset myasthenia gravis cohort. Front. Pediatr. 2022, 10, 996213. [Google Scholar] [CrossRef] [PubMed]
- Vandiedonck, C.; Beaurain, G.; Giraud, M.; Hue-Beauvais, C.; Eymard, B.; Tranchant, C.; Gajdos, P.; Dausset, J.; Garchon, H.-J. Pleiotropic effects of the 8.1 HLA haplotype in patients with autoimmune myasthenia gravis and thymus hyperplasia. Proc. Natl. Acad. Sci. USA 2004, 101, 15464–15469. [Google Scholar] [CrossRef] [PubMed]
- Popperud, T.H.; Viken, M.K.; Kerty, E.; Lie, B.A. Juvnile myasthenia gravis in Norway: HLA-DRB1*04:04 is positively associated with prepubertal onset. PLoS ONE 2017, 12, e0186383. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Sveinsson, O.; Thormar, G.; Granqvist, M.; Askling, J.; Lundberg, I.E.; Ye, W.; Hammarström, L.; Pirskanen, R.; Piehl, F. The autoimmune spectrum of myasthenia gravis: A Swedish population-based study. J. Int. Med. 2015, 77, 594–604. [Google Scholar] [CrossRef]
- Santos, E.; Bettencourt, A.; Duarte, S.; Gabriel, D.; Oliveira, V.; Da Silva, A.M.; Costa, P.P.; Lopes, C.; Gonçalves, G.; da Silva, B.M.; et al. Refractory myasthenia gravis: Characteristics of a Portuguese cohort. Muscle Nerve 2019, 60, 188–191. [Google Scholar] [CrossRef]
- Fekih-Mrissa, N.; Klai, S.; Zaouali, J.; Gritli, N.; Mrissa, R. Association of HLA-DR/DQ polymorphism with myasthenia gravis in Tunisian patients. Clin. Neurol. Neurosurg. 2013, 115, 32–36. [Google Scholar] [CrossRef]
- Feng, H.-Y.; Yang, L.-X.; Liu, W.-B.; Huang, X.; Qiu, L.; Li, Y. The HLA-B*4601-DRB1*0901 haplotype is positively correlated with juvenile ocular myasthenia gravis in a southern Chinese Han population. Neurol. Sci. 2015, 36, 1135–1140. [Google Scholar] [CrossRef]
- Matsuki, K.; Juji, T.; Tokunaga, K.; Takamizawa, M.; Maeda, H.; Soda, M.; Nomura, Y.; Segawa, M. HLA antigen in Japanese patients with myasthenia gravis. J. Clin. Investig. 1990, 86, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Shinomiya, N.; Nomura, Y.; Segawa, M. A variant of childhood-onset myasthenia gravis: HLA typing and clinical characteristics in Japan. Clin. Immunol. 2004, 110, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, K.; Kay, P.H.; Christiansen, F.T.; Saueracker, G.; Dawkins, R.L. Comparative mapping of the human major histocompatibility complex in different racial groups by pulsed field gel electrophoresis. Hum. Immnol. 1989, 26, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Kida, K.; Hayashi, M.; Yamada, I.; Matsuda, H.; Yoshinaga, J.; Takami, S.; Yashiki, S.; Sonoda, S. Heteropgeneity in myasthenia gravis: HLA phenotypes and autoantibody responses in ocular and generalized types. Ann. Neurol. 1987, 21, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Kida, K.; Yamada, I.; Matsuda, H.; Sonoda, S.; Inoue, H.; Shiga, S. Involvement of HLA in clinical courses of myasthenia gravis. J. Neuroimmunol. 1988, 18, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Perlo, V.P.; Poskanzer, D.C.; Schwab, R.S.; Viets, H.R.; Osserman, K.E.; Genkins, G. Myasthenia gravis: Evaluation of treatment in 1355 patients. Neurology 1966, 16, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Compston, D.A.S.; Vincent, A.; Newsom-Davis, J.; Batchelor, J.R. Clinical, pathological, HLA antigen and immunological evidence for disease heterogeneity in myasthenia gravis. Brain 1980, 103, 579–601. [Google Scholar] [CrossRef]
- Fambrough, D.M.; Drachman, D.B.; Satyamurti, S. Neuromuscular junction in myasthenia gravis: Decreased acetylcholine receptors. Science 1973, 182, 293–296. [Google Scholar] [CrossRef]
- Koneczny, I.; Herbst, R. Myasthenia gravis: Pathogenic effects of autoantibodies on neuromuscular architecture. Cells 2019, 8, 671. [Google Scholar] [CrossRef]
