

Research on Processing-Induced Chemical Variations in Polygonatum Cyrtonema Rhizome by Integrating Metabolomics and Glycomics

 , and
, and
Abstract
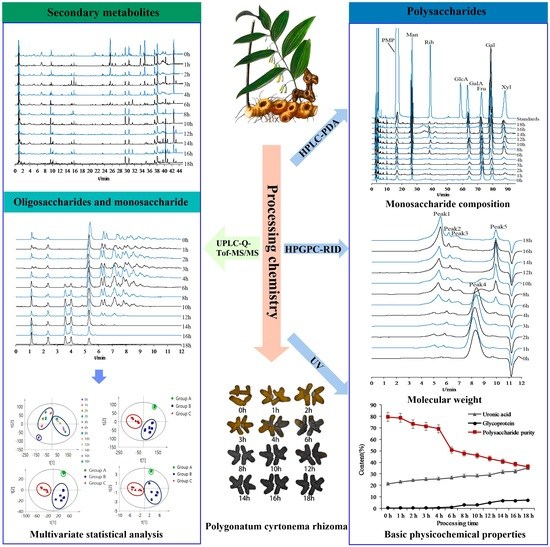
:
1. Introduction
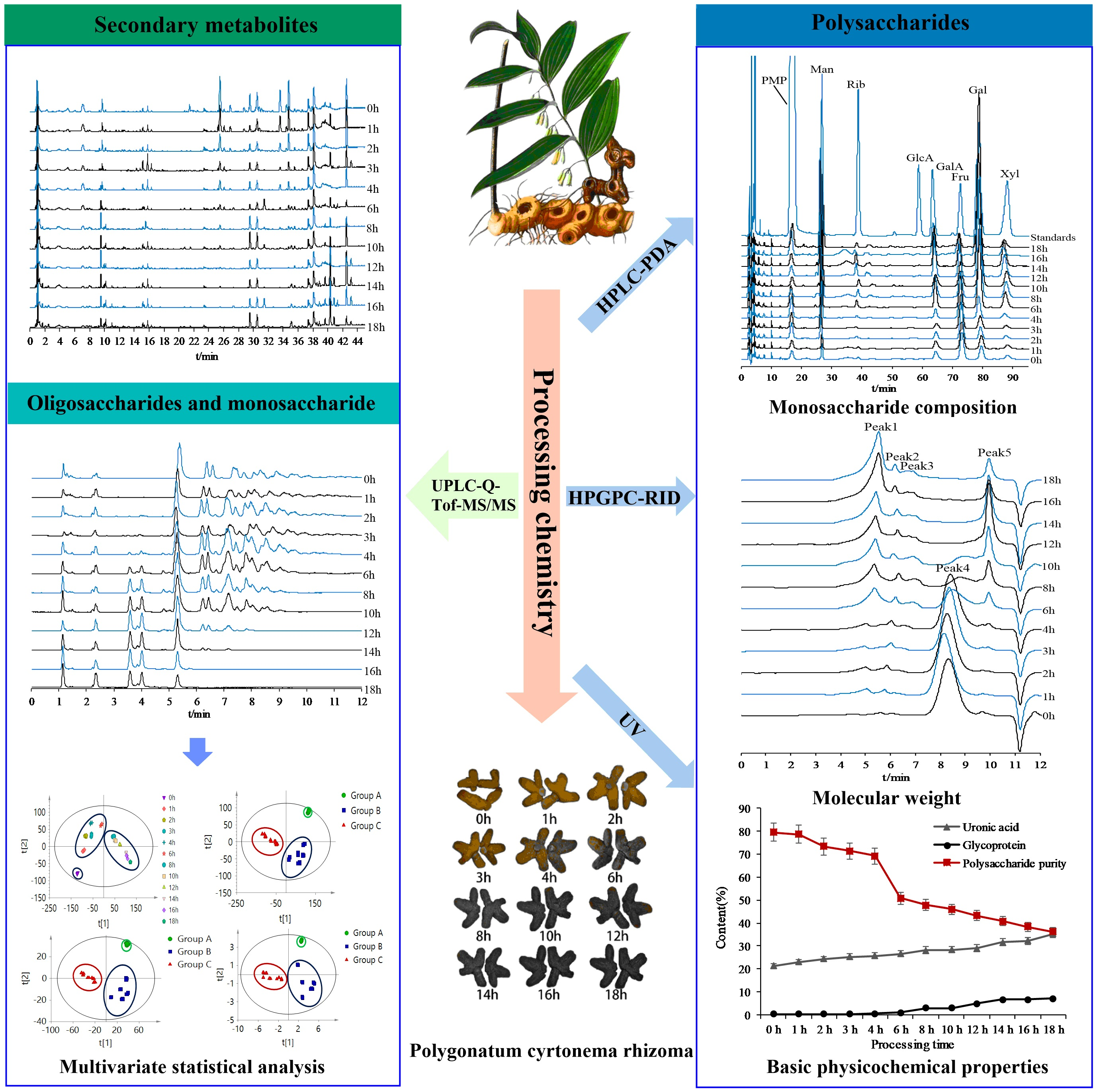
2. Results and Discussion
2.1. Method Optimization and Validation
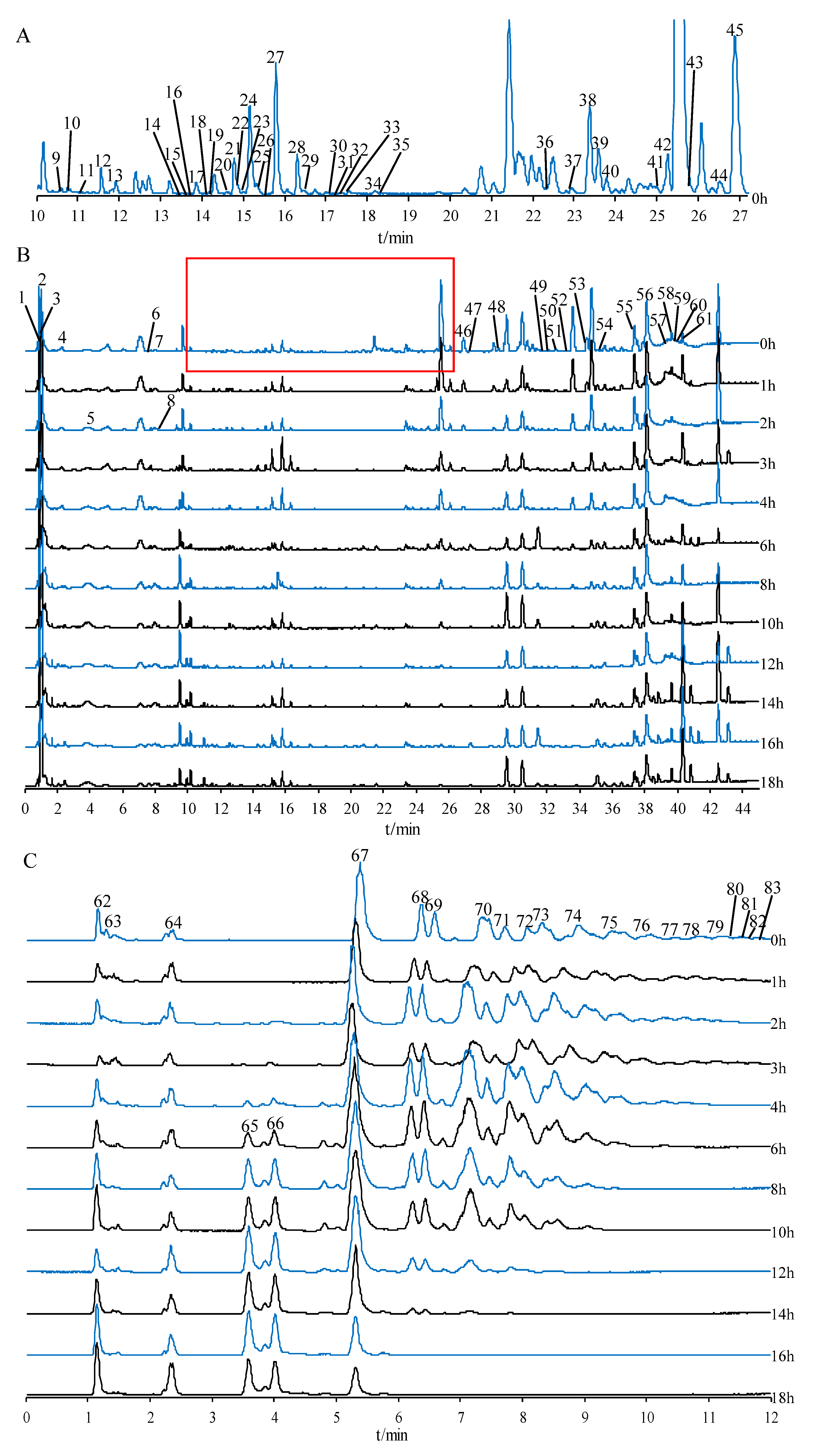
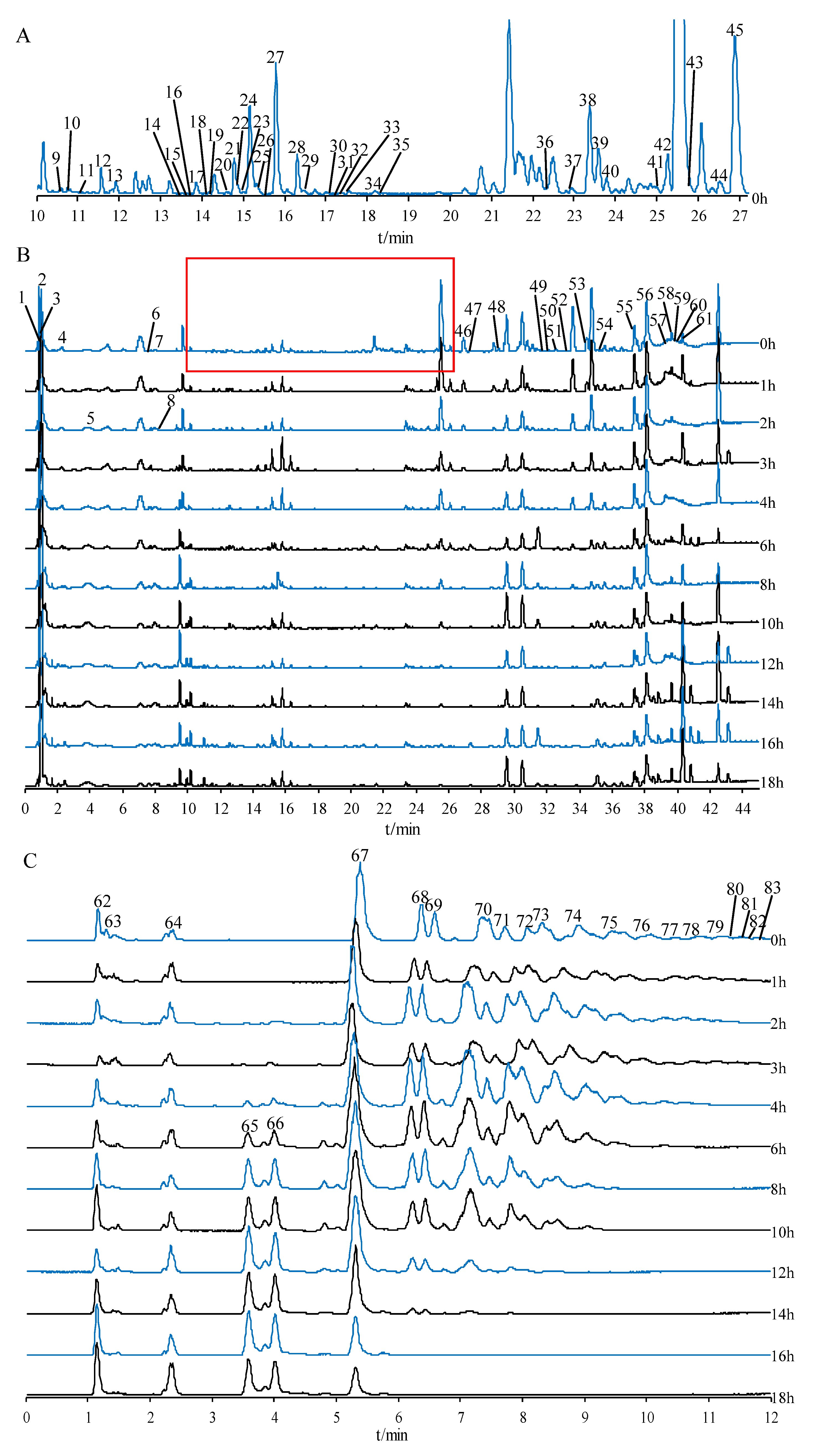
2.2. Compound Identification of Secondary Metabolites, Oligosaccharides, and Monosaccharides
2.2.1. Identification of Saponins
2.2.2. Identification of Flavonoids
2.2.3. Identification of Oligosaccharides and Monosaccharides
2.2.4. Identification of Other Compounds
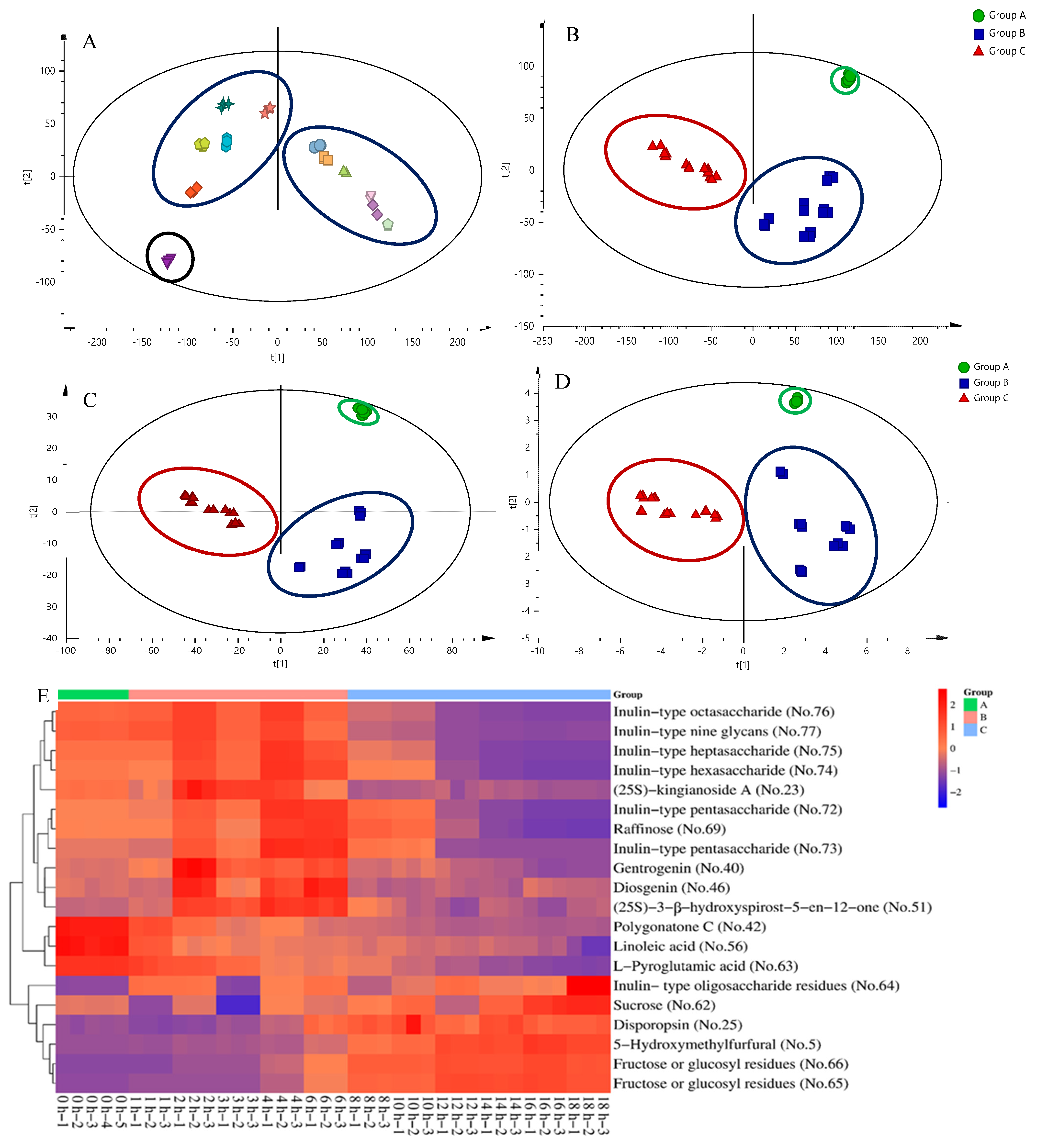
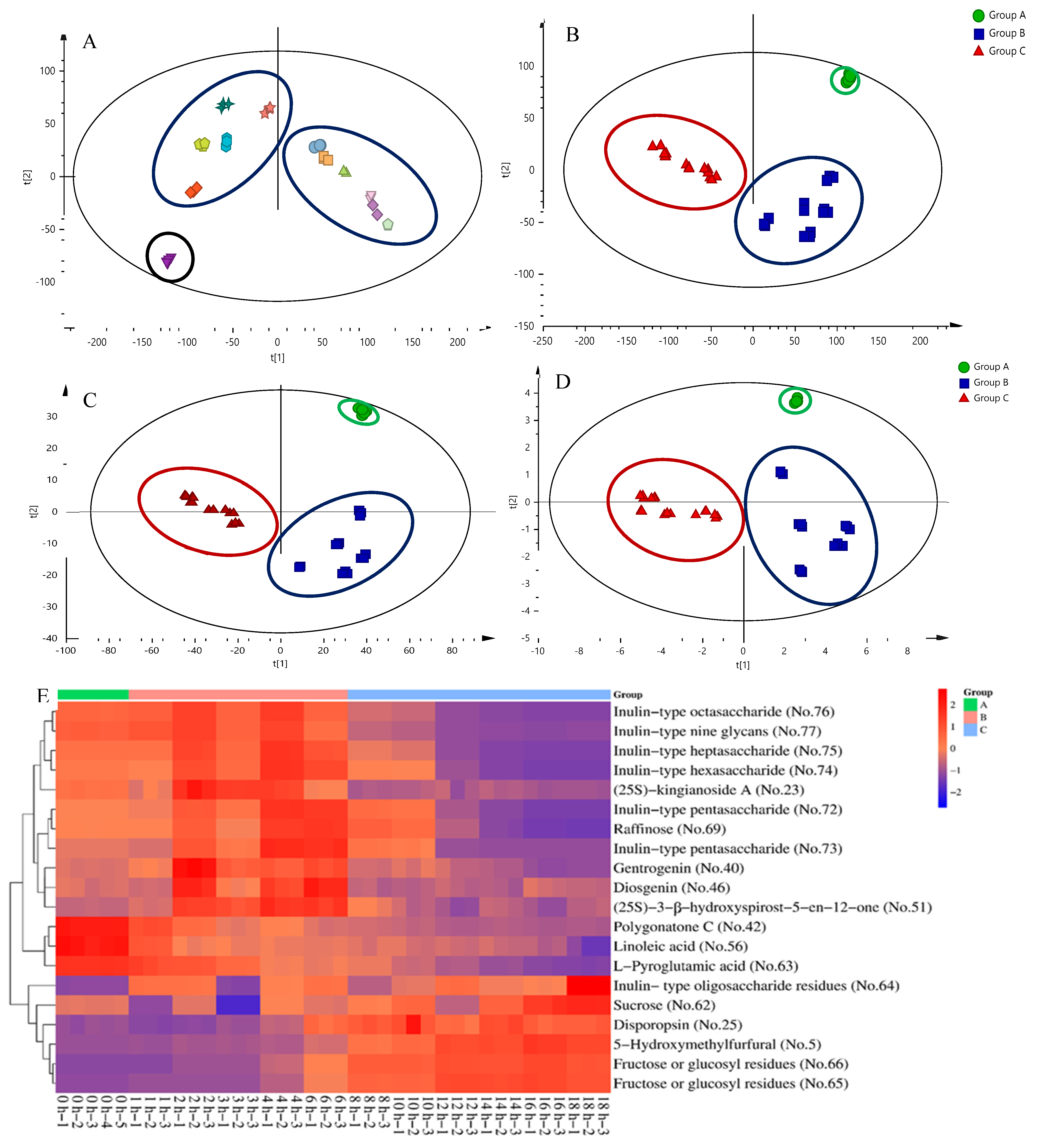
2.3. Screening of Differential Markers from Secondary Metabolites, Oligosaccharides, and Monosaccharides
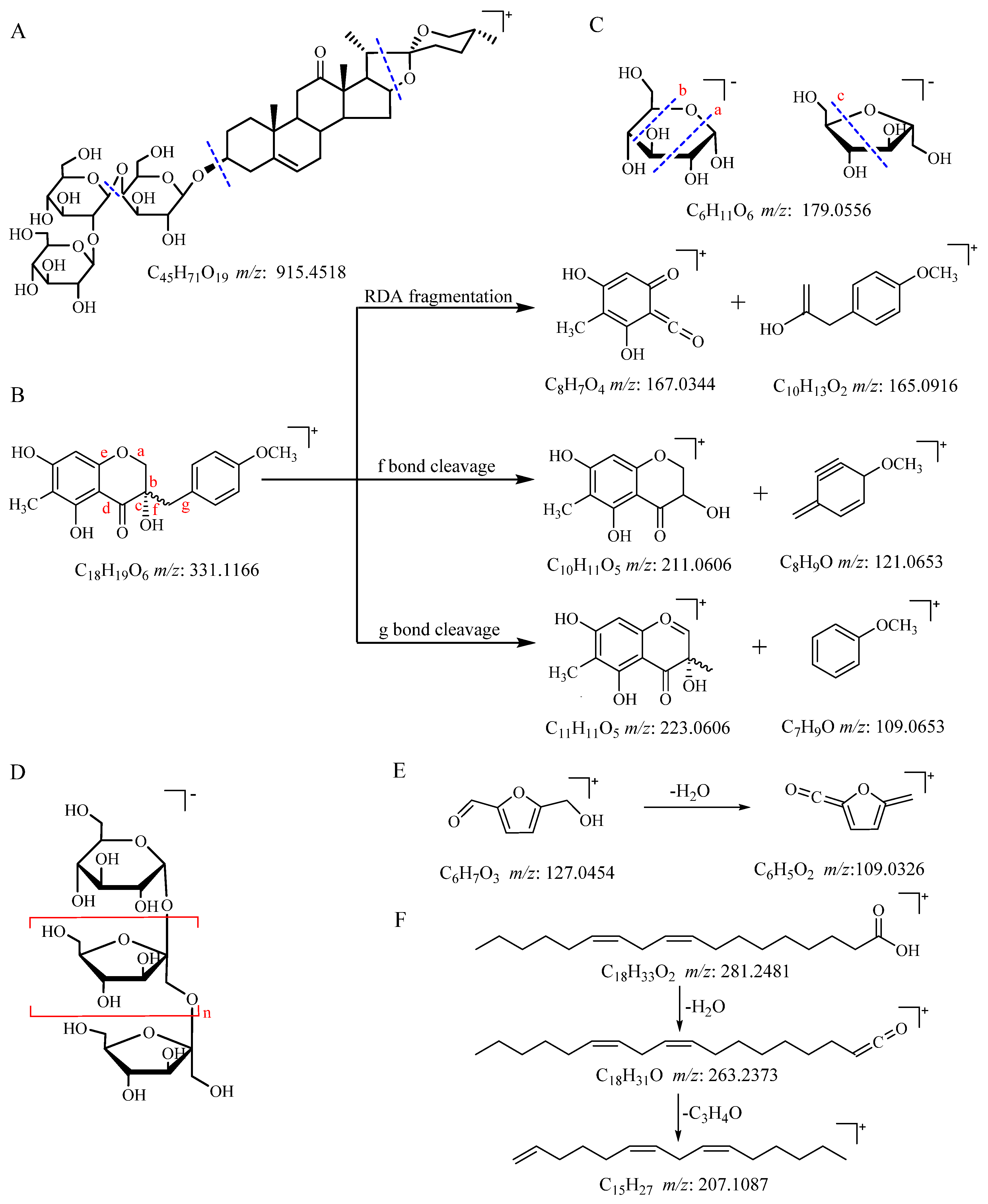
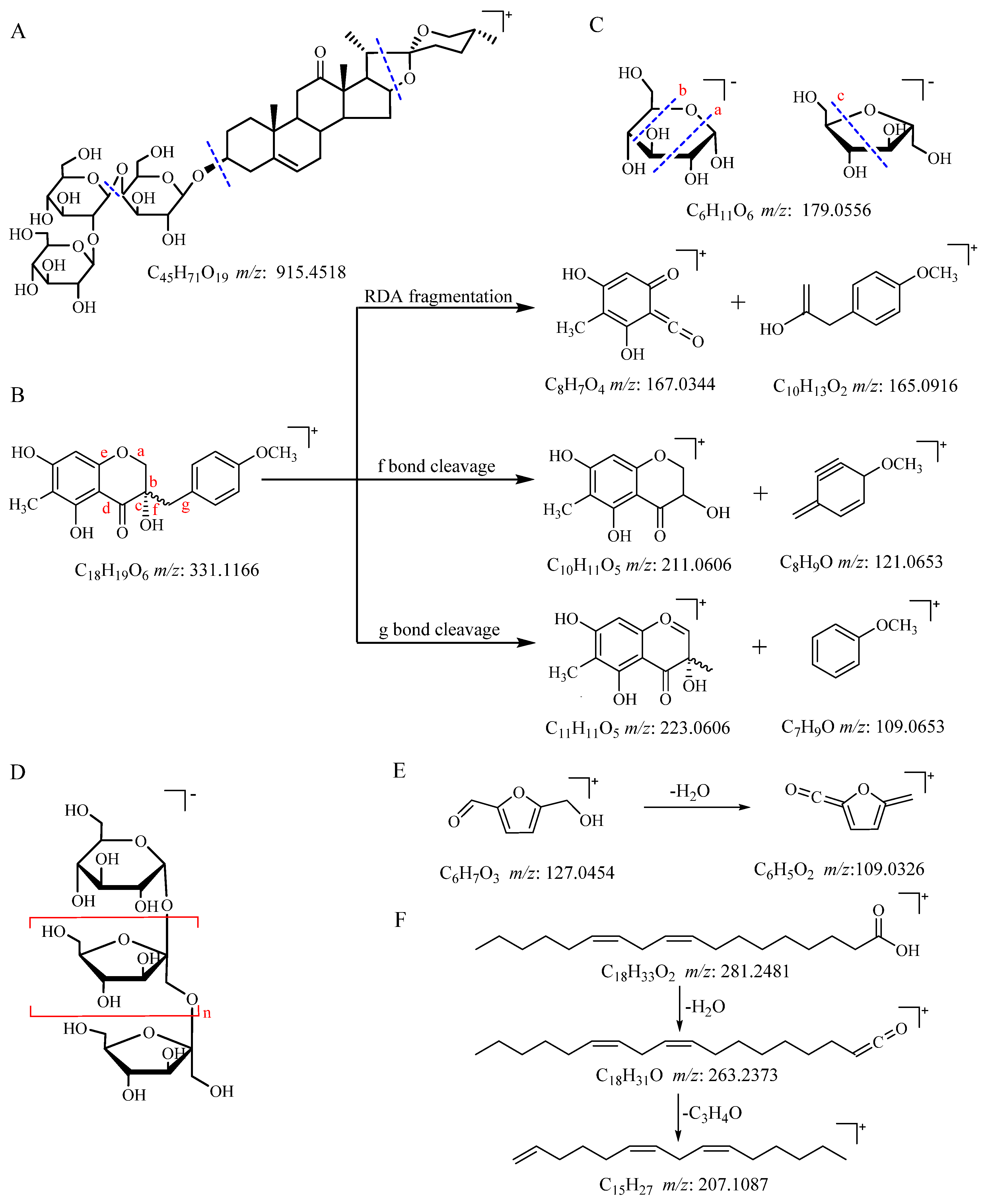
2.4. UPLC-QTOF-MS/MS Analysis of Secondary Metabolites, Oligosaccharides, and Monosaccharides
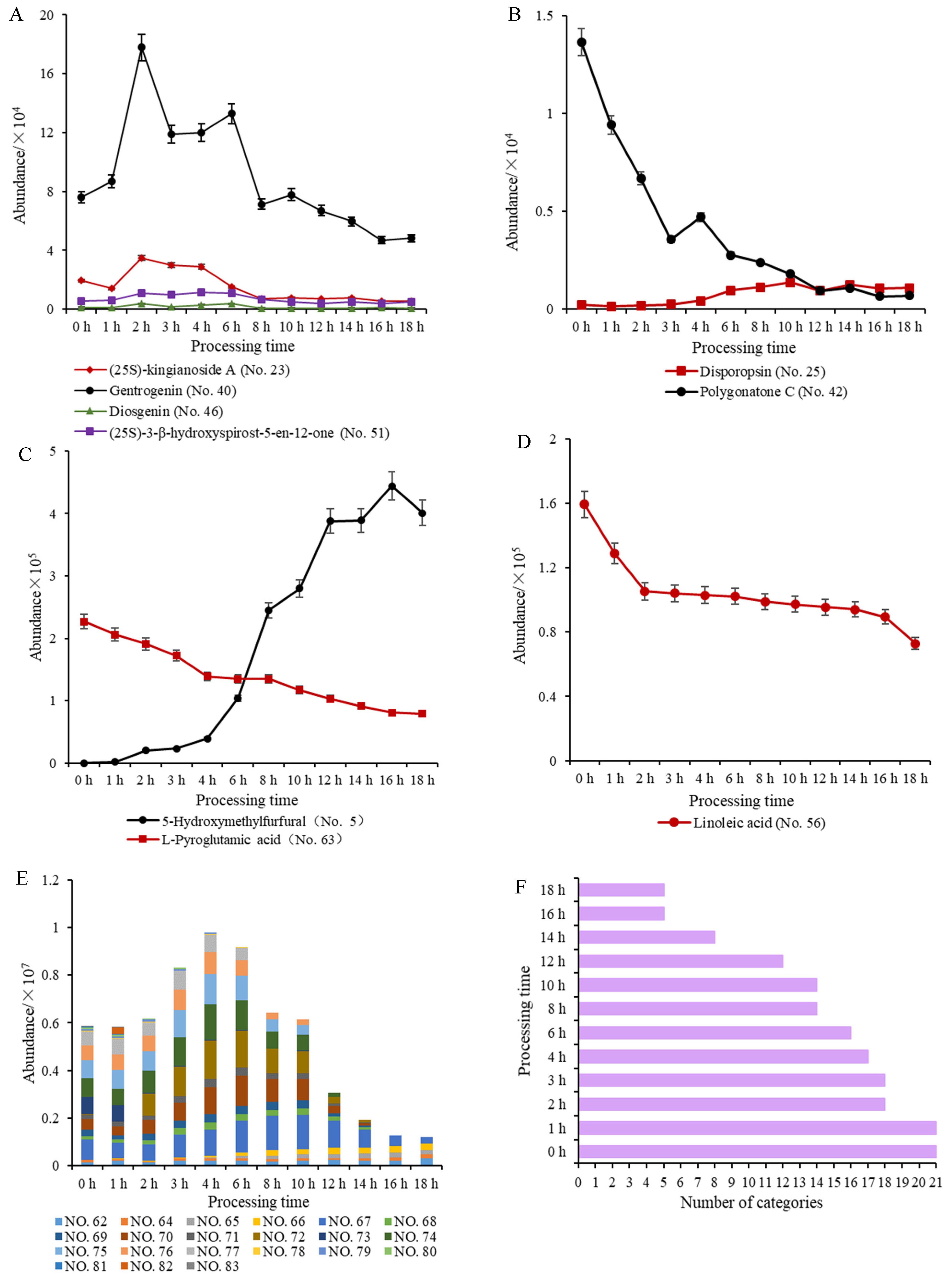
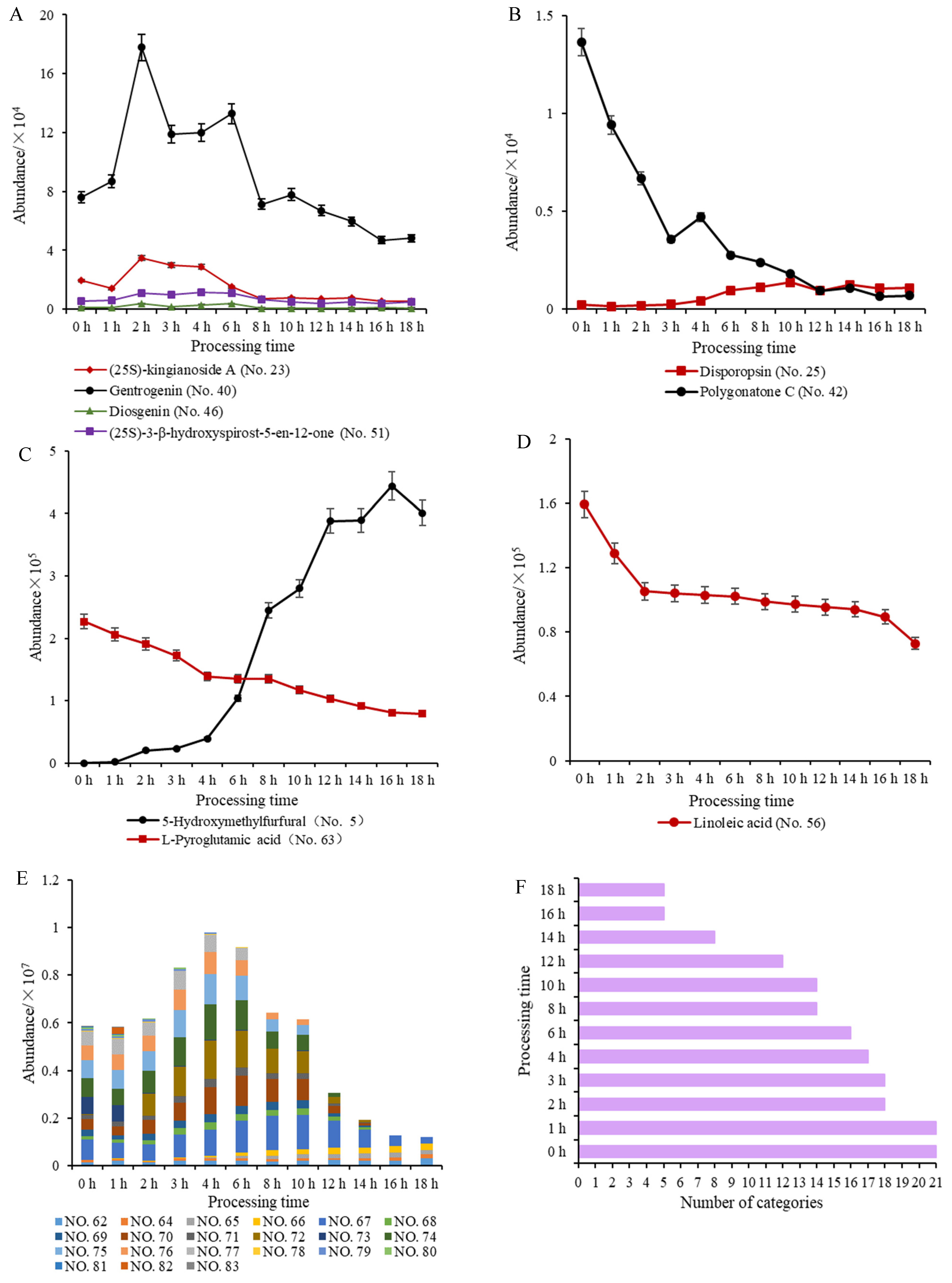
2.5. Qualitative and Quantitative Analysis of PCP during Processing
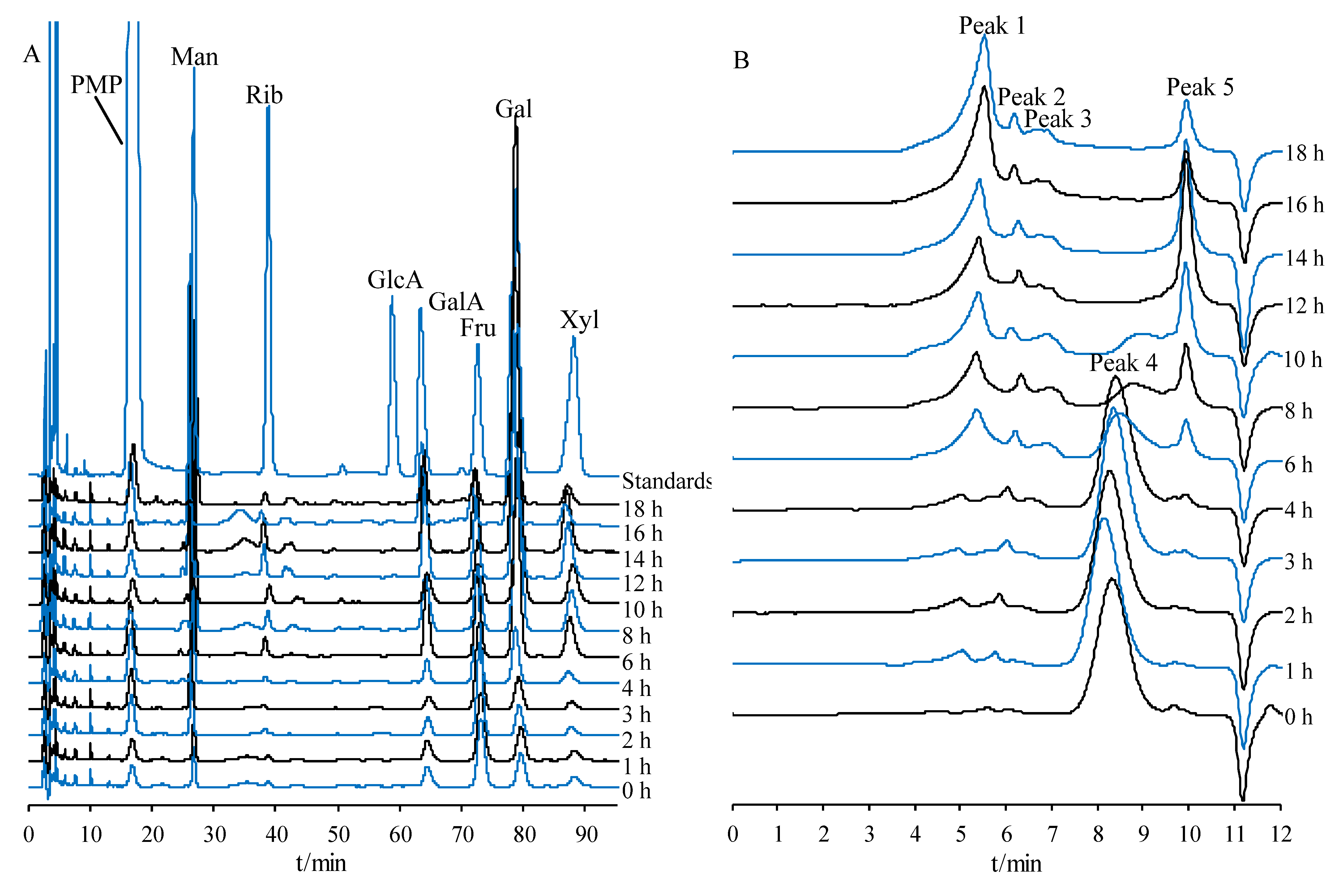
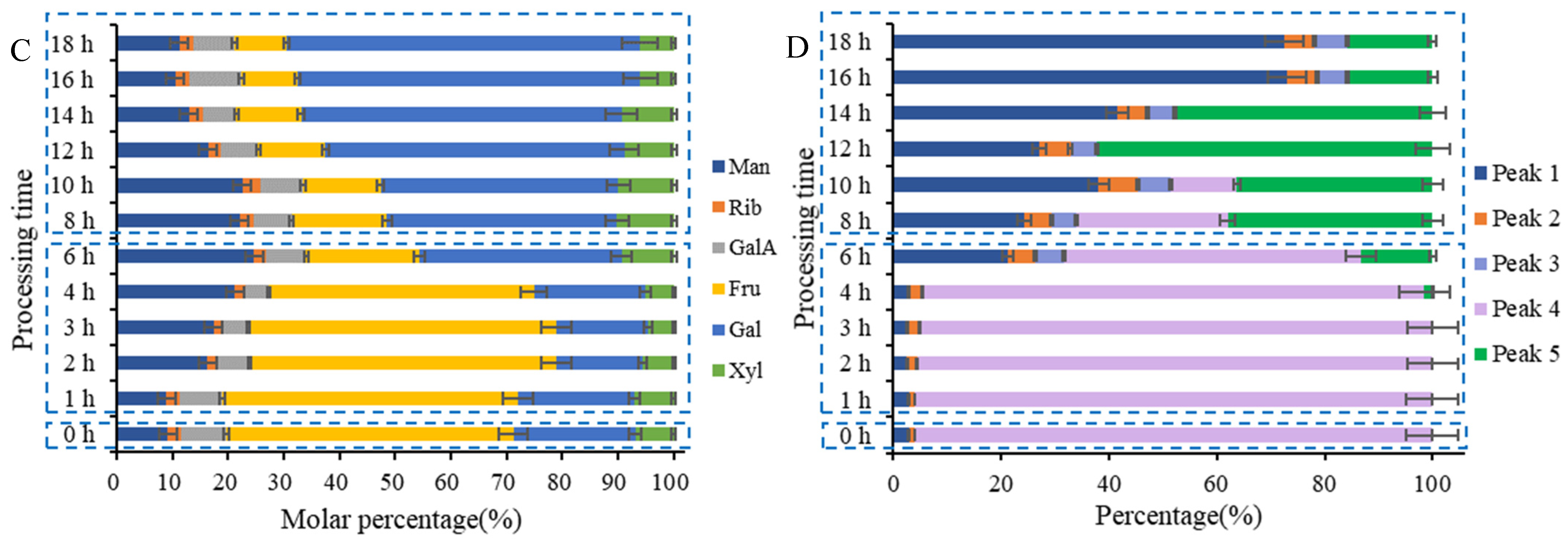
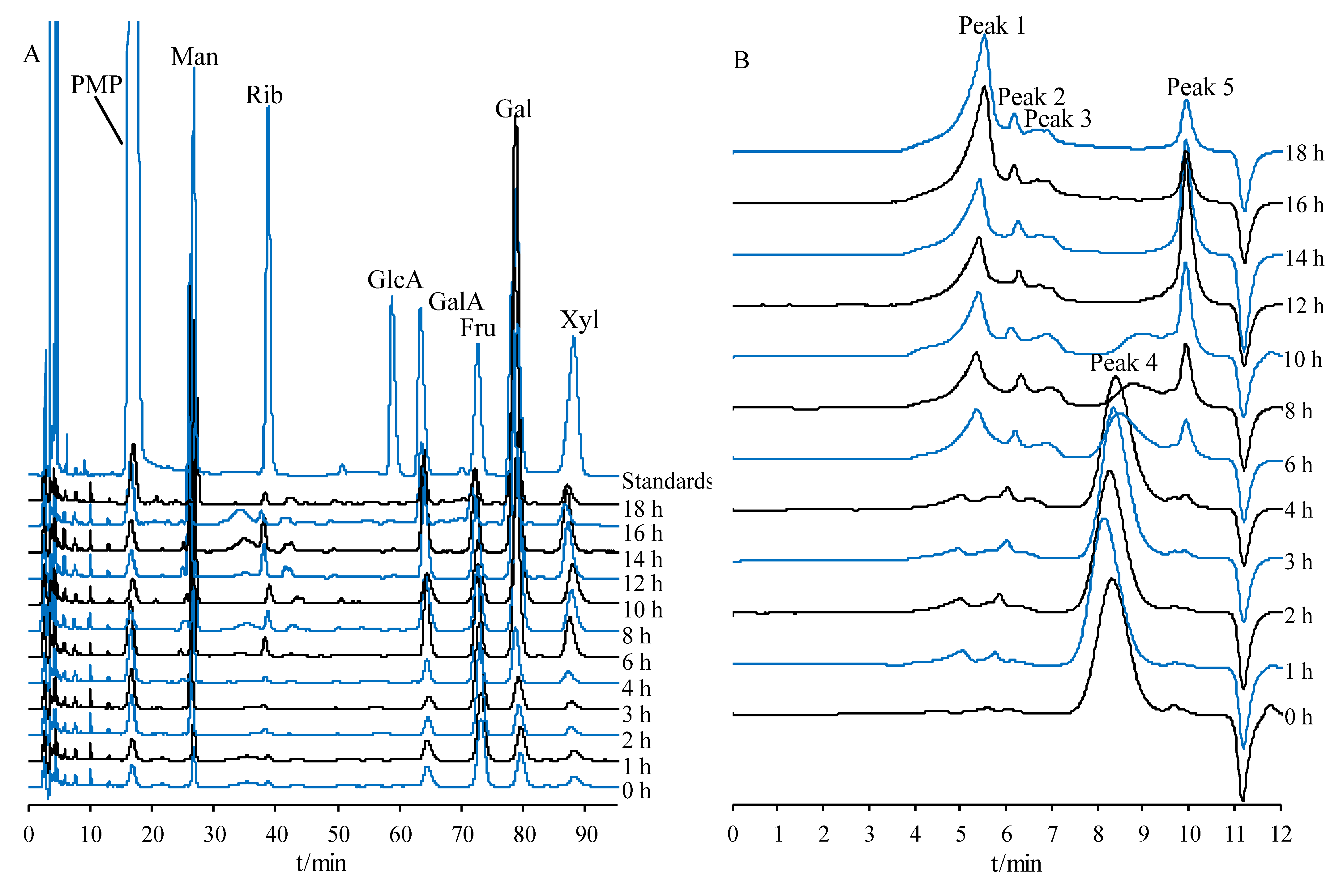
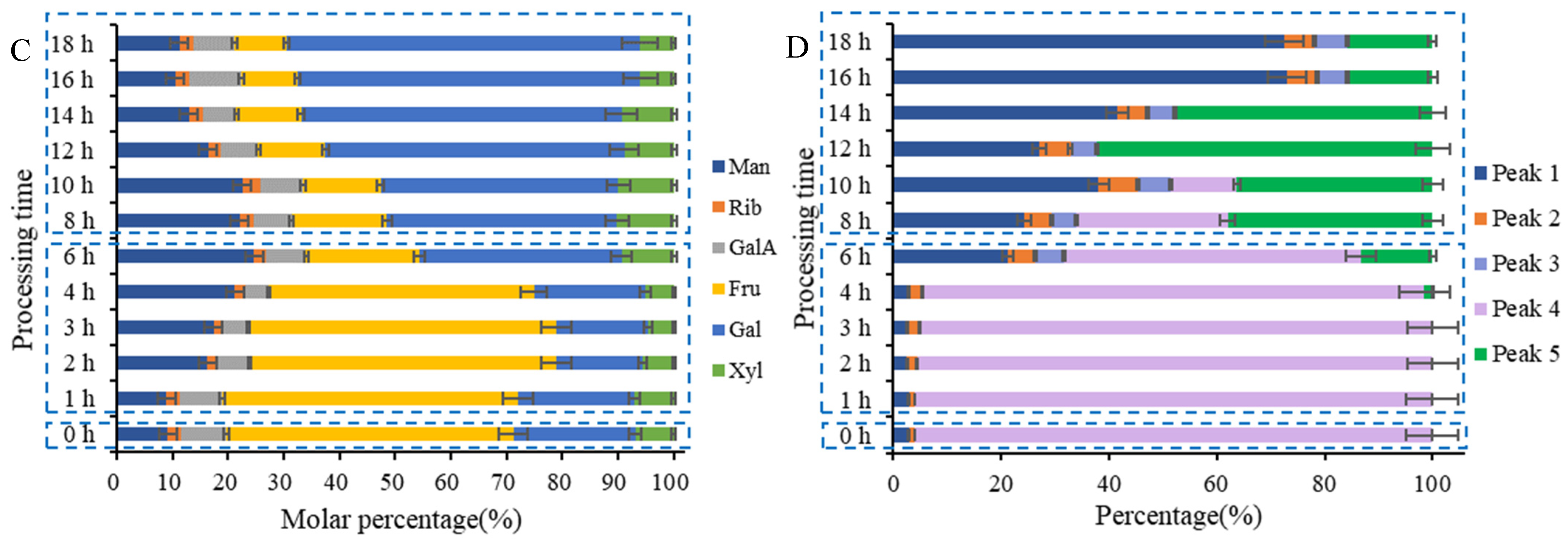
2.5.1. Monosaccharide Composition Variations in PCP during Processing
2.5.2. Molecular Weight Variations of PCP during Processing
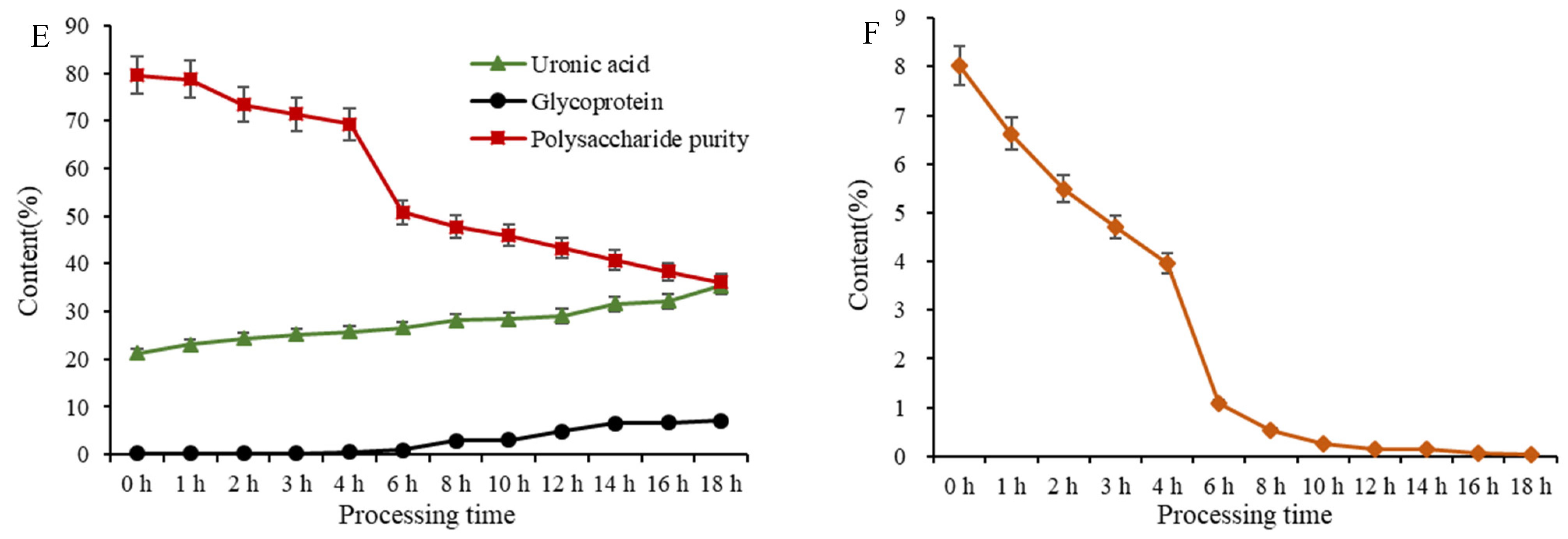
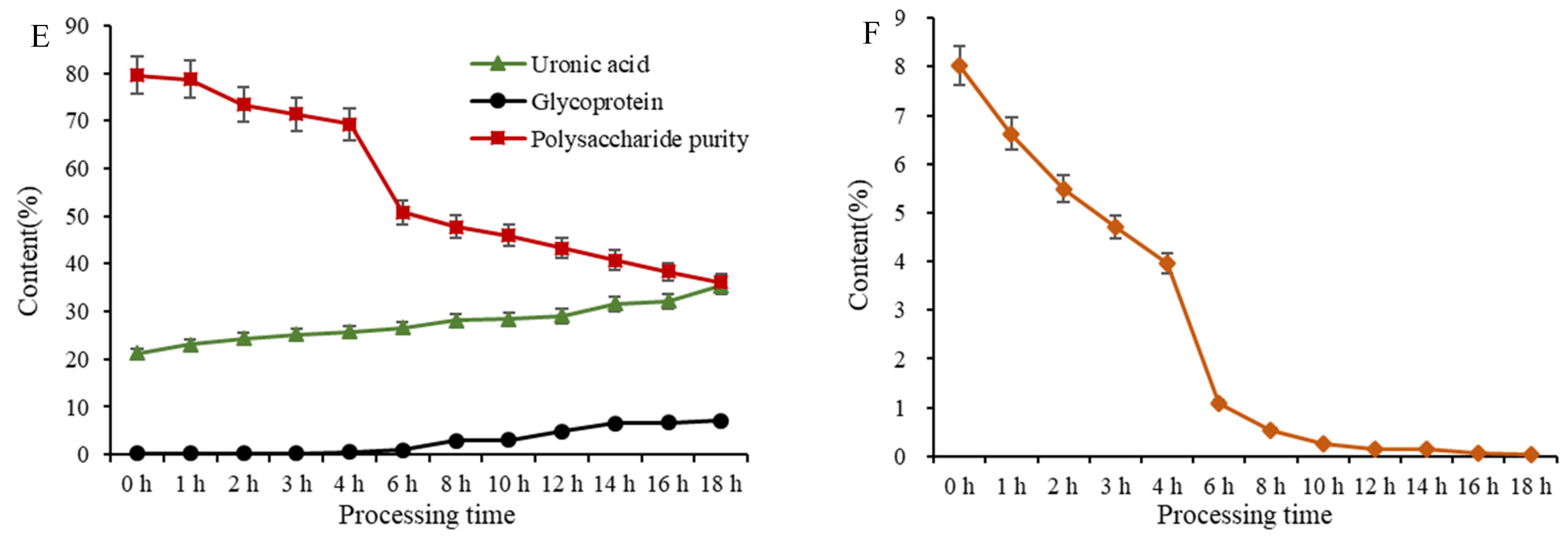
2.5.3. Glycoprotein, Uronic Acid, and Total Polysaccharide Content Variations of PCP during Processing
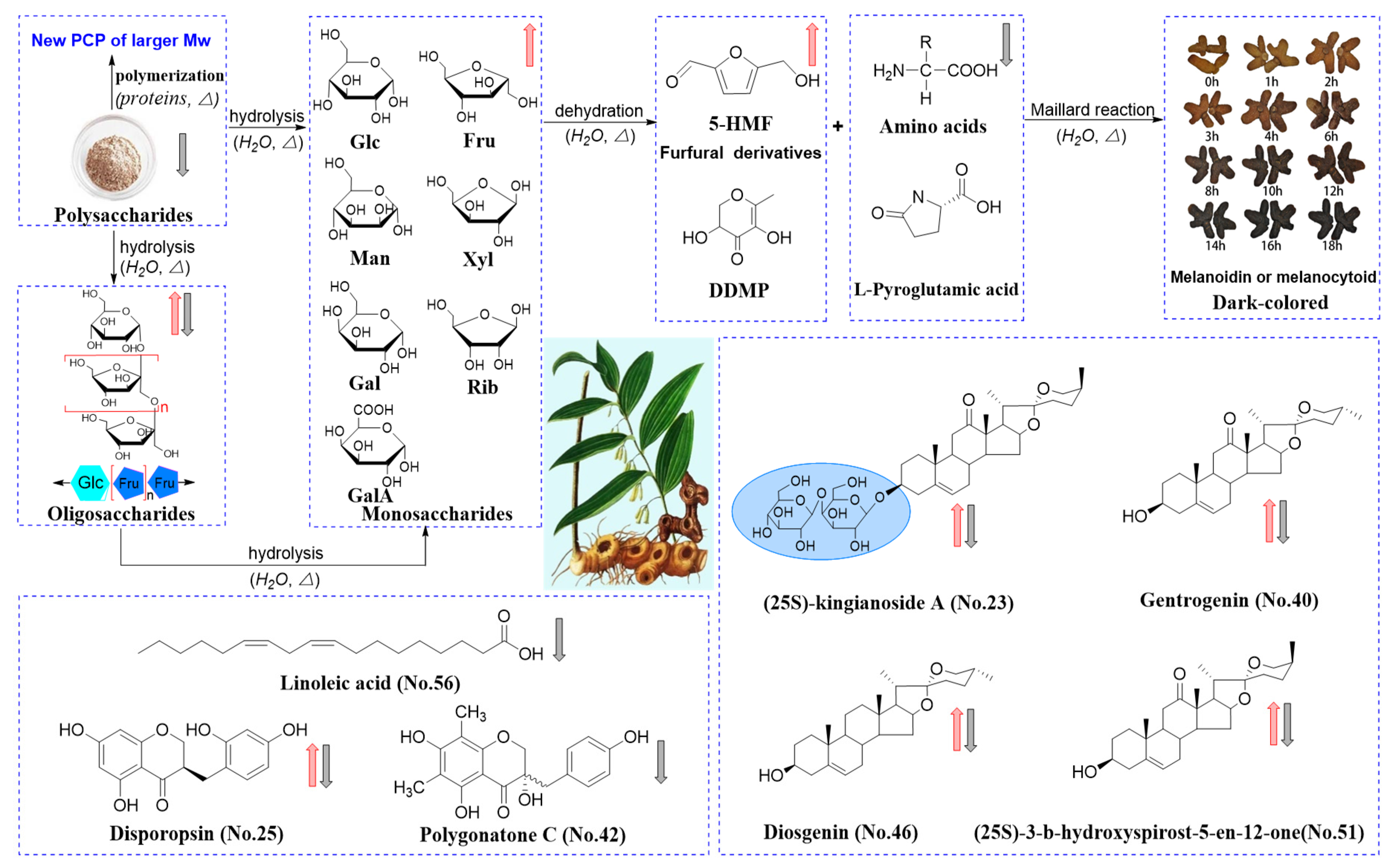
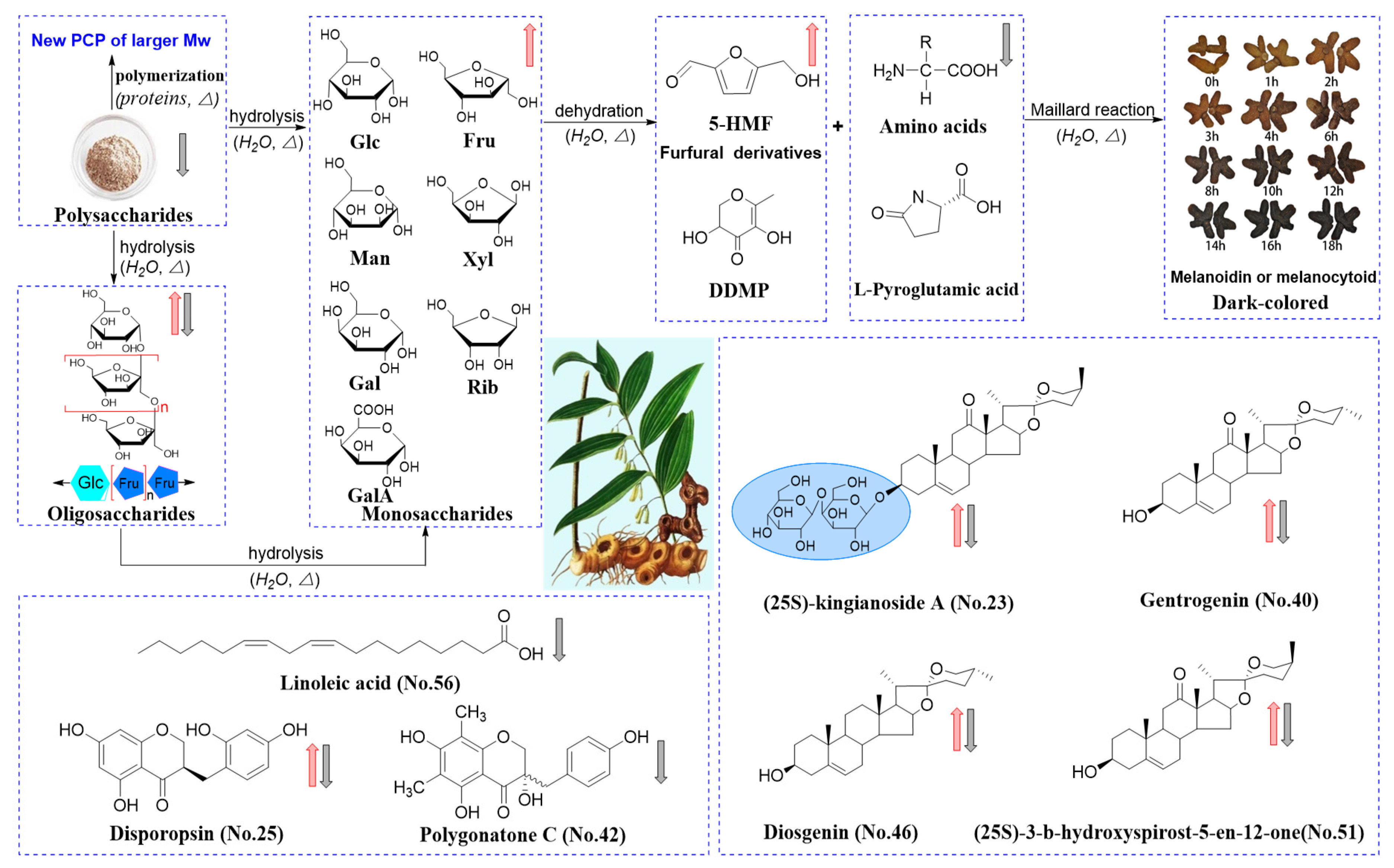
2.6. Processing-Induced Chemical Mechanisms
3. Materials and Methods
3.1. Reagents, Chemicals, and Materials
3.2. Sample Preparation
3.2.1. Sample Preparation with Varying PCR Processing Times
3.2.2. Secondary Metabolomics Sample Preparation
3.2.3. Glycomics Sample Preparation
3.3. Secondary Metabolomics UPLC-QTOF-MS/MS Analysis
3.3.1. UPLC Conditions
3.3.2. MS Conditions
3.4. UPLC-QTOF-MS/MS Analysis of Monosaccharides and Oligosaccharides
3.4.1. UPLC Conditions
3.4.2. MS Conditions
3.5. Multiple Chromatographic Techniques for the Analysis of Polysaccharides
3.5.1. HPGPC-RID Analysis of Polysaccharides
3.5.2. PMP-HPLC-PDA Analysis of Polysaccharides
3.5.3. Determination of Protein, Uronic Acid, and Total Polysaccharide Content by UV
3.6. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
References
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China, 11th ed.; China Medical Science Press: Beijing, China, 2020; Volume 1, pp. 319–320. [Google Scholar]
