
Preparation, Characterization and Study of the Dissociation of Naproxen from Its Chitosan Salt


Abstract
1. Introduction
2. Results and Discussion
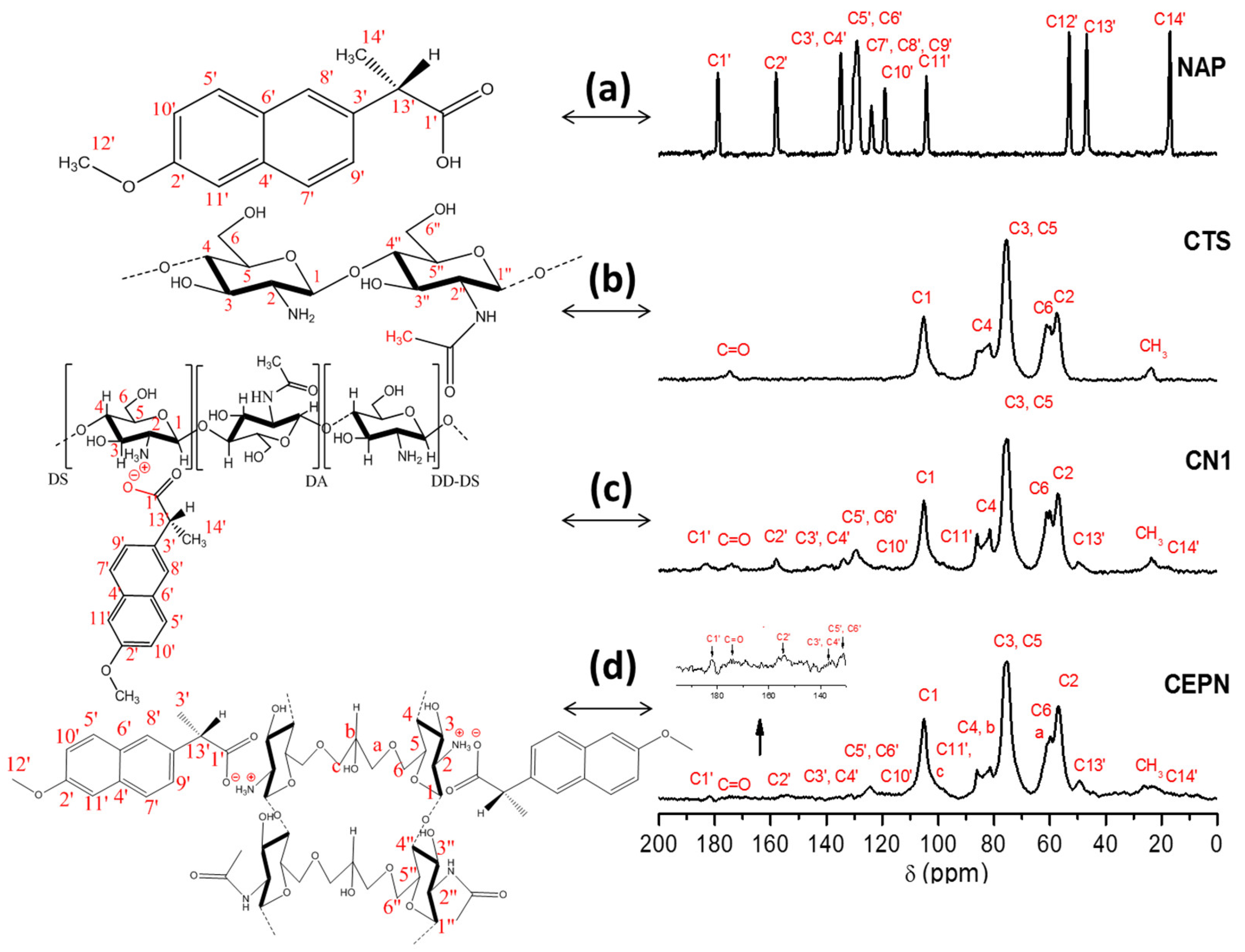
2.1. 13C NMR Spectroscopy
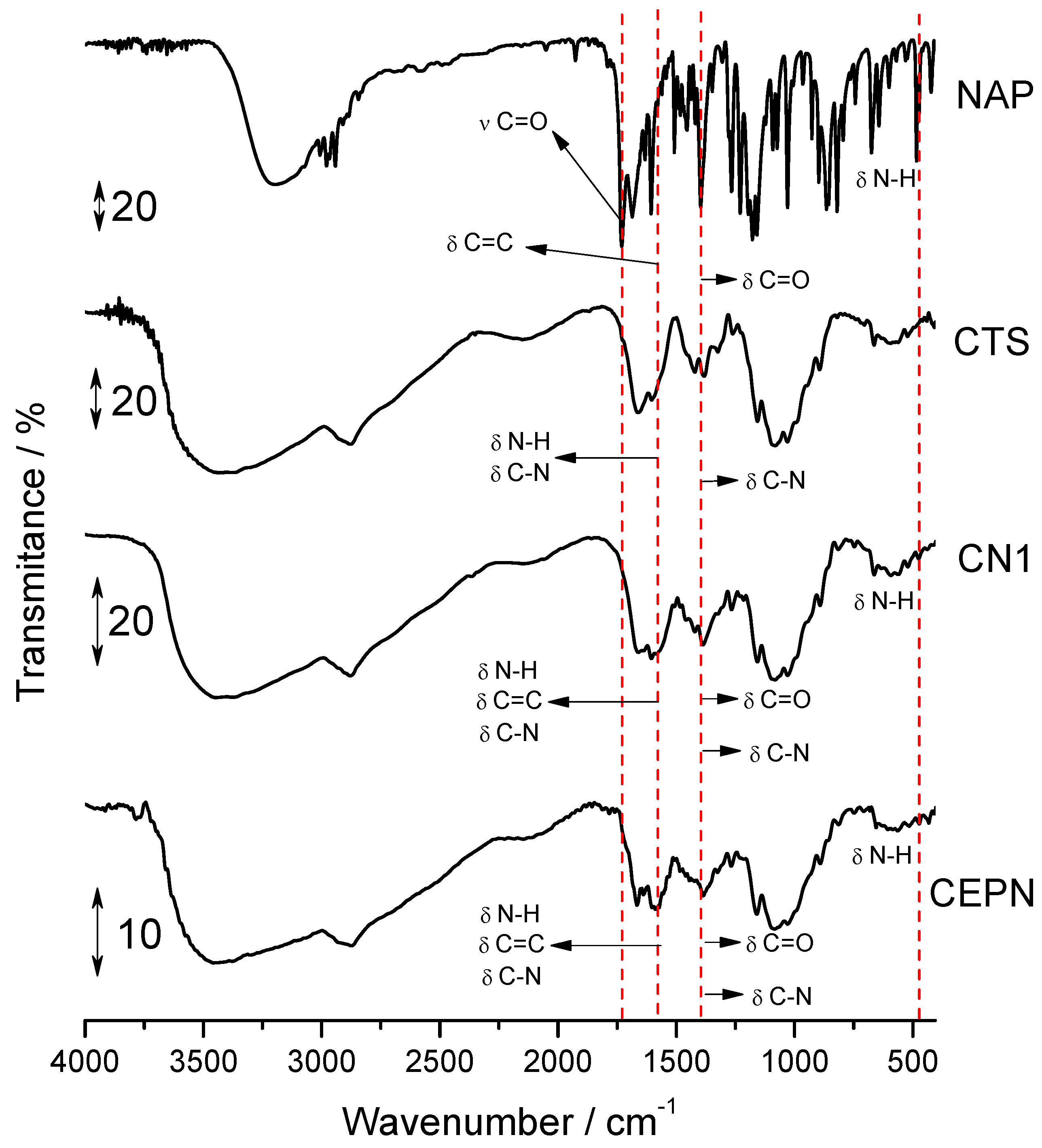
2.2. Infrared Spectroscopy
2.3. UV–Vis Diffuse Reflectance Spectroscopy
2.4. X-ray Diffraction
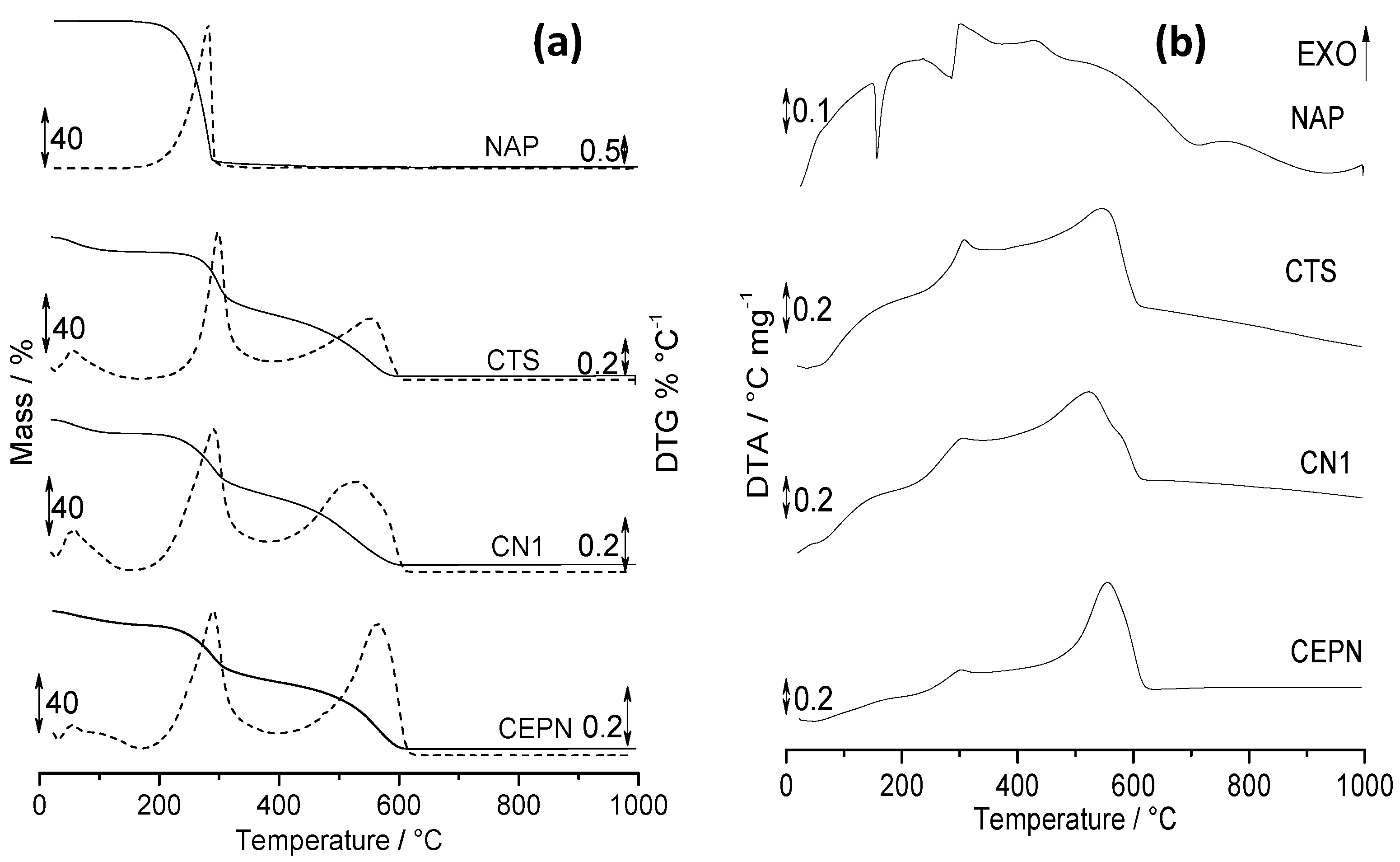
2.5. Thermal Analysis
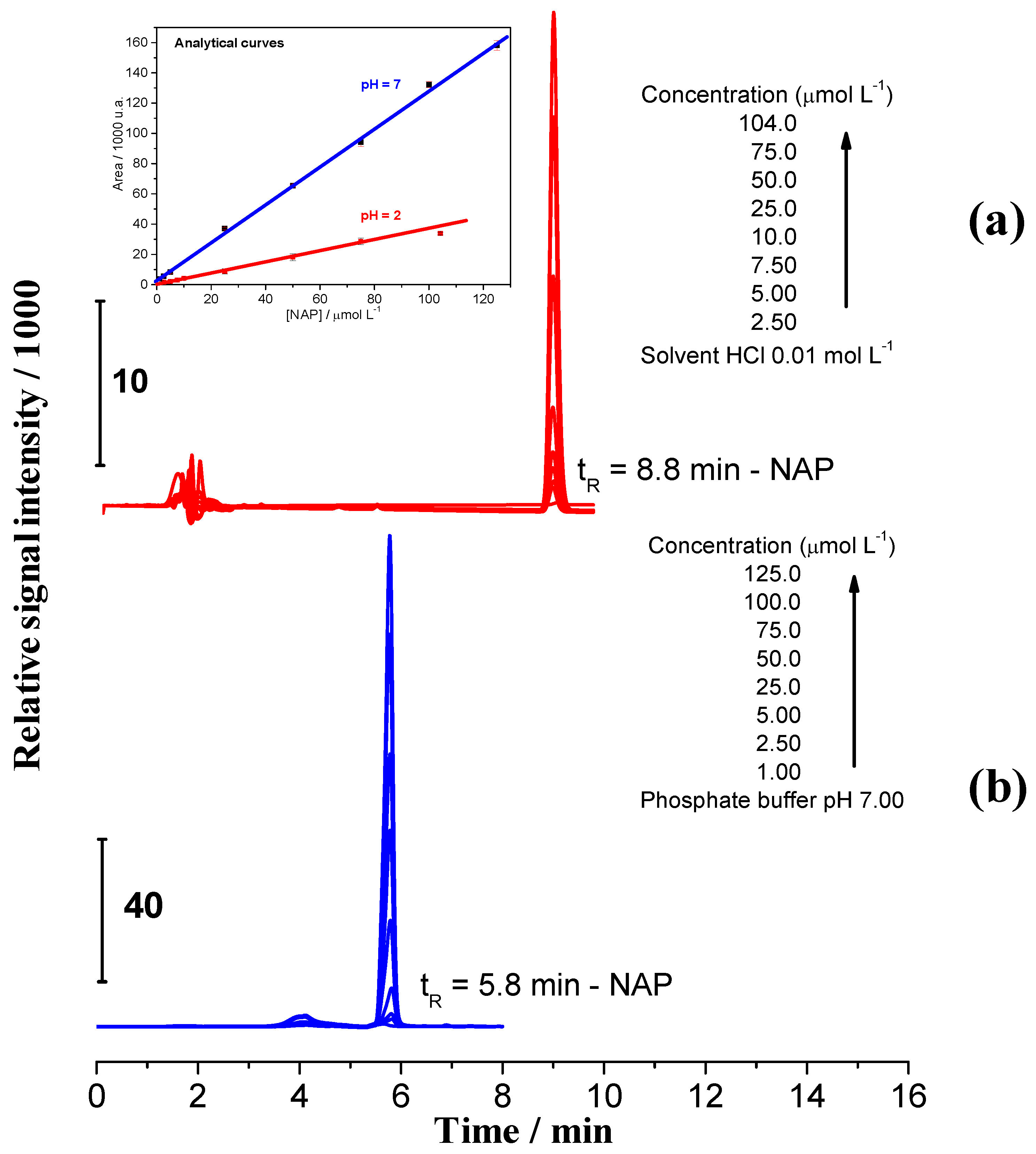
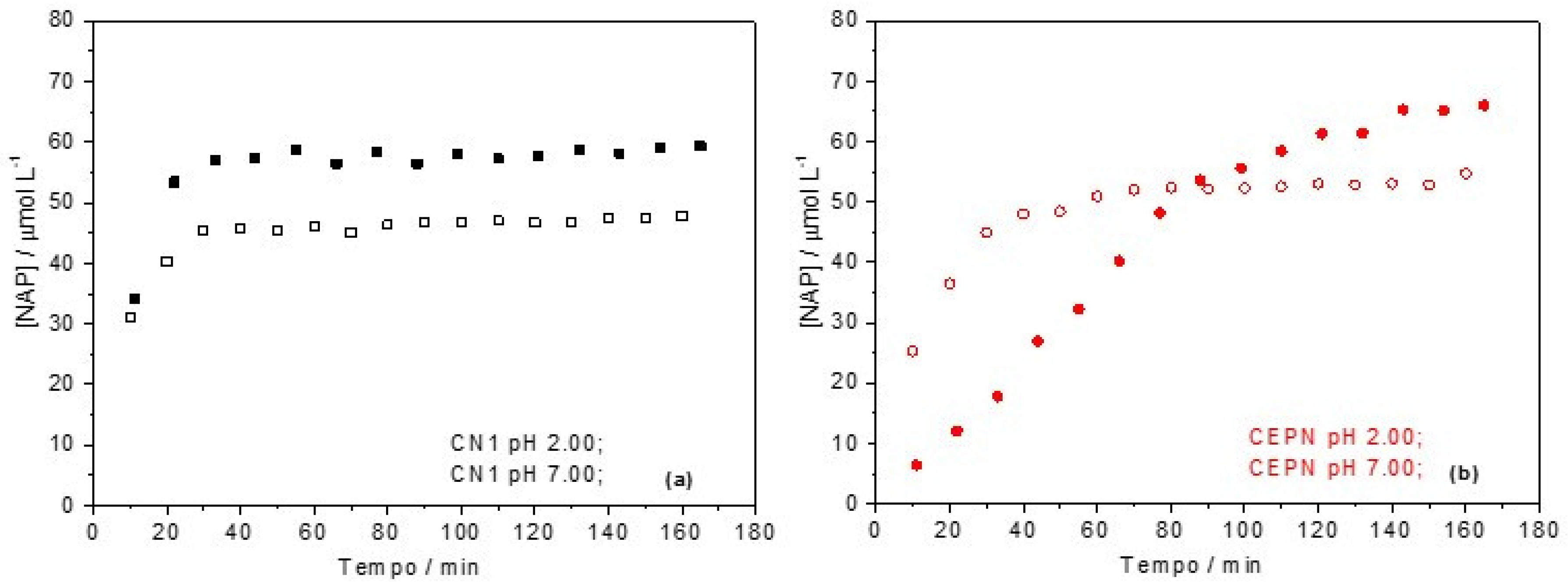
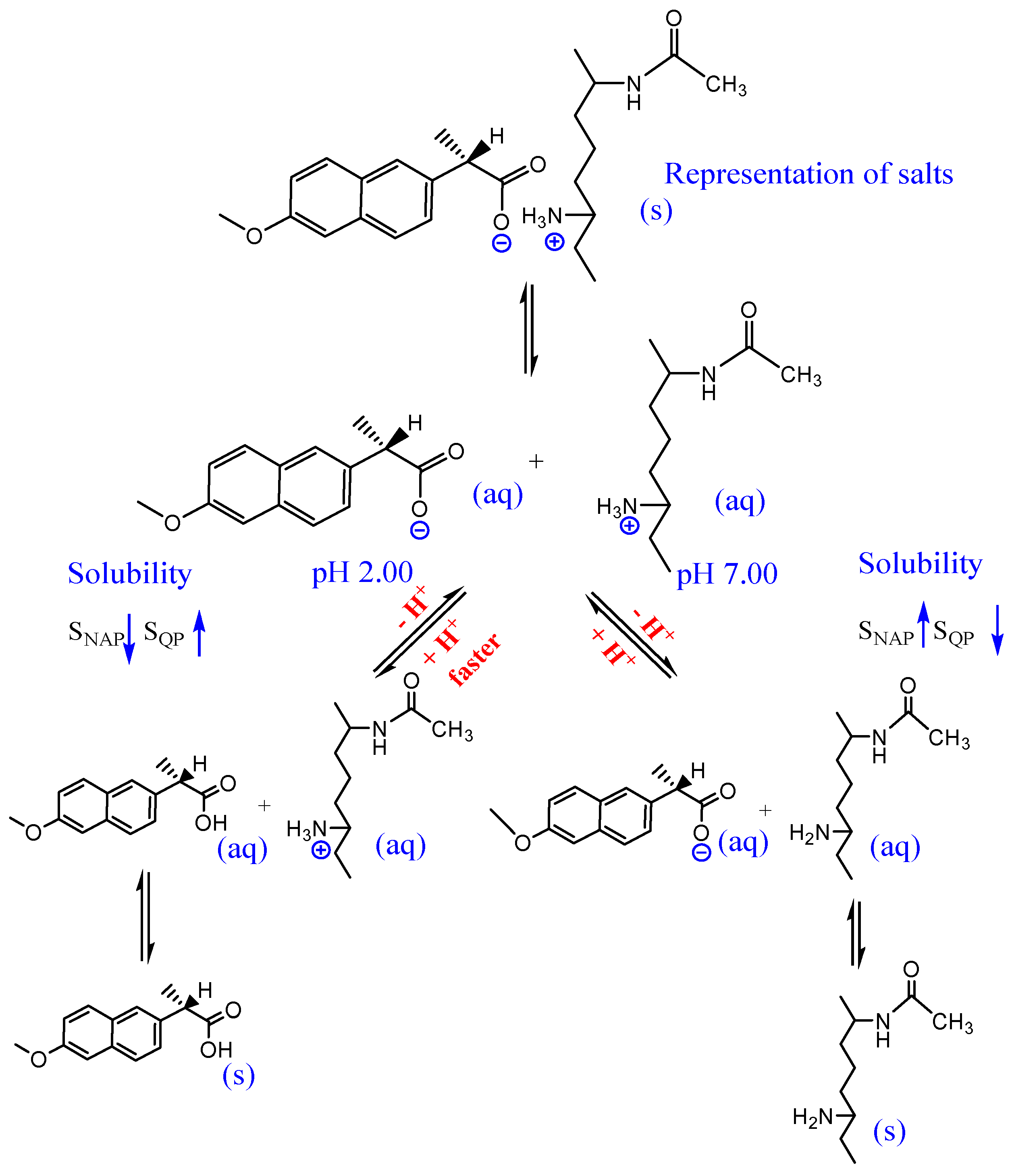
2.6. Dissociation Study Using High Performance Liquid Chromatography (HPLC)
3. Material and Methods
3.1. Reagents and Solutions
3.2. Chitosan Purification
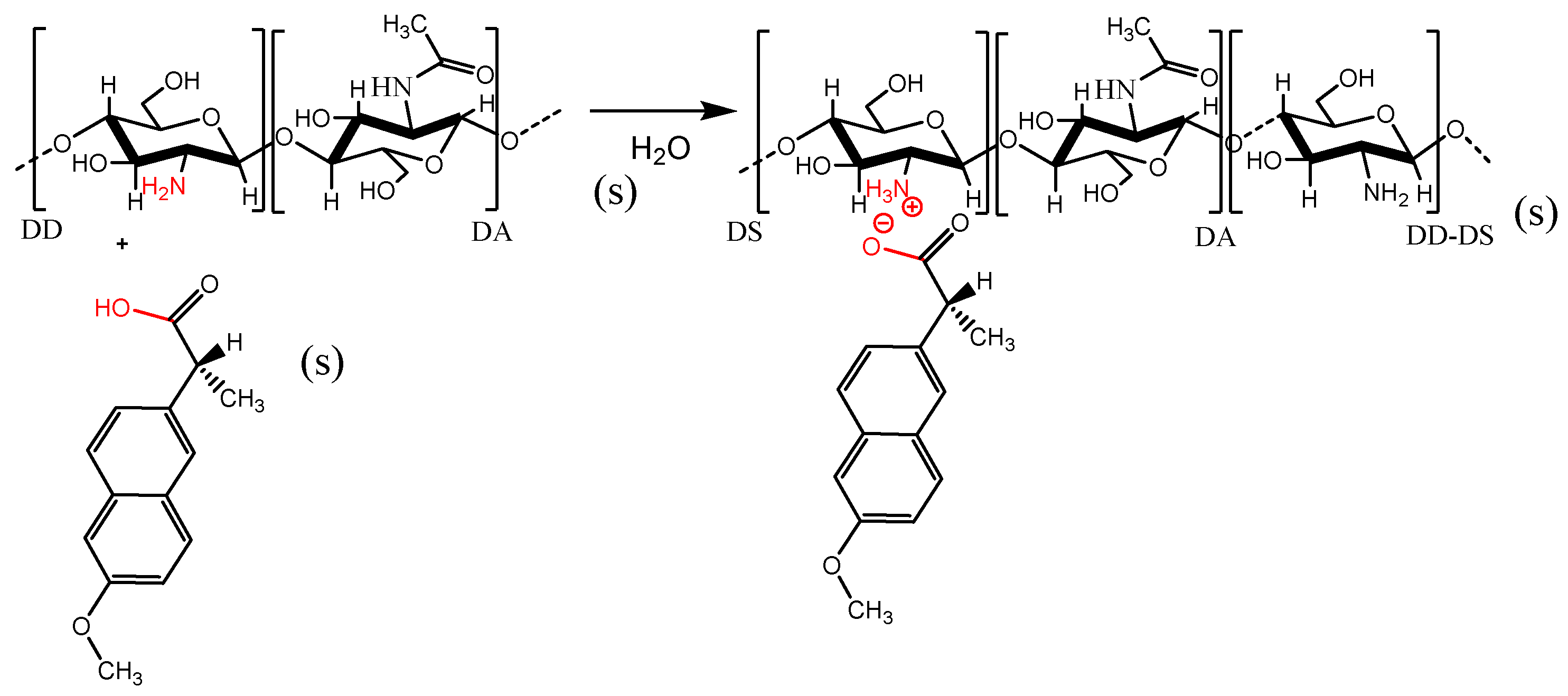
3.3. Reaction of Chitosan and Naproxen
3.4. CTS Reticulation
3.5. Study Regarding NAP–CTS and NAP–CEPN Salt Dissociation by High Performance Liquid Chromatography (HPLC)
3.6. Characterization
1H and 13C NMR Spectroscopy
3.7. Infrared Spectroscopy (FTIR)
UV–Vis Diffuse Reflectance Spectroscopy
3.8. X-ray Diffraction (XRD)
Thermal Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CEPN | Salt of naproxen and chitosan crosslinked with epichlorohydrin |
| CN | Salt of naproxen and chitosan |
| COX | Cyclo-oxygenase enzyme |
| CT | Chitin |
| CTS | Chitosan |
| CTS–NAP | Physical mixture of chitosan and naproxen |
| Degree of acetylation | |
| Degree of deacetylation | |
| Degree of substitution | |
| Id | Crystallinity index |