- Witzemann, V.; Brenner, H.-R.; Sakmann, B. Neural factors regulate AChR submit mRNAs at rat neuromuscular synapses. J. Cell Biol. 1991, 114, 125–141. [Google Scholar] [CrossRef]
- Takamori, M. Myasthenia gravis: From the viewpoint of pathogenicity focusing on acetylcholine receptor clustering, trans-synaptic homeostasis and synaptic stability. Front. Mol. Neurosci. 2020, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Cruz, P.M.; Cossins, J.; Beeson, D.; Vincent, A. The neuromuscular junction in health and disease: Molecular mechanisms governing synaptic formation and homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef]
- Ohkawara, B.; Ito, M.; Ohno, K. Secreted signaling molecules at the neuromuscular junction in physiology and pathology. Int. J. Mol. Sci. 2021, 22, 2455. [Google Scholar] [CrossRef] [PubMed]
- Almon, R.R.; Andrew, C.G.; Appel, S.H. Serum globulin in myasthenia gravis: Inhibition of a-bungarotoxin binding to acetylcholine receptors. Science 1974, 186, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.N.; Ringel, S.P.; Engel, W.K.; Daniels, M.P.; Vogel, Z. Myasthenia gravis: A serum factor blocking acetylcholine receptors of the human neuromuscular junction. Lancet 1975, 1, 607–608. [Google Scholar] [CrossRef]
- Engel, A.G.; Lindstrom, J.M.; Lambert, E.H.; Lennon, V.A. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and in its experimental autoimmune model. Neurology 1977, 27, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, S.J.; Seybold, M.E.; Lindstrom, M. Specificities of antibodies to acetylcholine receptors in sera from myasthenia gravis patients measured by monoclonal antibodies. Proc. Natl. Acad. Sci. USA 1982, 79, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Conti-Fine, B.M.; Milani, M.; Kaminski, H.J. Myasthenia gravis: Past, present, and future. J. Clin. Investig. 2006, 116, 2843–2854. [Google Scholar] [CrossRef]
- Drachman, D.B.; Adams, R.N.; Josifek, L.F.; Pestronk, A.; Stanley, E.F. Antibody-mediated mechanisms of ACh receptor loss in myasthenia gravis: Clinical relevance. Ann. N. Y. Acad. Sci. 1981, 377, 175–188. [Google Scholar] [CrossRef]
- Kao, I.; Drachman, D.B. Myasthenic immunoglobulin accelerates acetylcholine receptor degradation. Science 1977, 196, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.M.; Drachman, D.B. Anti-acetylcholine receptor antibodies directed against the α-bunbgarotoxin binding site induce a unique form of experimental myasthenia. Proc. Natl. Acad. Sci. USA 1983, 80, 4089–4093. [Google Scholar] [CrossRef]
- Whiting, P.J.; Vincent, A.; Newsom-Davis, J. Acetylcholine receptor antibody characteristics in myasthenia gravis. Fractionation of alpha-bungarotoxin binding site antibodies and their relationship to IgG subclass. J. Neuroimmunol. 1983, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef] [PubMed]
- Tuzun, E.; Christadoss, P. Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmun. Rev. 2013, 12, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Sahashi, K.; Engel, A.G.; Lambert, E.H.; Howard, F.M., Jr. Ultrastructural localization of the terminal and lytic ninth complement component (C9) at the motor end-plate in myasthenia gravis. J. Neuropathol. Exp. Neurol. 1980, 39, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Tsujihata, M.; Yoshimura, T.; Satoh, A.; Kinoshita, I.; Matsuo, H.; Mori, M.; Nagataki, S. Diagnostic significance of IgG, C3, and C9 at the limb muscle motor end-plate in minimal myasthenia gravis. Neurology 1989, 39, 1359–1363. [Google Scholar] [CrossRef]
- Lennon, V.A.; Seybold, M.E.; Lindstrom, J.M.; Cochrane, C.; Ulevitch, R. Role of complement in the pathogenesis of experimental autoimmune myasthenia gravis. J. Exp. Med. 1978, 147, 973–983. [Google Scholar] [CrossRef]