- Yan, H.L.; Lu, J.M.; Wang, Y.F.; Gu, W.; Yang, X.X.; Yu, J. Intake of total saponins and polysaccharides from Polygonatum kingianum affects the gut microbiota in diabetic rats. Phytomedicine 2017, 26, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.M.; Wang, Y.F.; Yan, H.L.; Lin, P.; Gu, W.; Yu, J. Antidiabetic effect of total saponins from Polygonatum kingianum in streptozotocin-induced diabetic rats. J. Ethnopharmacol. 2016, 179, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Li, C.; Yu, Y.; Chen, W.; Ma, T.; Zhou, Z. Antihyperglycemic and antihyperlipidemic activities of protodioscin in a high-fat diet and streptozotocin-induced diabetic rats. RSC Adv. 2016, 6, 88640–88646. [Google Scholar] [CrossRef]
- Du, L.; Nong, M.N.; Zhao, J.M.; Peng, X.M.; Zong, S.H.; Zeng, G.F. Polygonatum sibiricum polysaccharide inhibits osteoporosis by promoting osteoblast formation and blocking osteoclastogenesis through Wnt/β-catenin signalling pathway. Sci. Rep. 2016, 6, 32261. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhao, C.C.; Li, X.; Gao, Q.Z.; Huang, L.Q.; Xiao, P.G. The genus Polygonatum: A review of ethnopharmacology, phytochemistry and pharmacology. J. Ethnopharmacol. 2018, 214, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.X.; Zeng, X.C.; Zhang, R.; Liu, J.; Zhang, W.C.; Zhao, Y.J.; Zhang, X.Y.; Wu, Z.X.; Tan, Y.H.; Wu, Y.Y.; et al. Dioscin suppresses hepatocellular carcinoma tumor growth by inducing apoptosis and regulation of TP53, BAX, BCL2 and cleaved CASP3. Phytomedicine 2016, 23, 1329–1336. [Google Scholar] [CrossRef]
- Zhao, S.; Niu, F.; Xu, C.Y.; Liu, Y.; Ye, L.; Bi, G.B.; Chen, L.; Tian, G.; Nie, T.H. Diosgenin prevents bone loss on retinoic acid-induced osteoporosis in rats. Ir. J. Med. Sci. 2016, 185, 1–7. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Li, J. Chemical constituents of the genus Polygonatum and their role in medicinal treatment. Nat. Prod. Commun. 2014, 10, 683–688. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, F.; Jin, Q.; Song, X.C.; Hu, Z.F.; Zhu, D.N.; Yu, B.Y. Homoisoflavonoids from the fibrous roots of Polygonatum odoratum with glucose uptake-stimulatory activity in 3T3-L1 adipocytes. J. Nat. Prod. 2010, 73, 548–552. [Google Scholar] [CrossRef]
- Liu, F.; Liu, Y.H.; Meng, Y.W.; Yang, M.; He, K. Structure of polysaccharide from Polygonatum cyrtonema Hua and the antiherpetic activity of its hydrolyzed fragments. Antivir. Res. 2004, 63, 183–189. [Google Scholar] [CrossRef]
- He, L.L.; Yan, B.X.; Yao, C.Y.; Chen, X.Y.; Li, L.W.; Wu, Y.J.; Song, Z.J.; Song, S.S.; Zhang, Z.F.; Luo, P. Oligosaccharides from Polygonatum Cyrtonema Hua: Structural characterization and treatment of LPS-induced peritonitis in mice. Carbohyd. Polym. 2020, 255, 117392. [Google Scholar] [CrossRef]
- Zhao, P.; Li, X.; Wang, Y.; Yan, L.; Guo, L.; Huang, L.; Gao, W. Characterisation and saccharide mapping of polysaccharides from four common Polygonatum spp. Carbohyd. Polym. 2020, 233, 115836. [Google Scholar] [CrossRef]