| Kpart | Partition constant |
| Average viscometric molar mass | |
| NAP | Naproxen |
| [NAP] | Equilibrium concentration of naproxen |
| NSAID | Non-steroidal anti-inflammatory drug |
| PVP | Polyvinylpyrrolidone |
| Sd | Standard deviation |
| tR | PRetention time |
References
- Facchinatto, W.M.; dos Santos, D.M.; Fiamingo, A.; Bernardes-Filho, R.; Campana-Filho, S.P.; de Azevedo, E.R.; Colnago, L.A. Evaluation of chitosan crystallinity: A high-resolution solid-state NMR spectroscopy approach. Carbohydr. Polym. 2020, 250, 116891. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, H.F.G.; Francisco, D.S.; Ferreira, A.P.G.; Cavalheiro, E.T.G. A new look towards the thermal decomposition of chitins and chitosans with different degrees of deacetylation by coupled TG-FTIR. Carbohydr. Polym. 2019, 225, 115232. [Google Scholar] [CrossRef]
- Kaya, M.; Mujtaba, M.; Ehrlich, H.; Salaberria, A.M.; Baran, T.; Amemiya, C.T.; Galli, R.; Akyuz, L.; Sargin, I.; Labidi, J. On chemistry of γ-chitin. Carbohydr. Polym. 2017, 176, 177–186. [Google Scholar] [CrossRef]
- Rasmussen, R.S.; Morrissey, M.T. Marine biotechnology for production of food ingredientes. Adv. Food Nutr. Res. 2007, 52, 237–292. [Google Scholar] [PubMed]
- Muzzarelli, R.A.A. Chitin; Pergamon Press: Oxford, UK, 1977; Volume 309. [Google Scholar]
