
The Effect of Aflatoxin B1 on Tumor-Related Genes and Phenotypic Characters of MCF7 and MCF10A Cells

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Results
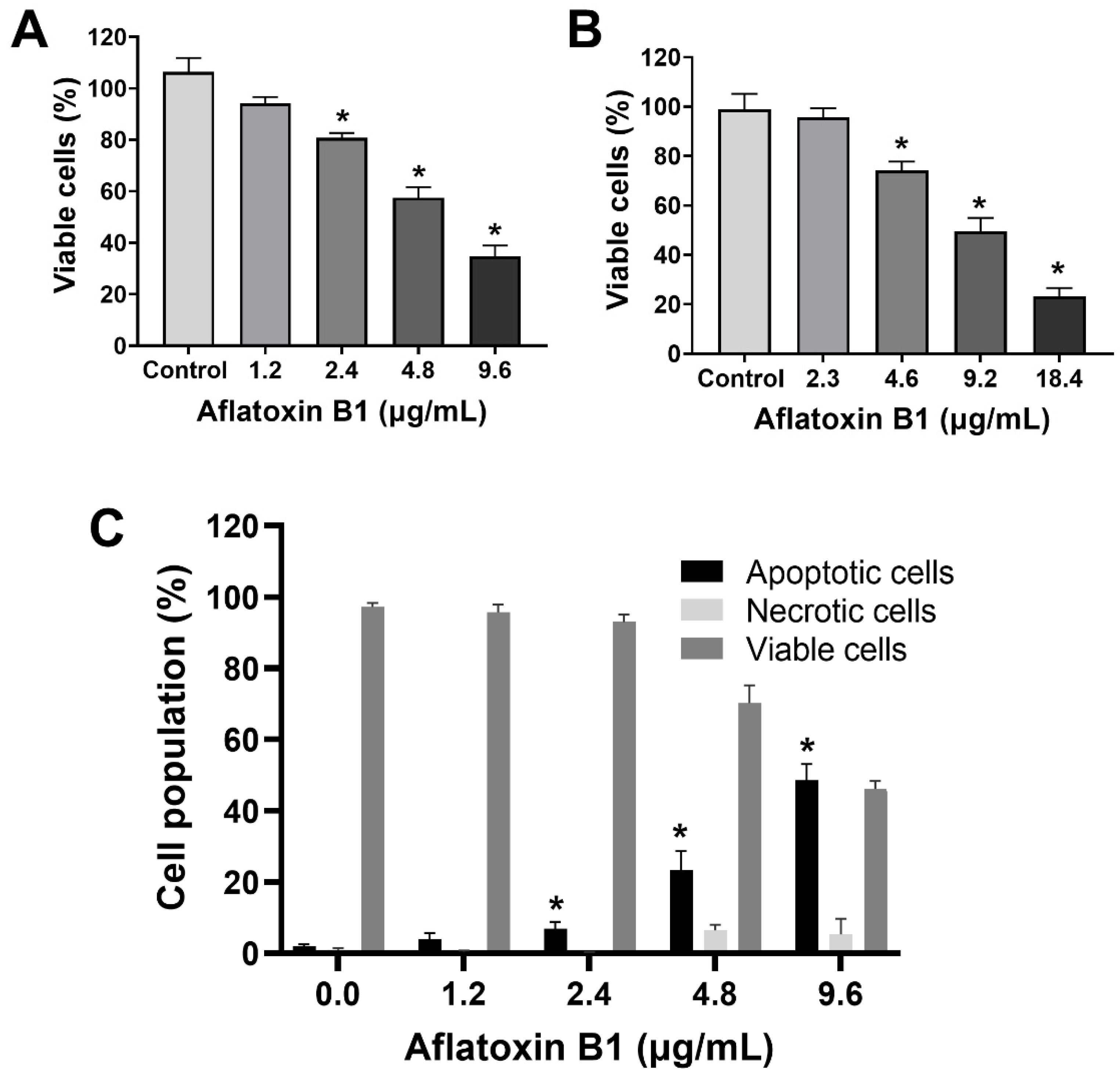
2.1. AB1-Induced Cytotoxicity and Apoptotic Cell Death with Reduction in Viability of MCF7 and MCF10A Cell Lines
2.2. MCF7 and MCF10A Cells Showed a Slight Decrease in Reactive Oxygen Species Levels Post Treatment of AB1
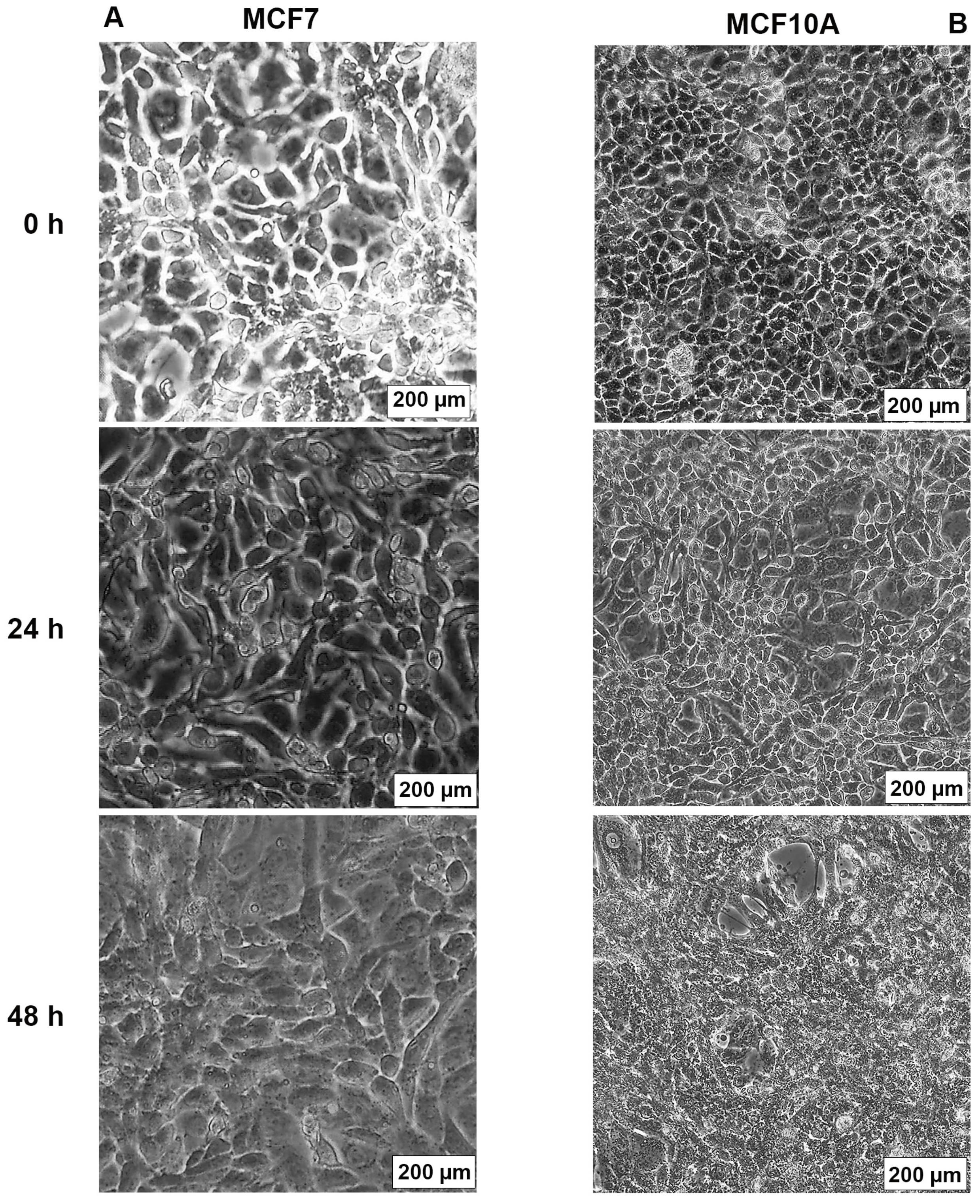
2.3. AB1 Caused Hypertrophy and Cell-Cycle Arrest but Increased Cell Migration Rates in MCF7 and MCF10A cells
2.4. Gene Expression Levels of Different Tumour-Related Cell Signaling Pathways
2.5. Gene Expression Levels of Different Cell Signaling Pathways following AB1 Treatment in MCF7 Cells
2.5.1. AB1 Increased the Gene Expressions of BRCA2 and p53 but Reduced the Expression of BRCA1 in MCF7 Cells
2.5.2. AB1 Reduced the Gene Expressions of HER1 and HER2 but Increased the Expression of cMyc in MCF7 Cells
2.5.3. AB1 Increased the Gene Expressions of WNT3A and MAPK1 but Reduced the Expressions of CCND1 and MAPK3 in MCF7 Cells
2.5.4. AB1 Increased the Gene Expressions of BCL2 and DAPK1 but Reduced the Expressions of MCL1 and Casp9 in MCF7 Cells
2.6. Gene Expression Levels of Different Cell Signaling Pathways following AB1 Treatment in MCF10A Cells
2.6.1. AB1 Reduced the Gene Expressions of BRCA1, BRCA2, and p53 in MCF10A Cells
2.6.2. AB1 Reduced the Gene Expressions of HER1, HER2, and cMyc in MCF10A Cells
2.6.3. AB1 Reduced the Gene Expressions of CCND1 and MAPK3 but Increased the Expression of MAPK1 in MCF10A Cells
2.6.4. AB1 Increased the Gene Expressions of BCL2, MCL1, DAPK1, Casp8, and Casp9 in MCF10A Cells
3. Discussion
3.1. Gene Expression of Tumor-Suppressing Genes Indicates Damage to the MCF7 and MCF10A Genome
3.2. Gene Expression of Oncogenes Indicates an Increase in MCF7 and MCF10A Invasiveness Post-Treatment with AB1 through Interfering with Proliferation Activities
3.3. AB1 Altered Cell Cycle Progression in MCF7 and MCF10A Cells
3.4. AB1 Promotes Evading Apoptosis, Increasing Cell Viability and Invasiveness
4. Materials and Methods
4.1. Cell Lines, Culture Media, Seeding Density, and Toxin Concentration
4.2. Measurement of Cytotoxicity, Cell Viability and Apoptosis
4.3. Measurement of Reactive Oxygen Species (ROS) Levels
4.4. Measurement Cell Morphology, Cell Migration Measurement, and Cell Cycle Progression
4.5. Measurement of mRNA and Gene Expression Levels
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmed Adam, M.A.; Tabana, Y.M.; Musa, K.B.; Sandai, D.A. Effects of different mycotoxins on humans, cell genome and their involvement in cancer. Oncol. Rep. 2017, 37, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Bbosa, G.S.; Kitya, D.; Lubega, A.; Ogwal-Okeng, J.; Anokbonggo, W.W.; Kyegombe, D.B. Review of the biological and health effects of aflatoxins on body organs and body systems. Aflatoxins-Recent Adv. Future Prospect. 2013, 12, 239–265. [Google Scholar] [CrossRef]