- Yelithao, K.; Surayot, U.; Lee, J.H.; You, S. Raw264.7 cell activating glucomannans extracted from rhizoma of Polygonatum sibiricum. Prev. Nutr. Food Sci. 2016, 21, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.F.; Zhang, Z.Y.; Lu, L.; Xiao, D.Q.; Xiong, C.X.; Zhao, Y.X.; Zong, S.H. Protective effects of Polygonatum sibiricum polysaccharide on ovariectomy-induced bone loss in rats. J. Ethnopharmacol. 2011, 136, 224–229. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, S.C.; Pen, G.Q.; Chen, D.; Han, C.; Miao, C.R.; Lu, B.J.; Su, C.; Feng, S.L.; Li, W.; et al. Potential ocular protection and dynamic observation of Polygonatum sibiricum polysaccharide against streptozocin-induced diabetic rats’ model. Exp. Biol. Med. 2017, 242, 92–101. [Google Scholar] [CrossRef]
- Wang, J.; Yue, Y.D.; Tang, F.; Tao, W.Q. Comparative analysis of volatile fractions in Polygonati Rhizoma and its processed products by GC-MS. China J. Chin. Mater. Med. 2011, 36, 2187–2191. [Google Scholar] [CrossRef]
- Jin, J.; Lao, J.; Zhou, R.R.; He, W.; Qin, Y.; Zhong, C.; Xie, J.; Liu, H.; Wan, D.; Zhang, S.H.; et al. Simultaneous identification and dynamic analysis of saccharides during steam processing of rhizomas of Polygonatum cyrtonema by HPLC-QTOF-MS/MS. Molecules 2018, 23, 2855. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.T.; Zhang, H.; Li, Y.; Liu, Y.; Dai, W.; Fang, J.; Cao, C.; Die, Y.; Liu, Q.; Wang, C.L.; et al. Physicochemical properties and immunological activities of polysaccharides from both crude and wine-processed Polygonatum sibiricum. Int. J. Biol. Macromol. 2019, 143, 255–264. [Google Scholar] [CrossRef]
- Wu, W.J.; Huang, N.W.; Huang, J.P.; Wang, L.L.; Wu, L.L.; Wang, Q.; Zhao, H.J. Effects of the steaming process on the structural properties and immunological activities of polysaccharides from Polygonatum cyrtonema. J. Funct. Foods 2021, 88, 104866. [Google Scholar] [CrossRef]
- Zhou, Q.; Wu, P.; Zhang, X.L.; Zhang, Y.X.; Zhang, Z.H.; Wang, Y. Effects of Polygonatum cyrtonema and braising of Polygonatum cyrtonema on gastrointestinal motility and hormonal readiness in spleen-deficiency mouse. Lishizhen Med. Mater. Med. Res. 2021, 32, 1061–1064. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2001, 19, 173. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.X.; Xu, G.W. Metabolomics and traditional Chinese medicine. Trend. Anal. Chem. 2014, 61, 207–214. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wang, Z.G.; Sun, W.J.; Wang, P.; Wang, X.J. Metabolomics: Towards understanding traditional Chinese medicine. Planta Med. 2010, 76, 2026–2035. [Google Scholar] [CrossRef]
- Hu, D.J.; Cheong, K.L.; Zhao, J.; Li, S.P. Chromatography in characterization of polysaccharides from medicinal plants and fungi. J. Sep. Sci. 2013, 36, 1–19. [Google Scholar] [CrossRef]
- Hart, G.W.; Copeland, R.J. Glycomics hits the big time. Cell 2010, 143, 672–676. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.J.; Wang, Z.X.; Cui, F.; Zhang, Q.N.; Song, P.P.; Li, B.; Tang, Z.S.; Hu, F.D.; Shi, X.F. Characterization of chemical composition variations in raw and processed Codonopsis Radix by integrating metabolomics and glycomics based on multiple chromatography-mass spectrometry technology. J. Sep. Sci. 2022, 45, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xu, J.D.; Zhou, S.S.; Mao, Q.; Kong, M.; Shen, H.; Li, X.Y.; Duan, S.M.; Xu, J.; Li, S.L. Integrating targeted glycomics and untargeted metabolomics to investigate the processing chemistry of herbal medicines, a case study on Rehmanniae Radix. J. Chromatogr. A 2016, 1472, 74–87. [Google Scholar] [CrossRef]