- Barbosa, H.F.G.; Attjioui, M.; Leitão, A.; Moerschbacher, B.M.; Cavalheiro, E.T.G. Characterization, solubility and biological activity of amphihilic biopolymeric Schiff bases synthesized using chitosans. Carbohydr. Polym. 2019, 220, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vadivel, T.; Dhamodaran, M. Synthesis, characterization and antibacterial studies of ruthenium (III) complexes derived from chitosan schiff base. Int. J. Biol. Macromol. 2016, 90, 44–52. [Google Scholar] [CrossRef]
- Antony, R.; David, S.T.; Karuppasamy, K.; Sanjeev, G.; Balakumar, S. Synthesis, spectroscopic and catalytic studies of Cu (II), Co (II) and Ni (II) complexes immobilized on Schiff base modified chitosan. J. Mol. Struct. 2013, 1050, 53–60. [Google Scholar] [CrossRef]
- Majeti, N.V.; Kumar, R. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 26, 1–27. [Google Scholar]
- Agnirotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Prabaharan, M. Chitosan-based nanopaticles for tumor-targed drug delivery. Int. J. Biol. Macromol. 2015, 72, 1313–1322. [Google Scholar] [CrossRef]
- Dos Santos, J.E.; Dockal, E.R.; Cavalheiro, E.T.G. Synthesis and characterization of Schiff bases from chitosan and salicylaldehyde derivatives. Carbohydr. Polym. 2005, 60, 277–282. [Google Scholar] [CrossRef]
- Hirai, A.; Odani, H.; Nakajima, A. Determination of degree of deacetilation of chitosan by 1H NMR spectroscopy. Polym. Bull. 1991, 26, 87–94. [Google Scholar] [CrossRef]
- Guinesi, L.S.; Cavalheiro, E.T.G. The use of DSC curves to determine the acetylation degree of chitin/chitosan samples. Thermochim. Acta 2006, 444, 128–133. [Google Scholar] [CrossRef]