- Al-Fakih, A.A. Overview on the fungal metabolites involved in mycopathy. Open J. Med. Microbiol. 2014, 2014, 43243. [Google Scholar] [CrossRef]
- Chu, F.S. Mycotoxins: Food contamination, mechanism, carcinogenic potential and preventive measures. Mutat. Res. Genet. Toxicol. 1991, 259, 291–306. [Google Scholar] [CrossRef]
- Jelinek, C.F.; Pohland, A.E.; Wood, G.E. Worldwide occurrence of mycotoxins in foods and feeds—An update. J. Assoc. Off. Anal. Chem. 1989, 72, 223–230. [Google Scholar] [CrossRef]
- Lane, K.S. Aflatoxin, tobacco, ammonia and the p53 tumor-suppressor gene: Cancer’s missing link. Med. Gen. Med. 1999, 1, E10. [Google Scholar]
- Rustom, I.Y. Aflatoxin in food and feed: Occurrence, legislation and inactivation by physical methods. Food Chem. 1997, 59, 57–67. [Google Scholar] [CrossRef]
- Tabata, S.; Kamimura, H.; Ibe, A.; Hashimoto, H.; Iida, M.; Tamura, Y.; Nishima, T. Aflatoxin contamination in foods and foodstuffs in Tokyo: 1986–1990. J. AOAC Int. 1993, 76, 32–35. [Google Scholar] [CrossRef]
- Bedoya-Serna, C.M.; Michelin, E.C.; Massocco, M.M.; Carrion, L.C.; Godoy, S.H.; Lima, C.G.; Ceccarelli, P.S.; Yasui, G.S.; Rottinghaus, G.E.; Sousa, R.L. Effects of dietary aflatoxin B1 on accumulation and performance in matrinxã fish (Brycon cephalus). PLoS ONE 2018, 13, e0201812. [Google Scholar] [CrossRef]
- Rasooly, R.; Hernlem, B.; He, X.; Friedman, M. Non-linear relationships between aflatoxin B1 levels and the biological response of monkey kidney vero cells. Toxins 2013, 5, 1447–1461. [Google Scholar] [CrossRef]
- Fasullo, M. Cellular Responses to Aflatoxin-Associated DNA Adducts. In DNA Repair—An Update; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Aguilar, F.; Hussain, S.P.; Cerutti, P. Aflatoxin B1 induces the transversion of G--> T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 8586–8590. [Google Scholar] [CrossRef] [PubMed]
- Gasco, M.; Shami, S.; Crook, T. The p53 pathway in breast cancer. Breast Cancer Res. 2002, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hosteller, R.; Cleary, K.; Signer, S.H.; Davidson, N.; Baylin, S.; Devilee, P. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Mehrgou, A.; Akouchekian, M. The importance of BRCA1 and BRCA2 genes mutations in breast cancer development. Med. J. Islam. Repub. Iran 2016, 30, 369. [Google Scholar]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1-and BRCA2-associated hereditary breast and ovarian cancer. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Deng, Z.-L.; Ma, Y. Aflatoxin sufferer and p53 gene mutation in hepatocellular carcinoma. World J. Gastroenterol. 1998, 4, 28. [Google Scholar] [CrossRef]
- Li, H.; Xing, L.; Zhang, M.; Wang, J.; Zheng, N. The toxic effects of aflatoxin B1 and aflatoxin M1 on kidney through regulating L-proline and downstream apoptosis. BioMed Res. Int. 2018, 2018, 9074861. [Google Scholar] [CrossRef]