- Donaldson, J.O.; Penn, A.S.; Lisak, R.P.; Abramski, O.; Brenner, T.; Schotland, D.L. Antiacetylcholine receptor antibody in neonatal myasthenia gravis. Am. J. Dis. Child. 1981, 135, 222–226. [Google Scholar] [CrossRef]
- Namba, T.; Brown, S.B.; Grob, D. Neonatal myasthenia gravis: Report of two cases and review of the literature. Pediatrics 1970, 45, 488–504. [Google Scholar] [CrossRef]
- Lefvert, A.K.; Osterman, P.O. Newborn infants to myasthenic mothers: A clinical study and an investigation of acetylcholine receptor antibodies in 17 children. Neurology 1983, 33, 133–138. [Google Scholar] [CrossRef]
- Tzartos, S.J.; Efthimiadis, A.; Morel, E.; Eymard, B.; Bach, J.F. Neonatal myasthenia gravis: Antigenic specificities of antibodies in sera from mothers and their infants. Clin. Exp. Immunol. 1990, 80, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E. Myasthenia gravis can have consequences for pregnancy and the developing child. Front. Neurol. 2020, 11, 554. [Google Scholar] [CrossRef]
- Samuels, P.; Bussel, J.B.; Braitman, L.E.; Tomaski, A.; Druzin, M.L.; Mennuti, M.T.; Cines, D.B. Estimation of the risk of Thrombocytonia in the offspring of pregnant women with presumed immune thrombocytopenic purpura. N. Engl. J. Med. 1990, 323, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, K.; Harada, Y.; Fujimoto, T.; Kuramotop, A.; Ikeda, Y.; Akatsuka, J.; Dan, K.; Omine, M.; Mizoguchi, H. Nationwide study of idiopathic thrombocytopenic purpura in pregnant women and the clinical influence on neonates. Int. J. Hematol. 2002, 75, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Newland, C.; Brueton, L.; Beeson, D.; Riemersma, S.; Huson, S.M.; Newsom-Davis, J. Arthrogryposis multiplex congenita with maternal autoantibodies specific for a fetal antigen. Lancet 1995, 346, 24–25. [Google Scholar] [CrossRef]
- Riemersma, S.; Vincent, A.; Beeson, D.; Newland, C.; Hawke, S.; Garabedian, B.V.-D.; Eymard, B.; Newsom-Davis, J. Association of arthrogryposis multiplex congenita with maternal antibodies inhibiting fetal acetylcholine receptor function. J. Clin. Investig. 1996, 98, 2358–2363. [Google Scholar] [CrossRef]
- Saxena, A.; Stevens, J.; Cetin, H.; Koneczny, I.; Webster, R.; Lazaridis, K.; Tzartos, S.; Vrolix, K.; Nogales-Gadea, G.; Machiels, B.; et al. Characterization of an anti-fetal AChR monoclonal antibopdy isolated from a myasthenia gravis patient. Sci. Rep. 2017, 7, 14426. [Google Scholar] [CrossRef]
- Hoch, W.; McConville, J.; Helms, S.; Newsom-Davis, J.; Melms, A.; Vincent, A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nature Med. 2001, 7, 365–368. [Google Scholar] [CrossRef]
- McConville, J.; Farrugia, M.E.; Beeson, D.; Kishore, U.; Mercalfe, R.; Newsom-Davis, J.; Vincent, A. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann. Neurol. 2014, 55, 580–584. [Google Scholar] [CrossRef]
- Sanders, D.B.; Ei-Salem, K.; Massey, J.M.; McConville, J.; Vincent, A. Clinical aspects of MuSK antibody positive seronegative MG. Neurology 2003, 60, 1978–1980. [Google Scholar] [CrossRef]
- Zagar, M.; Vranjes, D.; Sostarko, M.; Vorgrinc, Z.; Bilic, E.; Cepe, M.T. Myasthenia gravis patients with anti-MuSK antibodies. Coll. Antropol. 2009, 33, 1151–1154. [Google Scholar]