- Yip, K.M.; Xu, J.; Zhou, S.S.; Lau, Y.M.; Chen, Q.L.; Tang, Y.C.; Yang, Z.J.; Yao, Z.P.; Ding, P.; Chen, H.B.; et al. Characterization of chemical component variations in different growth years and tissues of Morindae Officinalis Radix by integrating metabolomics and glycomics. J. Agric. Food Chem. 2019, 67, 7304–7314. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.A.; Ge, A.H.; Zhang, L.; Li, J.; An, M.R.; Cao, J.; He, J.; Gao, X.M.; Chang, Y.X. Influence of different processing times on the quality of Polygoni Multiflora Radix by metabolomics based on ultra high performance liquid chromatography with quadrupole time-of-flight mass spectrometry. J. Sep. Sci. 2017, 40, 1928–1941. [Google Scholar] [CrossRef]
- Xue, S.J.; Wang, L.L.; Chen, S.Q.; Cheng, Y.X. Simultaneous analysis of saccharides between fresh and processed Radix Rehmanniae by HPLC and UHPLC-LTQ-Orbitrap-MS with multivariate statistical analysis. Molecules 2018, 23, 541. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.R.; Lou, Z.Y.; Ding, X.; Li, X.; Qi, Y.P.; Zhu, Z.Y.; Chai, Y.F. Global characterization of neutral saccharides in crude and processed Radix Rehmanniae by hydrophilic interaction liquid chromatography tandem electrospray ionization time-of-flight mass spectrometry. Food Chem. 2013, 141, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.J.; Kim, J. Identification and quantification of steroidal saponins in Polygonatum species by HPLC/ESI/MS. Arch. Pharmacal. Res. 2005, 28, 592–597. [Google Scholar] [CrossRef]
- Tomoda, M.; Satoh, N.; Sugiyama, A. Isolation and characterization of fructans from Polygonatum odoratum var. japonicum rhizomas. Chem. Pharm. Bull. 1973, 21, 1806–1810. [Google Scholar] [CrossRef]
- Tang, L.Y.; Wu, H.W.; Zhou, X.D.; Xu, Y.L.; Zhou, G.H.; Wang, T.; Kou, Z.Z.; Wang, Z.J. Discrimination of Semen cassia from two related species based on the multivariate analysis of high-performance liquid chromatography fingerprints. J. Sep. Sci. 2015, 38, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.K.; Han, X.; Li, R.Y.; Niu, M.; Dong, Q.; Yang, M.; Wang, J.B.; Xiao, X.H. Investigation of the chemical markers for experiential quality evaluation of crudeaconite by UHPLC-Q-TOF-MS. J. Sep. Sci. 2016, 39, 4281–4289. [Google Scholar] [CrossRef]
- Hotelling, H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 498–520. [Google Scholar] [CrossRef]
- Perez-Enciso, M.; Tenenhaus, M. Prediction of clinical outcome with microarray data: A partial least squares discriminant analysis (PLS-DA) approach. Hum. Genet. 2003, 112, 581–592. [Google Scholar] [CrossRef]
- Rosatella, A.A.; Simeonov, S.P.; Frade, R.F.M.; Afonso, C.A.M. 5-hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011, 13, 754–793. [Google Scholar] [CrossRef]