- Turner, N.W.; Subrahmanyam, S.; Piletsky, S.A. Analytical methods for determination of mycotoxins: A review. Anal. Chim. Acta 2009, 632, 168–180. [Google Scholar] [CrossRef]
- Renzulli, C.; Galvano, F.; Pierdomenico, L.; Speroni, E.; Guerra, M. Effects of rosmarinic acid against aflatoxin B1 and ochratoxin-A-induced cell damage in a human hepatoma cell line (Hep G2). J. Appl. Toxicol. 2004, 24, 289–296. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Tozlovanu, M.; Tran, T.L.; Pfohl-Leszkowicz, A. Occurrence of aflatoxin B1, citrinin and ochratoxin A in rice in five provinces of the central region of Vietnam. Food Chem. 2007, 105, 42–47. [Google Scholar] [CrossRef]
- Villa, P.; Markaki, P. Aflatoxin B1 and ochratoxin A in breakfast cereals from Athens market: Occurrence and risk assessment. Food Control 2009, 20, 455–461. [Google Scholar] [CrossRef]
- Kabak, B. The fate of mycotoxins during thermal food processing. J. Sci. Food Agric. 2009, 89, 549–554. [Google Scholar] [CrossRef]
- Bullerman, L.B.; Bianchini, A. Stability of mycotoxins during food processing. Int. J. Food Microbiol. 2007, 119, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Roebuck, B.D.; Wogan, G.N.; Groopman, J.D. Aflatoxin: A 50-year odyssey of mechanistic and translational toxicology. Toxicol Sci. 2011, 120, S28–S48. [Google Scholar] [CrossRef]
- Gradelet, S.; Le Bon, A.-M.; Bergès, R.; Suschetet, M.; Astorg, P. Dietary carotenoids inhibit aflatoxin B1-induced liver preneoplastic foci and DNA damage in the rat: Role of the modulation of aflatoxin B1 metabolism. Carcinogenesis 1998, 19, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Golli-Bennour, E.E.; Kouidhi, B.; Bouslimi, A.; Abid-Essefi, S.; Hassen, W.; Bacha, H. Cytotoxicity and genotoxicity induced by aflatoxin B1, ochratoxin A, and their combination in cultured Vero cells. J. Biochem. Mol. Toxicol. 2010, 24, 42–50. [Google Scholar] [CrossRef]
- Gabliks, J.; Schaeffer, W.; Friedman, L.; Wogan, G. Effect of aflatoxin B1 on cell cultures. J. Bacteriol. 1965, 90, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Bedard, L.L.; Massey, T.E. Aflatoxin B1-induced DNA damage and its repair. Cancer Lett. 2006, 241, 174–183. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Meki, A.-R.M.; Abdel-Ghaffar, S.K.; El-Gibaly, I. Aflatoxin B1 induces apoptosis in rat liver: Protective effect of melatonin. Neuro Endocrinol. Lett. 2001, 22, 417–426. [Google Scholar]
- Mavrommatis, A.; Giamouri, E.; Tavrizelou, S.; Zacharioudaki, M.; Danezis, G.; Simitzis, P.E.; Zoidis, E.; Tsiplakou, E.; Pappas, A.C.; Georgiou, C.A. Impact of mycotoxins on animals’ oxidative status. Antioxidants 2021, 10, 214. [Google Scholar] [CrossRef]