- Brugnerotto, J.; Desbrieres, J.; Roberts, G.; Rinaudo, M. Characterization of chitosan by steric exclusion chromatography. Polymer 2001, 42, 9921–9927. [Google Scholar] [CrossRef]
- Rinaudo, M.; Milas, M.; Ledung, P. Characterization of chitosan—Influence of ionic strenght and degree of acetylation on chain expasion. Int. J. Biol. Macromol. 1993, 15, 281–285. [Google Scholar] [CrossRef]
- Peter, M.G. Applications and environmental aspects of chitin and chitosan. J. Macromol. Sci. Part A 1995, 32, 629–640. [Google Scholar] [CrossRef]
- Negm, N.A.; Hefni, H.H.H.; Abd-Elaal, A.A.A.; Badr, E.A.; Abou Kana, M.T.H. Advancement on modification of chitosan biopolymer and its potential applications. Int. J. Biol. Macromol. 2020, 152, 681–702. [Google Scholar] [CrossRef]
- Husain, S.; Al-Samadani, K.H.; Najeeb, S.; Zafar, M.S.; Khurshid, Z.; Zohaib, S.; Qasim, S.B. Chitosan Biomaterials for Current and Potential Dental Applications. Materials 2017, 10, 602. [Google Scholar] [CrossRef]
- Vakili, M.; Rafatullah, M.; Salamatinia, B.; Abdullah, A.Z.; Ibrahim, M.H.; Tan, K.B.; Gholami, Z.; Amouzgar, P. Application of chitosan and its derivatives as adsorbents for dye removal from water and wastewater: A review. Carbohydr. Polym. 2014, 113, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, P.S.; Selvakumar, D.; Kadirvelu, K.; Kumar, N.S. Chitosan as an environment friendly biomaterial—A review on recent modifications and applications. Int. J. Biol. Macromol. 2020, 150, 1072–1083. [Google Scholar] [CrossRef]