- Chan, K.H.; Lachance, D.H.; Harper, C.M.; Lennon, V.A. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve 2007, 36, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Gunaratne, P.; Gamage, R.; Riffsy, M.T.M.; Vincent, A. MuSK-antibody-positive myasthenia gravis in a south Asian population. J. Neurol. Sci. 2009, 284, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guan, Y.; Han, J.; Li, M.; Shi, M.; Deng, H. Regional features of MuSK antibody-positive myasthenia gravis in North China. Front. Neurol. 2020, 11, 516211. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Sung, J.J.; Cho, J.-Y.; Oh, D.-H.; Kim, H.-J.; Park, J.-H.; Lee, K.W.; Choi, Y.-C.; Vincent, A. MuSK antibody-positive, seronegative myasthenia gravis in Korea. J. Clin. Neurosci. 2006, 13, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Shigemoto, K.; Fujinami, A.; Maruyama, N.; Konishi, T.; Ohta, M. Clinical and experimental features of MuSK antibody positive MG in Japan. Eur. J. Neurol. 2007, 14, 1029–1034. [Google Scholar] [CrossRef]
- Murai, H.; Noda, T.; Himeno, E.; Kawano, Y.; Ohyagi, Y.; Shiraishi, H.; Motomura, M.; Kira, J. Infantile onset myasthenia gravis with MuSDK antibodies. Neurology 2006, 67, 174. [Google Scholar] [CrossRef]
- Takahashi, Y.; Sugiyama, M.; Ueda, Y.; Itoh, T.; Yagyu, K.; Shiraishi, H.; Ukeba-Terashita, Y.; Nakanishi, M.; Nagashima, T.; Imai, T.; et al. Childhood-onset anti-MuSK antibody positive myasthenia gravis demonstrates a distinct clinical course. Brain Dev. 2012, 34, 784–786. [Google Scholar] [CrossRef]
- Niks, E.H.; Kuks, J.B.M.; Roep, B.O.; Haasnoot, G.W.; Verduijn, W.; Ballieux, B.E.P.B.; De Baets, M.H.; Vincent, A.; Verschuuren, J.J.G.M. Strong association of MuSK antibody-positive myasthenia gravis and HLA-DR14-DQ5. Neurology 2006, 66, 1772–1774. [Google Scholar] [CrossRef]
- Kanai, T.; Uzawa, A.; Kawaguchi, N.; Sakamaki, T.; Yoshiyama, Y.; Himuro, K.; Oda, F.; Kuwabara, S. HLA-DRB1*14 and DQB1*05 are associated with Japanese anti-MuSK antibody-positive myasthenia gravis patients. J. Neurol. Sci. 2016, 363, 116–118. [Google Scholar] [CrossRef]
- Hong, Y.; Li, H.-F.; Romi, F.; Skeie, G.O.; Gilhus, N.E. HLA and MuSK-positive myasthenia gravis: A systemtic review and meta-analysis. Acta Neurol. Scand. 2018, 138, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Tsugawa, J.; Fukae, J.; Fukuhara, K.; Kawano, H.; Fujioka, S.; Tsuboi, Y. Myasthenia gravis with anti-muscle-specific tyrosine kinase antibody during pregnancy and risk of neonatal myasthenia gravis: A case report and review of the literature. Case Rep. Neurol. 2020, 12, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.L.H.; Mills, K.R.; Riordan-Eva, P.; Barnes, P.R.J.; Rose, M.R. Anti-MuSK antibodies in a case of ocular myasthenia gravis. J. Neurol. Neurosurg. Psychiatry 2005, 564, 075812. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hosaka, A.; Takuma, H.; Ohta, K.; Tamaoka, A. An ocular form of myasthenia gravis with a high titer of anti-MuSK antibodies during a long-term follow-up. Int. Med. 2012, 51, 3077–3079. [Google Scholar] [CrossRef][Green Version]
- Shiraishi, H.; Motomura, M.; Yoshimura, T.; Fukudome, T.; Fukuda, T.; Nakao, Y.; Tsujihata, M.; Vincent, A.; Eguchi, K. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody positive myasthenia gravis. Ann. Neurol. 2005, 57, 289–293. [Google Scholar] [CrossRef]
- Koneczny, I.; Cossins, J.; Waters, P.; Beeson, D.; Vincent, A. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1-3 can disperse preformed agrin-independent AChR clusters. PLoS ONE 2013, 8, e80695. [Google Scholar] [CrossRef]
- Meriggioli, M.N.; Sanders, D.B. Autoimmune myasthenia gravis: Emerging clinical and biological heterogeneity. Lancet Neurol. 2009, 8, 475–490. [Google Scholar] [CrossRef]
- Otsuka, K.; Ito, M.; Ohkawara, B.; Masuda, A.; Kawakami, Y.; Sahashi, K.; Nishida, H.; Mabuchi, N.; Takano, A.; Engel, A.G.; et al. Collagen Q and anti-MuSK autoantibody competitively suppress agrin/LRP4/MuSK signaling. Sci. Rep. 2015, 5, 13928. [Google Scholar] [CrossRef]