- Georgelis, N.; Fencil, K.; Richael, C.M. Validation of a rapid and sensitive HPLC/MS method for measuring sucrose, fructose and glucose in plant tissues. Food Chem. 2018, 262, 191–198. [Google Scholar] [CrossRef]
- Liu, S.C.; Yang, D.J.; Jin, S.Y.; Hsu, C.H.; Chen, S.L. Kinetics of color development, pH decreasing, and anti-oxidative activity reduction of Maillard reaction ingalactose/glycine model systems. Food Chem. 2008, 108, 533–541. [Google Scholar] [CrossRef]
- Chen, X.Q.; Ye, Y.; Cheng, H.; Jiang, Y.W.; Wu, Y.L. Thermal effects on the stability and antioxidant activity of an acid polysaccharide conjugate derived from green tea. J. Agric. Food Chem. 2009, 57, 5795–5798. [Google Scholar] [CrossRef]
- Melton, L.D.; Mindt, L.; Rees, D.A.; Sanderson, G.R. Covalent structure of the extracellular polysaccharide from Xanthomonas campestris: Evidence from partial hydrolysis studies. Carbohydr. Res. 1976, 46, 245–257. [Google Scholar] [CrossRef]
- Lan, G.S.; Chen, H.X.; Chen, S.H.; Tian, J.G. Chemical composition and physicochemical properties of dietary fiber from Polygonatum odoratum as affected by different processing methods. Food Res. Int. 2012, 49, 406–410. [Google Scholar] [CrossRef]
- Pierce, J.; Suelter, C.H. An evaluation of the Coomassie brilliant blue G-250 dye-binding method for quantitative protein determination. Anal. Biochem. 1977, 81, 478–480. [Google Scholar] [CrossRef]
- Selvendran, R.R.; March, J.F.; Ring, S.G. Determination of aldoses and uranic acid content of vegetable fiber. Anal. Biochem. 1979, 96, 282–292. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | RT/min | Component | m/z | Formula | MS/MS Fragmention | Adducts | C. |
|---|---|---|---|---|---|---|---|
| 1 | 0.87 | Polygonatone B | 347.1206 | C18H18O7 | 175.1194, 241.0712 | [M+H]+, [M+K]+ | e |
| 2 | 0.89 | (22S)-16β-[(α-L-rhamnopyranosyl)oxy]-3β,22-dihydroxycholest-5-en-1β-yl-α-L-rhamnopyranoside | 727.4660 | C39H66O12 | 313.2728 | [M+H]+ | a |
| 3 | 0.90 | (22S)-cholest-5-en-1β,3β,16β,22-tetrol-1-O-α-L-rhamnpyranosyl-16-O-β-D-glucopyranoside | 743.4617 | C39H66O13 | 184.0738 | [M+H]+ | a |
| 4 | 2.02 | (25R)-Polygodoside E | 593.3590 | C33H52O9 | 325.1237 | [M+H]+ | c |
| 5 | 4.23 | 5-Hydroxymethylfurfural (5-HMF) | 127.0454 | C6H6O3 | 343.0265, 99.0482, 109.0326 | [M+H] + , [3M+H-2H2O]+ | g |
| 6 | 7.50 | (25S)-3β,24-dihydroxy-spirostan-5-en-12-one-3-O-β-D-glucopyranosyl-(1→2)-β-D-glucopyranosyl-(1→4)-β-D-galactopyranoside | 948.4807 | C45H70O20 | 561.2966, 387.1856, 337.1713, 175.1193 | [M+NH4]+ | c |
| 7 | 8.51 | 5,7-dihydroxy-6-methyl-3-(2′,4′-dihydroxybenzyl)-chroman-4-one | 317.1113 | C17H16O6 | 310.1280, 141.9589, 195.0657 | [M+H]+ | e |
| 8 | 8.96 | 2,3-dihydro-3,5-dihydroxy-6-methyl-4(H)-pyran-4-one(DDMP) | 145.0537 | C6H8O4 | 109.0326, 81.0361 | [M+H]+ | g |
| 9 | 10.77 | (E)-3-(3′,4′-dihydroxybenzylidene)-5,7-dihydroxy-8-methoxy-6-methylchroman-4-one | 362.1216 | C18H16O7 | 331.1273 | [M+NH4]+ | e |
| 10 | 10.82 | (E)-5,7-dihydroxy-6,8-dimethyl-3-(4′-hydroxybenzylidene)-chroman-4-one | 330.1319 | C18H16O5 | 205.0969 | [M+NH4]+ | e |