- Hou, Y.-J.; Zhao, Y.-Y.; Xiong, B.; Cui, X.-S.; Kim, N.-H.; Xu, Y.-X.; Sun, S.-C. Mycotoxin-containing diet causes oxidative stress in the mouse. PLoS ONE 2013, 8, e60374. [Google Scholar] [CrossRef] [PubMed]
- Engin, A.B.; Engin, A. DNA damage checkpoint response to aflatoxin B1. Environ. Toxicol. Pharmacol. 2019, 65, 90–96. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Shi, X.; Tang, X.; Wang, Y.; Shen, F.; Zhang, Q.; Wang, C.; Jiang, M.; Liu, M.; Yu, L. Aflatoxin B1 induces reactive oxygen species-mediated autophagy and extracellular trap formation in macrophages. Front. Cell Infect. Microbiol. 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Sznarkowska, A.; Kostecka, A.; Meller, K.; Bielawski, K.P. Inhibition of cancer antioxidant defense by natural compounds. Oncotarget 2017, 8, 15996. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APCCdh1 by reactive oxygen species. Mol. Cell Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef]
- Agami, R.; Bernards, R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 2000, 102, 55–66. [Google Scholar] [CrossRef]
- Huang, B.; Chen, Q.; Wang, L.; Gao, X.; Zhu, W.; Mu, P.; Deng, Y. Aflatoxin B1 Induces Neurotoxicity through Reactive Oxygen Species Generation, DNA Damage, Apoptosis, and S-Phase Cell Cycle Arrest. Int. J. Mol. Sci. 2020, 21, 6517. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Cui, J.; Xing, L.; Shen, H.; Wu, S.; Lian, H.; Wang, J.; Yan, X.; Zhang, X. Ochratoxin A induces oxidative DNA damage and G1 phase arrest in human peripheral blood mononuclear cells in vitro. Toxicol. Lett. 2012, 211, 164–171. [Google Scholar] [CrossRef]
- Ricordy, R.; Gensabella, G.; Cacci, E.; Augusti-Tocco, G. Impairment of cell cycle progression by aflatoxin B1 in human cell lines. Mutagenesis 2002, 17, 241–249. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. In Cell Cycle Control; Springer: Berlin/Heidelberg, Germany, 2014; pp. 29–40. [Google Scholar] [CrossRef]
- Bonneton, C.; Sibarita, J.B.; Thiery, J.P. Relationship between cell migration and cell cycle during the initiation of epithelial to fibroblastoid transition. Cell Motil. Cytoskelet. 1999, 43, 288–295. [Google Scholar] [CrossRef]
- Fukui, R.; Amakawa, M.; Hoshiga, M.; Shibata, N.; Kohbayashi, E.; Seto, M.; Sasaki, Y.; Ueno, T.; Negoro, N.; Nakakoji, T. Increased migration in late G1 phase in cultured smooth muscle cells. Am. J. Physiol. Cell Physiol. 2000, 279, C999–C1007. [Google Scholar] [CrossRef] [PubMed]
- Prest, S.J.; Rees, R.C.; Murdoch, C.; Marshall, J.F.; Cooper, P.A.; Bibby, M.; Li, G.; Ali, S.A. Chemokines induce the cellular migration of MCF-7 human breast carcinoma cells: Subpopulations of tumour cells display positive and negative chemotaxis and differential in vivo growth potentials. Clin. Exp. Metastasis 1999, 17, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.; Basolo, F.; Albini, A.; Melchiori, A.; Watanabe, H.; Elliott, J.; Raz, A.; Parodi, S.; Russo, J. Increased invasive, chemotactic and locomotive abilities of c-Ha-ras-transformed human breast epithelial cells. Invasion Metastasis 1991, 11, 38–47. [Google Scholar] [PubMed]