- Rodriguez Cruz, P.M.; Al-Hjjar, M.; Huda, S.; Jacobson, L.; Woodhall, M.; Jayawant, S.; Buckley, C.; Hilton-Jones, D.; Beeson, D.; Vincent, A.; et al. Clinical features and diagnostic usefulness of antibodies to cultured acetylcholine receptors in the diagnosis of seronegative myasthenia gravis. JAMA Neurol. 2015, 72, 642–649. [Google Scholar] [CrossRef]
- Budhram, A. Fixed cell-based assays for autoantibody detection in myasthenia gravis: A diagnostic breakthrough. Lancet Reg. Health West. Pac. 2023, 38, 100876. [Google Scholar] [CrossRef]
- Pevzner, A.; Schoser, B.; Peters, K.; Cosma, N.-C.; Karakatsani, A.; Schalke, B.; Melms, A.; Kröger, S. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J. Neurol. 2012, 259, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, O.; Hamuro, J.; Motomura, M.; Yamanashi, Y. Autoantibodies to low-density lipoprotein receptor-related protein in myasthenia gravis. Ann. Neurol. 2011, 69, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Zhang, B.; Belimezi, M.; Ragheb, S.; Bealmear, B.; Lewis, R.A.; Xiong, W.-C.; Lisak, R.P.; Tzartos, S.J.; Mei, L. Autoantibodies to lipoprotein-related protein 4 patients with double-seronegative myasthenia gravis. Arch. Neurol. 2012, 69, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Lu, Y.; Zhang, B.; Figueiredo, D.; Bean, J.; Jung, J.; Wu, H.; Barik, A.; Yin, D.-M.; Xiong, W.-C.; et al. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J. Clin. Investig. 2013, 123, 5190–5202. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, C.; Melms, A.; Schoser, B.; Zhang, Y.; Meltoranta, J.; Risson, V.; Schaeffer, L.; Schalke, B.; Kröger, S. Anti-agrin autoantibodies in myasthenia gravis. Neurology 2014, 82, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shen, C.; Bealmear, B.; Ragheb, S.; Xiong, W.-C.; Lewis, R.A.; Lisak, R.P.; Mei, L. Autoantibodies to agrin in myasthenia gravis patients. PLoS ONE 2014, 9, e91816. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Liu, Z.; Fei, E.; Chen, W.; Lai, X.; Luo, B.; Chen, P.; Jing, H.; Pan, J.-X.; Rivner, M.H.; et al. Induction of anti-agrin antibodies causes myasthenia gravis in mice. Neuroscience 2018, 373, 113–121. [Google Scholar] [CrossRef]
- Rivner, M.H.; Quarles, B.M.; Pan, J.-X.; Yu, Z.; Howard, J.F.; Corse, A.; Dimachkie, M.M.; Jackson, C.; Vu, T.; Small, G.; et al. Clinical features of LRP4/agrin-antibody-positive myasthenia gravis: A multicenter study. Muscle Nerve 2020, 62, 333–343. [Google Scholar] [CrossRef]
- Kaminski, H.J. Seronegative myasthenia gravis—A vanishing disorder? JAMA Neurol. 2016, 73, 1055–1056. [Google Scholar] [CrossRef]
- Drachman, D.B. How to recognize an antibody-mediated autoimmune disease: Criteria. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1990, 68, 183–186. [Google Scholar]
- Shigemoto, K.; Kubo, S.; Maruyama, N.; Hato, N.; Yamada, H.; Jie, C.; Kobayashi, N.; Mominoki, K.; Abe, Y.; Ueda, N.; et al. Induction of myasthenia gravis by immunization against muscle-specific kinase. J. Clin. Investig. 2006, 116, 1016–1024. [Google Scholar] [CrossRef]
- Viegas, S.; Jacobson, L.; Waters, P.; Cossins, J.; Jacob, S.; Leite, M.I.; Webster, R.; Vincent, A. Passive and active immunization models of MuSK-Ab positive myasthenia: Electrophysiological evidence for pre and postsynaptic defects. Exp. Neurol. 2012, 234, 506–512. [Google Scholar] [CrossRef]