| 11 | 11.04 | (3R)-5,7-dihydroxy-6-methoxyl-8-methyl-3-(2′,4′-dihydroxybenzyl)-chroman-4-one | 364.1386 | C18H18O7 | 195.0916, 225.0763 | [M+NH4]+ | e |
| 12 | 11.76 | (2R)-5-(9H-pyrido [3,4-b]indol-1-yl)pentane-1,2,5-triol | 287.1391 | C16H18N2O3 | 286.1317 | [M+H]+ | g |
| 13 | 11.87 | (3R)-5,7-dihydroxy-3-(4′-hydroxybenzyl)-chroman-4-one | 304.1144 | C16H14O5 | 245.1856, 181.0501 | [M+NH4]+ | e |
| 14 | 13.64 | Isonarthogenin 3-O-β-D-glucopyranosyl-(1→4)-β-D-galactopyranoside | 755.4151 | C39H62O14 | 397.1967 | [M+H]+ | c |
| 15 | 13.74 | Rutin | 628.7041 | C27H30O16 | 611.1379, 465.1101 | [M+NH4]+ | e |
| 16 | 13.88 | (25S)-pratioside D1 | 915.4570 | C45H70O19 | 453.3537 | [M+H]+ | c |
| 17 | 13.95 | Polygonatoside C | 769.3996 | C39H60O15 | 632.3469 | [M+H]+ | c |
| 18 | 14.03 | (25S)-spirost-5-en-3β,14α-diol 3-O-β-D-glucopyranosyl- (1→4)-β-D-galactopyranoside | 755.4186 | C39H62O14 | 350.2867, 173.1277 | [M+H]+ | c |
| 19 | 14.08 | Polygonatoside A | 915.4570 | C45H70O19 | 611.3829, 429.2907 | [M+H]+ | c |
| 20 | 14.80 | Pratioside D1 | 915.4518 | C45H70O19 | 591.3515, 429.2989, 297.2206 | [M+H]+ | c |
| 21 | 14.83 | Pratioside C | 1079.5345 | C51H82O24 | 591.3515, 429.2989 | [M+H]+ | c |
| 22 | 14.95 | (25R)-furost-5-en-3β,22α,26-triol 26-O-β-D-glucopyranoside | 617.3658 | C33H54O9 | 427.2834, 120.0814 | [M+Na] +, [M+NH4]+ | b |
| 23 | 15.00 | (25S)-kingianoside A | 753.4026 | C39H60O14 | 591.3515, 573.3401, 429.2989, 297.2206 | [M+H]+ | c |
| 24 | 15.24 | Diosgenin 3-O-α-L-rhamnopyranosyl (1→4)-β-D-glucopyranoside | 740.4587 | C39H62O12 | 322.2485 | [M+NH4]+ | c |
| 25 | 15.55 | Disporopsin | 303.0871 | C16H14O6 | 201.0517, 181.0501, | [M+H]+ | e |
| 26 | 15.60 | Huangjinoside F | 793.3937 | C39H62O15 | 322.2487 | [M+Na]+ | c |
| 27 | 15.87 | (3β,22ξ,25R)-3-22-hydroxyfurost-5-en-26-yl-β-D-glucopyranoside | 903.4886 | C45H74O18 | 561.2966, 337.1713, 175.1193 | [M+H]+ | b |
| 28 | 16.32 | Huangjingenin | 464.3313 | C27H42O5 | 322.2481 | [M+NH4]+ | c |
| 29 | 16.34 | (3β,23S,25R)-23-hydroxy-12-oxospirost-5-en-3-yl-4-O-β-D-glucopyranosyl-β-D-galactopyranoside | 786.4236 | C39H60O15 | 387.1777, 147.0811 | [M+NH4]+ | c |
| 30 | 17.20 | Kingianoside B | 737.4040 | C39H60O13 | 453.3667, 211.0672 | [M+H]+ | c |
| 31 | 17.37 | (25S)-spirost-5-ene-3β-ol-3-O-β-D-galactopyranoside | 577.3695 | C33H52O8 | 253.1946 | [M+H]+ | c |
| 32 | 17.38 | Funkioside C | 739.4225 | C39H62O13 | 577.3708 | [M+H]+ | c |
| 33 | 17.40 | Polygonatosides A | 577.3708 | C33H52O8 | 415.3201, 253.1943 | [M+H]+ | c |
| 34 | 18.09 | Yamogenin 3-O-β-D-glucopyranosyl-(1→3)-[β-D-fucopyranosyl-(1→2)]-β-D-glucopyranosyl-(1→4)-β-D-galactopyranoside | 1047.5289 | C51H82O22 | 739.4229 | [M+H]+ | c |
| 35 | 18.32 | Kaempferol | 287.0705 | C15H10O6 | 258.0569, 168.0110 | [M+H]+ | e |
| 36 | 22.46 | (25R)-3β,14α-dihydroxy-spirost-5-ene | 431.3141 | C27H42O4 | 401.1942 | [M+H]+ | c |
| 37 | 22.98 | (6R,9R)-roseoside | 823.4065 | C20H32O8 | 279.1104 | [2M+Na]+ | g |