- Boonsongrit, Y.; Mitrevej, A.; Mueller, B.W. Chitosan drug binding by ionic interaction. Eur. J. Pharm. Biopharm. 2006, 62, 267–274. [Google Scholar] [CrossRef]
- Boonsongrit, Y.; Mueller, B.W.; Mitrevej, A. Characterization of drug–chitosan interaction by 1H NMR, FTIR and isothermal titration calorimetry. Eur. J. Pharm. Biopharm. 2008, 69, 388–395. [Google Scholar] [CrossRef]
- Jayakumar, R.; Prabaharan, M.; Sudheesh Kumar, P.T.; Nair, S.V.; Tamura, H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol. Adv. 2011, 29, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Andreica, B.I.; Cheng, X.; Marin, L. Quaternary ammonium salts of chitosan. A critical overview on the synthesis and properties generated by quaternization. Eur. Polym. J. 2020, 139, 110016. [Google Scholar] [CrossRef]
- Qandil, A.M.; Obaidat, A.A.; Ali, M.A.M.; Al-Taani, B.M.; Tashtoush, B.M.; Al-Jbour, N.D.; Al Remawi, M.M.; Al-Sou’od, K.A.; Badwan, A.A. Investigation of the interactions in complexes of low molecular weight chitosan with ibuprofen. J. Solut. Chem. 2009, 38, 695–712. [Google Scholar] [CrossRef]
- Imai, T.; Shiraishi, S.; Saitô, H.; Otagiri, M. Interaction of indomethacin with low molecular weight chitosan and improvements of some pharmaceutical properties of indomethacin by low molecular weight chitosans. Int. J. Pharm. 1991, 67, 11–20. [Google Scholar] [CrossRef]