- Ulsoy, C.; Cavus, F.; Yilmaz, V.; Tuzun, E. Immunization with recombinantly expressed LRP4 induces experimental autoimmune myasthenia gravis in C57BL/6 mice. Immunol. Investig. 2017, 46, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Motohashi, N.; Takashima, R.; Kishi, M.; Nishimune, H.; Shigemoto, K. Immunization of mice with LRP4 induces myasthenia similar to MuSK-associated myasthenia gravis. Exp. Neurol. 2017, 297, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, M.; Jing, H.; Chen, P.; Cao, R.; Pan, J.; Luo, B.; Yu, Y.; Quarles, B.M.; Xiong, W.; et al. Characterization of LRP4/Agrin antibodies from a patient with myasthenia gravis. Neurology 2021, 97, e975–e987. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.-M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndrome: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Parr, J.R.; Andrew, M.J.; Finnis, M.; Beeson, D.; Vincent, A.; Jayawant, S. How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch. Dis. Child. 2014, 99, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Nakata, T.; Tanaka, M.; Shen, X.-M.; Ito, M.; Iwata, S.; Okuno, T.; Nomura, Y.; Ando, N.; Ishigaki, K.; et al. Congenital myasthenic syndrome in Japan: Ethnically unique mutations in muscle nicotinic acetylcholine receptor subunits. Neuromuscul. Disord. 2015, 25, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Ohkawara, B.; Shen, X.-M.; Selcen, D.; Engel, A.G. Clinical and pathologic features of congenital myasthenic syndromes caused by 35 genes—A comprehensive review. Int. J. Mol. Sci. 2023, 24, 3730. [Google Scholar] [CrossRef]
- Hayashi, M.; Kida, K.; Yamada, I.; Morimoto, T.; Matsuda, H.; Mimaki, T.; Yabuuchi, H. Anti-acetylcholine receptor antibody in juvenile and adult myasthenia gravis. Acta Paediatr. Jpn. 1986, 28, 781–787. [Google Scholar]
- Zao, G.; Wang, X.; Yu, X.; Zhang, X.; Guan, Y.; Jiang, J. Clinical application of clustered-AChR for the detection SNMG. Sci. Rep. 2015, 5, 10193. [Google Scholar] [CrossRef] [PubMed]
- Oda, K. Differences in acetylcholine receptor-antibody interactions between extraocular and extremity muscle fibers. Ann. N. Y. Acad. Sci. 1993, 681, 238–255. [Google Scholar] [CrossRef]
- Kaminski, H.J.; Maas, E.; Spiegel, P.; Ruff, R.L. Why are eye muscles frequently involved in myasthenia gravis? Neurology 1990, 40, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.M.; Manfredi, A.A.; Conti-Tronconi, B.M. The ‘embrionic’ gamma subunit of the nicotinic acetylcholine receptor is expressed in adult extraocular muscle. Neurology 1993, 43, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Missias, A.C.; Chu, G.C.; Klocke, B.J.; Sanes, J.R.; Merlie, J.P. Maturation of the acetylcholine receptor in skeletal muscle: Regulation of the AChR γ-to-ϵ switch. Dev. Biol. 1996, 179, 223–238. [Google Scholar] [CrossRef]
- Oda, K. Motor innervation and acetylcholine receptor distribution of human extraocular muscle fibers. J. Neurol. Sci. 1986, 74, 125–133. [Google Scholar] [CrossRef]
- Kaminski, H.J.; Kusner, L.L.; Block, C.H. Expression of acetylcholine receptor isoforms at extraocular muscle endplates. Investig. Ophthalmol. Vis. Sci. 1996, 37, 345–351. [Google Scholar]
- Ruff, R.L.; Lennon, V.A. How myasthenia gravis alters the safety factor for neuromuscular transmission? J. Neuroimmunol. 2008, 201–202, 13–20. [Google Scholar] [CrossRef]
- Serra, A.; Ruff, R.; Leigh, R.J. Neuromuscular transmission failure in myasthenia gravis: Decrement of safety factor and susceptibility of extraocular muscle. Ann. N. Y. Acad. Sci. 2012, 1275, 129–135. [Google Scholar] [CrossRef]
- Heckmann, J.M.; Europa, T.A.; Soni, A.J.; Nel, M. The epidemiology and phenotypes of ocular manifestations in childhood and juvenile myasthenia gravis: A review. Front. Neurol. 2022, 13, 834212. [Google Scholar] [CrossRef]