| 38 | 23.27 | Spiroakyroside | 1092.5147 | C51H78O24 | 591.3509 | [M+Na]+, [M+NH4]+ | c |
| 39 | 23.50 | Polygodoside A | 1064.5237 | C50H78O23 | 952.4227, 369.1949, 284.2569 | [M+NH4]+ | c |
| 40 | 23.78 | Gentrogenin | 429.2991 | C27H40O4 | 297.2206 | [M+H]+ | c |
| 41 | 25.21 | (3R)-5,7-dihydroxyl-6,8-dimethyl-3-(4′-hydroxylbenzyl)-chroman-4-one | 315.1216 | C18H18O5 | 209.0807,209.0814 | [M+H]+ | e |
| 42 | 25.51 | Polygonatone C | 331.1176 | C18H18O6 | 209.0807, 195.0657 | [M+H]+ | e |
| 43 | 25.63 | (20α,22R,25S)-Spirosta-5-ene-3β-ol | 415.2259 | C27H42O3 | 384.2042 | [M+H]+ | b |
| 44 | 26.72 | ( ± )-5,7-dihydroxy-6,8-dimethyl-3-(2′-hydroxy-4′-methoxybenzyl)-chroman-4-one | 345.1320 | C19H20O6 | 272.2940, 209.0814 | [M+H]+ | e |
| 45 | 27.07 | Polygonatoside E′ | 1080.5530 | C51H82O23 | 577.3644, 184.0728 | [M+NH4]+ | c |
| 46 | 27.41 | Diosgenin | 415.3173 | C27H42O3 | 279.2312, 397.3086 | [M+H]+ | c |
| 47 | 27.42 | Yamogenin 3-O-β-lycotetraoside | 1055.4988 | C50H80O22 | 659.2311, 437.1931 | [M+Na]+ | c |
| 48 | 28.76 | (3R)-5,7-dihydroxy-3-(2′-hydroxy-4′-methoxybenzyl)- chroman-4-one | 317.1115 | C17H16O6 | 290.6156, 181.0501 | [M+H]+ | e |
| 49 | 31.97 | Polygonatumoside C | 1193.5678 | C56H88O27 | 901.3682, 561.2792 | [M+H]+ | a |
| 50 | 32.12 | Yamogenin 3-O-β-D-glucopyranosyl-(1→4)-β-D-xylopyranoside | 731.3993 | C38H60O12 | 277.2160 | [M+Na]+ | c |
| 51 | 32.94 | (25S)-3-β-hydroxyspirost-5-en-12-one | 429.2986 | C27H40O4 | 285.1771, 327.2636 | [M+H]+ | c |
| 52 | 33.23 | (3R)-5,7-dihydroxyl-6-methyl-8-methoxyl-3-(4′-hydroxylbenzyl)-chroman-4-one | 348.1500 | C18H18O6 | 149.0236, 225.0763 | [M+NH4]+ | e |
| 53 | 34.43 | (3R)-5,7-dihydroxy-8-methyl-3-(4′-hydroxybenzyl)-chroman-4-one | 301.1052 | C17H16O5 | 285.0838, 195.0657 | [M+H]+ | e |
| 54 | 35.54 | (E)-3-(3′,4′-dihydroxybenzylidene)-5,7-dihydroxy-6,8-dimethylchroman-4-one | 329.1692 | C18H16O6 | 273.1056, 209.0814 | [M+H]+ | e |
| 55 | 37.54 | (3S)-3,5,7-trihydroxy-6-methyl-3-(4′-methoxybenzyl) chroman-4-one | 331.1166 | C18H18O6 | 223.0606, 211.0606, 167.0344 | [M+H]+ | e |
| 56 | 37.89 | Linoleic acid | 263.2373 | C18H32O2 | 207.1087 | [M+H-H2O]+ | g |
| 57 | 39.41 | Dioscin | 869.4951 | C45H72O16 | 760.5807, 321.3148 | [M+H]+ | c |
| 58 | 39.8 | Huangjinoside I | 774.4222 | C38H60O15 | 184.0739 | [M+NH4]+ | c |
| 59 | 39.95 | 9,19-cyclolart-25-en-3,24 (R)-diol | 481.3506 | C30H50O2 | 227.1273 | [M+K]+ | d |
| 60 | 40.32 | Funkioside B | 631.3796 | C34H56O9 | 507.3658 | [M+Na]+ | b |
| 61 | 40.34 | (E)-8,11,12-trihydroxyoctadec-9-enoic acid | 295.2262 | C18H34O5 | 122.1032 | [M+H-2H2O]+ | g |
| 62 | 1.16 | D-fructose | 341.2071 | C6H12O6 | 160.9872, 143.0502, 119.0528, 89.0275 | [2M-H-H2O]− | f |
| 63 | 1.22 | L-Pyroglutamic acid | 128.0348 | C5H7NO3 | 96.9626 | [M-H]− | g |
| 64 | 2.37 | Saccharide residues | 191.0302 | C10H7O4 | 111.0501, 191.0245 | [M-H]− | f |
| 65 | 3.59 | Fructose or glucosyl residues | 323.1146 | C12H20O10 | 161.0547, 143.0428 | [2M-2H2O-H]− | f |