- Saitô, H.; Tabeta, R.; Ogawa, K. High-resolution solid-state 13C NMR study of chitosan and its salts with acids: Conformational characterization of polymorphs and helical structures as viewed from the conformation-dependent 13C chemical shifts. Macromolecules 1987, 20, 2424–2430. [Google Scholar] [CrossRef]
- Al-Shammary, F.J.; Aziz Mian, N.A.; Saleem Mian, M. Naproxen. Anal. Profiles Drug Subst. Excip. 1992, 21, 345–373. [Google Scholar]
- Medeiros, R.S.; Ferreira, A.P.G.; Cavalheiro, E.T.G. Thermal behavior of naproxen and ketoprofen nonsteroidal anti-inflammatory drugs. J. Therm. Anal. Calorim. 2020, 142, 849–859. [Google Scholar] [CrossRef]
- Corti, G.; Maestrelli, F.; Cirri, M.; Mura, P. Dissolution and permeation proprieties of naproxen from solid-state systems with chitosan. Drug Deliv. 2008, 15, 303–312. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mura, P.; Zerrouk, N.; Mennini, N.; Maestrelli, F.; Chemtob, C. Development and characterization of naproxen-chitosan solid systems with improved drug dissolution properties. Eur. J. Pharm. Sci. 2003, 19, 67–75. [Google Scholar] [CrossRef]
- Bhise, K.S.; Dhumal, R.S.; Paradkar, A.R.; Kadam, S.S. Effect of drying methods on swelling, erosion and drug release from chitosan-naproxen sodium complexes. Pharm. Sci. Tech. 2008, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zerrouk, N.; Mennini, N.; Maestrelli, F.; Chemtob, C.; Mura, P. Comparison of the effect of chitosan and polyvinylpyrrolidone on dissolution properties and analgesic effect of naproxen. Eur. J. Pharm. Biopharm. 2004, 57, 93–99. [Google Scholar] [CrossRef]