- Thapa, P.; Farber, D.L. The role of the thymus in the immune response. Thorac. Surg. Clin. 2019, 29, 123–1312. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Ohigashi, I.; Takahama, Y. Thymus machinery for T-cell selection. Int. Immunol. 2019, 31, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Thome, J.J.C.; Grinshpun, B.; Kumar, B.V.; Kubota, M.; Ohmura, Y.; Lerner, H.; Sempowski, G.D.; Shen, Y.; Farber, D.L. Long-term maintenance of human naïve T cell through in situ homeostasis in lymphoid tissue sites. Sci. Immunol. 2016, 1, eaah6506. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.N.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Immunosenescence and human vaccine immune responses. Immun. Ageing 2019, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Berman, P.W.; Patrick, J. Experimental myasthenia gravis: A murine system. J. Exp. Med. 1980, 151, 204–223. [Google Scholar] [CrossRef] [PubMed]
- Christadoss, P.; Lennon, V.A.; Krco, C.J.; David, C.S. Genetic control of experimental autoimmune myasthenia gravis in mice III. Ia molecules mediate cellular immune responsiveness to acetylcholine receptors. J. Immunol. 1982, 128, 1141–1144. [Google Scholar] [CrossRef]
- McIntyre, K.R.; Seidman, J.G. Nucleotide sequence of mutant I-Abbm12 gene is evidence for genetic exchange between mouse immune response genes. Nature 1984, 308, 551–553. [Google Scholar] [CrossRef]
- Christadoss, P.; Lindstrom, J.M.; Melvold, R.W.; Talal, N. Mutation at I-A beta chain prevents experimental autoimmune myasthenia gravis. Immunogenet 1985, 21, 33–38. [Google Scholar] [CrossRef]
- Bellone, M.; Ostlie, N.; Lei, S.; Wu, X.-D.; Conti-Tronconi, B.M. The I-Abm12 mutation, which confers resistance to experimental myasthenia gravis, drastically affects the epitope repertoire of murine CD4+ cells sensitized to nicotinic acetylcholine receptor. J. Immunol. 1991, 147, 1484–1491. [Google Scholar] [CrossRef]
- Infante, A.J.; Thompson, P.A.; Krolick, K.A.; Wall, K.A. Determinant selection in murine experimental autoimmune myasthenia gravis: Effect of the bm12 mutation on T cell recognition of acetylcholine receptor epitopes. J. Immunol. 1991, 146, 2977–2982. [Google Scholar] [CrossRef]
- Wu, X.; Tuzun, E.; Saini, S.S.; Wang, J.; Li, J.; Aguilera-Aguirre, L.; Huda, R.; Christadoss, P. Ocular myasthenia gravis induced by human acetylcholine receptor e subunit immunization in HLA-DR3 transgenic mice. Immunol. Lett. 2015, 168, 306–3012. [Google Scholar] [CrossRef]
- Hayashi, M.; Matsuda, O.; Ishida, Y.; Kida, K. Change of immunological parameters in the clinical course of a myasthenia gravis patient with chronic graft-versus-host disease. Acta Paediatr. Jpn. 1996, 38, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P., III; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Shapiro, M.S.; Brunner, N.G.; Grob, D. Myasthenia gravis occurring in twins. J. Neurol. Neurosurg. Psychiatr. 1971, 34, 531–534. [Google Scholar] [CrossRef] [PubMed][Green Version]
- O’Connell, K.; Ramdas, S.; Palace, J. Management of juvenile myasthenia gravis. Front. Neurol. 2020, 11, 743. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M. Childhood myasthenia gravis in Japan: Pathophysiology and treatment options. Clin. Exp. Neuroimmunol. 2023, 14, 185–194. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Skeie, G.O.; Romi, F.; Lazaridis, K.; Zisimopoulou, P.; Tzartos, S. Myasthenia gravis—Autoantibody characteristics and their implications for therapy. Nat. Rev. Neurol. 2016, 12, 259–268. [Google Scholar] [CrossRef]
- Hayashi, M.; Kida, K.; Yoshinaga, J. Possible distinct pathogenesis in low responder myasthenia gravis: Association of soluble interleukin-2 receptor with acetylcholine receptor antibody titre or abnormal thymus. J. Neurol. Neurosurg. Psychiatr. 1996, 61, 207–208. [Google Scholar] [CrossRef][Green Version]
- Wiesendanger, M.; D’Alessandri, A. Myasthenia gravis mit fokaler infiltration der endplattenzone. Acta Neuropathol. 1963, 2, 246–252. [Google Scholar] [CrossRef]