| 66 | 4.02 | Fructose or glucosyl residues | 323.1146 | C12H20O10 | 161.0547, 89.0295 | [2M-2H2O-H]− | f |
| 67 | 5.31 | Sucrose | 341.1202 | C12H22O11 | 179.0613 | [M-H]− | f |
| 68 | 6.26 | 1-Kestose | 503.1743 | C18H32O16 | 341.1201, 179.0613 | [M-H]− | f |
| 69 | 6.47 | Raffinose | 503.1743 | C18H32O16 | 341.1239, 323.1072 | [M-H]− | f |
| 70 | 7.23 | Nistose | 665.2316 | C24H42O21 | 485.1704, 341.1201 | [M-H]− | f |
| 71 | 7.54 | Inulin-type tetrasaccharide | 665.2316 | C24H42O21 | 485.1704, 341.1201 | [M-H]− | f |
| 72 | 7.90 | Inulin-type pentasaccharide | 827.2936 | C30H52O26 | 545.1896, 341.1201 | [M-H]− | f |
| 73 | 8.11 | Inulin-type pentasaccharide | 827.2936 | C30H52O26 | 341.1201, 179.0613 | [M-H]− | f |
| 74 | 8.66 | Inulin-type hexasaccharide | 989.3560 | C36H62O31 | 179.0613 | [M-H]− | f |
| 75 | 9.18 | Inulin-type heptasaccharide | 1151.4080 | C42H72O36 | 989.3560 | [M-H]− | f |
| 76 | 9.73 | Inulin-type octasaccharide | 1313.4729 | C48H82O41 | 1151.4149, 656.2355 | [M-H]− | f |
| 77 | 10.07 | Inulin-type nine glycans | 1475.5381 | C54H92O46 | 1313.4729, 737.2625 | [M-H]− | f |
| 78 | 10.44 | Inulin-type ten glycans | 1637.5312 | C60H102O51 | 818.2943, 737.2625 | [M-H]− | f |
| 79 | 10.79 | Inulin-type eleven glycans | 1799.5812 | C66H112O56 | 899.3264, 818.2943 | [M-H]− | f |
| 80 | 11.08 | Inulin-type twelve glycans | 1961.6432 | C72H122O61 | 980.3521, 899.3264 | [M-H]− | f |
| 81 | 11.35 | Inulin-type thirteen glycans | 2123.6914 | C78H132O66 | 1061.3872, 980.3585 | [M-H]− | f |
| 82 | 11.57 | Inulin-type fourteen glycans | 2285.7421 | C84H142O71 | 1142.9181, 1061.3872 | [M-H]− | f |
| 83 | 11.78 | Inulin-type fifteen glycans | 2447.8032 | C90H152O76 | 1223.9530, 1142.4196 | [M-H]− | f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, T.; Wu, T.; Gao, P.; Wang, L.; Yang, X.; Chen, X.; Chen, Y.; Yue, C.; Liang, K.; Tang, L.; et al. Research on Processing-Induced Chemical Variations in Polygonatum Cyrtonema Rhizome by Integrating Metabolomics and Glycomics. Molecules 2022, 27, 5869. https://doi.org/10.3390/molecules27185869
Jiang T, Wu T, Gao P, Wang L, Yang X, Chen X, Chen Y, Yue C, Liang K, Tang L, et al. Research on Processing-Induced Chemical Variations in Polygonatum Cyrtonema Rhizome by Integrating Metabolomics and Glycomics. Molecules. 2022; 27(18):5869. https://doi.org/10.3390/molecules27185869
Chicago/Turabian StyleJiang, Tong, Tong Wu, Peiyun Gao, Lixia Wang, Xiaoyun Yang, Xiaoxu Chen, Yingying Chen, Chunyu Yue, Keqing Liang, Liying Tang, and et al. 2022. "Research on Processing-Induced Chemical Variations in Polygonatum Cyrtonema Rhizome by Integrating Metabolomics and Glycomics" Molecules 27, no. 18: 5869. https://doi.org/10.3390/molecules27185869
APA StyleJiang, T., Wu, T., Gao, P., Wang, L., Yang, X., Chen, X., Chen, Y., Yue, C., Liang, K., Tang, L., & Wang, Z. (2022). Research on Processing-Induced Chemical Variations in Polygonatum Cyrtonema Rhizome by Integrating Metabolomics and Glycomics. Molecules, 27(18), 5869. https://doi.org/10.3390/molecules27185869