- Maestrelli, F.; Zerrouk, N.; Chemtob, C.; Mura, P. Influence of chitosan and its glutamate and hydrochloride salts on naproxen dissolution rate and permeation across Caco-2 cells. Int. J. Pharm. 2004, 271, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Rege, P.R.; Shukla, D.J.; Block, L.H. Chitinosan-drug complexes: Effect of electrolyte on naproxen release In Vitro. Int. J. Pharm. 2003, 250, 259–272. [Google Scholar] [CrossRef]
- Perchyonok, V.T.; Reher, V.; Zhang, S.; Grobler, S.R.; Oberholzer, T.G.; Massey, W. Insights and relative effect of aspirin, naproxen and ibuprofen containing hydrogels: From design to performance as a functional dual capacity restorative material and build in free radical defense: In-Vitro studies. Open J. Stomatol. 2014, 4, 73–83. [Google Scholar] [CrossRef]
- Gouda, M.; Elayaan, U.; Youssef, M.M. Synthesis and biological activity of drug delivery system based on chitosan nanocapsules. Adv. Nanopart. 2014, 3, 148–158. [Google Scholar] [CrossRef][Green Version]
- Pereira, F.S.; Lanfredi, S.; González, E.R.P.; da Silva Agostini, D.L.; Gomes, H.M.; Medeiros, R.S. Thermal and morphological study of chitosan metal complexes. J. Therm. Anal. Calorim. 2017, 129, 291–301. [Google Scholar] [CrossRef]