- Monni, O.; Joensuu, H.; Franssila, K.; Klefstrom, J.; Alitalo, K.; Knuutila, S. BCL2 overexpression associated with chromosomal amplification in diffuse large B-cell lymphoma. Blood J. Am. Soc. Hematol. 1997, 90, 1168–1174. [Google Scholar] [CrossRef]
- Koehler, B.C.; Scherr, A.-L.; Lorenz, S.; Urbanik, T.; Kautz, N.; Elssner, C.; Welte, S.; Bermejo, J.L.; Jäger, D.; Schulze-Bergkamen, H. Beyond cell death–antiapoptotic Bcl-2 proteins regulate migration and invasion of colorectal cancer cells in vitro. PLoS ONE 2013, 8, e76446. [Google Scholar] [CrossRef] [PubMed]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Marmorstein, L.Y.; Ouchi, T.; Aaronson, S.A. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc. Natl. Acad. Sci. USA 1998, 95, 13869–13874. [Google Scholar] [CrossRef]
- Rajan, J.V.; Wang, M.; Marquis, S.T.; Chodosh, L.A. Brca2 is coordinately regulated with Brca1 during proliferation and differentiation in mammary epithelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 13078–13083. [Google Scholar] [CrossRef]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J. Cell Sci. 2001, 114, 3591–3598. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007, 8, 235–244. [Google Scholar] [CrossRef]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef]
- Schmidt, E.V. The role of c-myc in cellular growth control. Oncogene 1999, 18, 2988–2996. [Google Scholar] [CrossRef]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Witton, C.J.; Reeves, J.R.; Going, J.J.; Cooke, T.G.; Bartlett, J.M. Expression of the HER1–4 family of receptor tyrosine kinases in breast cancer. J. Pathol. 2003, 200, 290–297. [Google Scholar] [CrossRef]
- Smela, M.E.; Currier, S.S.; Bailey, E.A.; Essigmann, J.M. The chemistry and biology of aflatoxin B1: From mutational spectrometry to carcinogenesis. Carcinogenesis 2001, 22, 535–545. [Google Scholar] [CrossRef]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef]
- Kohrman, A.Q.; Matus, D.Q. Divide or conquer: Cell cycle regulation of invasive behavior. Trends Cell Biol. 2017, 27, 12–25. [Google Scholar] [CrossRef]
- Howe, L.R.; Brown, A.M. Wnt signaling and breast cancer. Cancer Biol. Ther. 2004, 3, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Wang, R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2006, 20, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Fu, H.; Liu, Q.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008, 582, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Brown, M.K.; Steptoe, A.L.; Wani, S.; Chan, W.L.; Forrest, A.R.; Kolle, G.; Gabrielli, B.; Grimmond, S.M. The miR-17-5p microRNA is a key regulator of the G1/S phase cell cycle transition. Genome Biol. 2008, 9, R127. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.M.; Katayama, C.D.; Pagès, G.; Pouysségur, J.; Hedrick, S.M. The role of erk1 and erk2 in multiple stages of T cell development. Immunity 2005, 23, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-g.; Zhou, W.; Han, T.; Du, S.-q.; Li, Z.-h.; Zhang, Z.; Shan, G.-y.; Kong, C.-z. MiR-378 suppresses prostate cancer cell growth through downregulation of MAPK1 in vitro and in vivo. Tumor Biol. 2016, 37, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.A.; Yellen, P.; Xu, L.; Saqcena, M. Regulation of G1 cell cycle progression: Distinguishing the restriction point from a nutrient-sensing cell growth checkpoint (s). Genes Cancer 2010, 1, 1124–1131. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Kim, C.N.; Wang, X.; Huang, Y.; Ibrado, A.M.; Liu, L.; Fang, G.; Bhalla, K. Overexpression of Bcl-xL inhibits Ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res. 1997, 57, 3115–3120. [Google Scholar]
- Gerber, H.-P.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 1998, 273, 13313–13316. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Byun, H.-J.; Kim, B.-R.; Lee, K.H.; Park, S.-Y.; Rho, S.B. DAPk1 inhibits NF-κB activation through TNF-α and INF-γ-induced apoptosis. Cell Signal. 2012, 24, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Xu, L.; Wang, P. MiR-191 inhibits TNF-α induced apoptosis of ovarian endometriosis and endometrioid carcinoma cells by targeting DAPK1. Int. J. Clin. Exp. Pathol. 2015, 8, 4933. [Google Scholar] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, L.; Lobito, A.; Chan, F.K.-M.; Dale, J.; Sneller, M.; Yao, X.; Puck, J.M.; Straus, S.E.; Lenardo, M.J. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 1999, 98, 47–58. [Google Scholar] [CrossRef]
- Olsson, M.; Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 2011, 18, 1441–1449. [Google Scholar] [CrossRef]
- Calmon, M.F.; Colombo, J.; Carvalho, F.; Souza, F.P.; José Filho, F.; Fukuyama, E.E.; Camargo, A.A.; Caballero, O.L.; Tajara, E.H.; Cordeiro, J.A. Methylation profile of genes CDKN2A (p14 and p16), DAPK1, CDH1, and ADAM23 in head and neck cancer. Cancer Genet. Cytogenet. 2007, 173, 31–37. [Google Scholar] [CrossRef]
- Christoph, F.; Kempkensteffen, C.; Weikert, S.; Köllermann, J.; Krause, H.; Miller, K.; Schostak, M.; Schrader, M. Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in vitro effects of demethylating agents in bladder and kidney cancer. Br. J. Cancer 2006, 95, 1701–1707. [Google Scholar] [CrossRef]
- Qu, Y.; Han, B.; Yu, Y.; Yao, W.; Bose, S.; Karlan, B.Y.; Giuliano, A.E.; Cui, X. Evaluation of MCF10A as a reliable model for normal human mammary epithelial cells. PLoS ONE 2015, 10, e0131285. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef] [PubMed]
- ATCC. MCF-7 Breast Adenocarcinoma SOP: Thawing, Propagating and Cryopreserving. Available online: https://www.atcc.org/products/htb-22 (accessed on 14 July 2022).