- Fenichel, G.M.; Shy, G.M. Muscle biopsy experience in myasthenia gravis. Arch. Neurol. 1963, 9, 237–243. [Google Scholar] [CrossRef]
- Fenichel, G.M. Muscle lesions in myasthenia gravis. Ann. N. Y. Acad. Sci. 1966, 135, 60–67. [Google Scholar] [CrossRef]
- Pascuzzi, R.M.; Campa, J.F. Lymphorrhage localized to the muscle end-plate in myasthenia gravis. Arch. Pathol. Lab. Med. 1988, 112, 934–937. [Google Scholar]
- Maselli, R.A.; Richman, D.P.; Wollmann, R.L. Inflammation at the neuromuscular junction in myasthenia gravis. Neurology 1991, 41, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Engel, A.G. Myasthenia gravis: Quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the endplate in 30 patients. Neurology 1993, 43, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Hans, J.G.H. Oosterhuis. Animal model of myasthenia gravis. In Myasthenia Gravis; Glaser, G.H., Barbeau, A., Eds.; Clinical Neurology Neurosurgery Monographs; Churchill Livingstone: Edinburgh, UK; Melbourne, Australia; New York, NY, USA, 1984; Volume 5, pp. 131–141. [Google Scholar]
- Lindstrom, J.M.; Engel, A.G.; Seybold, M.E.; Lennon, V.A.; Lambert, E.H. Pathological mechanisms in experimental autoimmune myasthenia gravis. II. Passive transfer of experimental autoimmune myasthenia gravis in rats with anti-acetylcholine receptor antibodies. J. Exp. Med. 1976, 144, 739–753. [Google Scholar] [CrossRef]
- Gomez, C.M.; Wollmann, R.L.; Richman, D.P. Induction of morphologic changes of both acute and chronic experimental myasthenia by monoclonal antibody directed against acetylcholine receptor. Acta Neuropathol. 1984, 63, 131–143. [Google Scholar] [CrossRef]
- Zhou, Y.; Kaminski, H.J.; Gong, B.; Cheng, G.; Feuerman, J.M.; Kusner, L. RNA expression analysis of passive transfer myasthenia supports extraocular muscle as a unique immunological environment. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4348–4359. [Google Scholar] [CrossRef] [PubMed][Green Version]



{kind=link}
{kind=link}
| Author (Reference) | Year | Nation | Number of Patients | OMG | Onset Age (yr) | OMG to GMG | Spontaneous Remission | |
|---|---|---|---|---|---|---|---|---|
| Asia | Murai [15] | 2011 | Japan | 268 | 80% | <10 | ||
| Lee HN [19] | 2016 | Korea | 88 | 97% | <18 | |||
| Huang X [16] | 2013 | China | 327 | 75% | <18 | 19.9% | 3.4% | |
| Gui [17] | 2015 | China | 424 | 83% | <14 | 11.8% | ||
| Yang L [25] | 2022 | China | 343 | 96% | <14 | 13.4% | ||
| Chou CC [18] | 2019 | Taiwan | 54 | 83% | <20 | 4.8% | 24.1% | |
| Western | Popperud [21] | 2017 | Norway | 63 | 59% | <18 | ||
| Mansukhani [23] | 2019 | USA | 146 | 23% | <19 | 31.3% | ||
| Vecchio [24] | 2020 | UK | 74 | 51% | <16 | 23% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashi, M. Pathophysiology of Childhood-Onset Myasthenia: Abnormalities of Neuromuscular Junction and Autoimmunity and Its Background. Pathophysiology 2023, 30, 599-617. https://doi.org/10.3390/pathophysiology30040043
Hayashi M. Pathophysiology of Childhood-Onset Myasthenia: Abnormalities of Neuromuscular Junction and Autoimmunity and Its Background. Pathophysiology. 2023; 30(4):599-617. https://doi.org/10.3390/pathophysiology30040043
Chicago/Turabian StyleHayashi, Masatoshi. 2023. "Pathophysiology of Childhood-Onset Myasthenia: Abnormalities of Neuromuscular Junction and Autoimmunity and Its Background" Pathophysiology 30, no. 4: 599-617. https://doi.org/10.3390/pathophysiology30040043
APA StyleHayashi, M. (2023). Pathophysiology of Childhood-Onset Myasthenia: Abnormalities of Neuromuscular Junction and Autoimmunity and Its Background. Pathophysiology, 30(4), 599-617. https://doi.org/10.3390/pathophysiology30040043