- Dönmez, Y.; Gündüz, U. Reversal of multidrug resistance by small interfering RNA (siRNA) in doxorubicin-resistant MCF-7 breast cancer cells. Biomed. Pharmacother. 2011, 65, 85–89. [Google Scholar] [CrossRef]
- Yoshino, H.; Enokida, H.; Chiyomaru, T.; Tatarano, S.; Hidaka, H.; Yamasaki, T.; Gotannda, T.; Tachiwada, T.; Nohata, N.; Yamane, T. Tumor suppressive microRNA-1 mediated novel apoptosis pathways through direct inhibition of splicing factor serine/arginine-rich 9 (SRSF9/SRp30c) in bladder cancer. Biochem. Biophys. Res. Commun. 2012, 417, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell viability assays. In Assay Guidance Manual [Internet]; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2016. [Google Scholar]
- Kwan, Y.P.; Saito, T.; Ibrahim, D.; Al-Hassan, F.M.S.; Ein Oon, C.; Chen, Y.; Jothy, S.L.; Kanwar, J.R.; Sasidharan, S. Evaluation of the cytotoxicity, cell-cycle arrest, and apoptotic induction by Euphorbia hirta in MCF-7 breast cancer cells. Pharm. Biol. 2016, 54, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Wang, W.; Jin, X.; Wang, Z.; Ji, Z.; Meng, G. Silibinin, a natural flavonoid, induces autophagy via ROS-dependent mitochondrial dysfunction and loss of ATP involving BNIP3 in human MCF7 breast cancer cells. Oncol. Rep. 2015, 33, 2711–2718. [Google Scholar] [CrossRef]
- Han, J.; Talorete, T.P.; Yamada, P.; Isoda, H. Anti-proliferative and apoptotic effects of oleuropein and hydroxytyrosol on human breast cancer MCF-7 cells. Cytotechnology 2009, 59, 45–53. [Google Scholar] [CrossRef]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In vitro cell migration and invasion assays. J. Vis. Exp. 2014, 88, e51046. [Google Scholar] [CrossRef]
- Agelaki, S.; Spiliotaki, M.; Markomanolaki, H.; Kallergi, G.; Mavroudis, D.; Georgoulias, V.; Stournaras, C. Caveolin-1 regulates EGFR signalling in MCF-7 breast cancer cells and enhances gefitinib-induced tumor cell inhibition. Cancer Biol. Ther. 2009, 8, 1470–1477. [Google Scholar] [CrossRef][Green Version]
- Al-Asmari, A.K.; Albalawi, S.M.; Athar, M.T.; Khan, A.Q.; Al-Shahrani, H.; Islam, M. Moringa oleifera as an anti-cancer agent against breast and colorectal cancer cell lines. PLoS ONE 2015, 10, e0135814. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Hu, S.-S.; Dong, Q.; Cai, J.-X.; Zhang, W.-P.; Sun, J.-Y.; Wang, T.-T.; Xie, J.; He, H.-R.; Xing, J.-F. Establishment of paclitaxel-resistant breast cancer cell line and nude mice models, and underlying multidrug resistance mechanisms in vitro and in vivo. Asian Pac. J. Cancer Prev. 2013, 14, 6135–6140. [Google Scholar] [CrossRef] [PubMed]
- Hunakova, L.; Macejova, D.; Toporova, L.; Brtko, J. Anticancer effects of tributyltin chloride and triphenyltin chloride in human breast cancer cell lines MCF-7 and MDA-MB-231. Tumor Biol. 2016, 37, 6701–6708. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequences |
|---|---|
| BRCA1 | FW-5′-TCGTATTCTGAGAGGCTGCTG-3′ RE-5′-TCTTCAACGCGAAGAGCAGA-3′ |
| BRCA2 | FW-5′-GTTGTGAAAAAAACAGGACTTG-3′ RE-5′-CAGTCTTTAGTTGGGGTGGA-3′ |
| p53 | FW-5′-AGGCCTTGGAACTCAAGGAT-3′ RE-5′-CCCTTTTTGGACTTCAGGTG-3′ |
| HER1 | FW-5′-CAGCGCTACCTTGTCATTCA-3′ RE-5′-TGCACTCAGAGAGCTCAGGA-3′ |
| HER2 | FW-5′-AAAGGCCCAAGACTCTCTCC-3′ RE-5′-CAAGTACTCGGGGTTCTCCA-3′ |
| cMyc | FW-5′-TGAGGAGACACCGCCCAC-3′ RE-5′-CAACATCGATTTCTTCCTCATCTTC-3′ |
| BCL2 | FW-5′-GAACTGGGGGAGGATTGTGG-3′ RE-5′-GCCGGTTCAGGTACTCAGTC-3′ |
| MCL1 | FW-5′-TTCCAGTAAGGAGTCGGGGT-3′ RE-5′-TGGCCAAAAGTCGCCCTC-3′ |
| CCND1 | FW-5′-TTCAAATGTGTGCAGAAGGA’3 RE-5′-GGGATGGTCTCCTTCATCTT-3′ |
| WNT3A | FW-5′-GTGTTCCACTGGTGCTGCTA-3′ RE-5′-CCCTGCCTTCAGGTAGGAGT-3′ |
| MAPK1 | FW-5′-CAGTTCTTGACCCCTGGTCC-3′ RE-5′-TACATACTGCCGCAGGTCAC-3′ |
| MAPK3 | FW-5′-TATGACCACGTGCGCAAGAC-3′ RE-5′-GACATTCTCATGGCGGAAGC-3′ |
| DAPK1 | FW-5′-TGGAGAGAGATTGCTCCCAGT-3′ RE-5′-CACAACCGCAAACTGTCCAC-3′ |
| Casp8 | FW-5′-CTGGTCTGAAGGCTGGTTGT-3′ RE-5′-CAGGCTCAGGAACTTGAGGG-3′ |
| Casp9 | Fw-5′-CAGGCCCCATATGATCGAGG-3′ Re-5′-TCGACAACTTTGCTGCTTGC-3′ |
| Β-Actin | FW-5′-AGAGCTACGAGCTGCCTGAC-3′ RE-5′-AGCACTGTGTTGGCGTACAG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adam, M.A.A.; Kamal, L.Z.M.; Kanakal, M.; Babu, D.; Dahham, S.S.; Tabana, Y.; Lok, B.; Bermoy, B.M.; Yunus, M.A.; Than, L.T.L.; et al. The Effect of Aflatoxin B1 on Tumor-Related Genes and Phenotypic Characters of MCF7 and MCF10A Cells. Int. J. Mol. Sci. 2022, 23, 11856. https://doi.org/10.3390/ijms231911856
Adam MAA, Kamal LZM, Kanakal M, Babu D, Dahham SS, Tabana Y, Lok B, Bermoy BM, Yunus MA, Than LTL, et al. The Effect of Aflatoxin B1 on Tumor-Related Genes and Phenotypic Characters of MCF7 and MCF10A Cells. International Journal of Molecular Sciences. 2022; 23(19):11856. https://doi.org/10.3390/ijms231911856
Chicago/Turabian StyleAdam, Mowaffaq Adam Ahmed, Laina Zarisa Muhd Kamal, Mahibub Kanakal, Dinesh Babu, Saad Sabbar Dahham, Yasser Tabana, Bronwyn Lok, Brittany M. Bermoy, Muhammad Amir Yunus, Leslie Thian Lung Than, and et al. 2022. "The Effect of Aflatoxin B1 on Tumor-Related Genes and Phenotypic Characters of MCF7 and MCF10A Cells" International Journal of Molecular Sciences 23, no. 19: 11856. https://doi.org/10.3390/ijms231911856
APA StyleAdam, M. A. A., Kamal, L. Z. M., Kanakal, M., Babu, D., Dahham, S. S., Tabana, Y., Lok, B., Bermoy, B. M., Yunus, M. A., Than, L. T. L., Barakat, K., & Sandai, D. (2022). The Effect of Aflatoxin B1 on Tumor-Related Genes and Phenotypic Characters of MCF7 and MCF10A Cells. International Journal of Molecular Sciences, 23(19), 11856. https://doi.org/10.3390/ijms231911856