- Carignani, E.; Borsacchi, S.; Bradley, J.P.; Brown, S.P.; Geppi, M. Strong intermolecular ring current influence on 1H chemical shifts in two crystalline forms of naproxen: A combined solid-state NMR and DFT study. J. Phys. Chem. C 2013, 117, 17731–17740. [Google Scholar] [CrossRef]
- Chen, A.H.; Liu, S.C.; Chen, C.Y.; Chen, C.Y. Comparative adsortion of Cu (II), Zn (II), and Pb (II) ions in aqueous solution on the crosslinked chitosan with epichlorohydrin. J. Hazad. Mater. 2008, 154, 184–191. [Google Scholar] [CrossRef]
- Duarte, M.L.; Ferreira, M.C.; Marvão, M.R.; Rocha, J. An optimised method to determine the degree of acetylation of chitin and chitosan by FTIR spectroscopy. Int. J. Biol. Macromol. 2002, 31, 1–8. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C.; Morril, T.C. Identificação Espectrométrica de Compostos Orgânicos, 5th ed.; Guanabara Koogan: Rio de Janeiro, Brazil, 1994. [Google Scholar]
- Guilherme, V.A.; Ribeiro, L.N.M.; Alcântara, A.C.S.; Castro, S.R.; Rodrigues da Silva, G.H.; da Silva, C.G.; Breitkreitz, M.C.; Clemente-Napimoga, J.; Macedo, C.G.; Abdalla, H.B.; et al. Improved efficacy of naproxen-loaded NLC for temporomandibular joint administration. Sci. Rep. 2019, 9, 11160. [Google Scholar] [CrossRef] [PubMed]
- Bannach, G.; Arcaro, R.; Ferroni, D.C.; Siqueira, A.B.; Treu-Filho, O.; Ionashiro, M.; Schnitzler, E. Thermoanalytical study of some anti-inflammatory analgesic agents. J. Therm. Anal. Calorim. 2010, 102, 163–170. [Google Scholar] [CrossRef]
- Silva, R.C.; Andrade Junior, M.A.; Cestari, A.R. Adsorção de Cr (VI) em esferas reticuladas de quitosana: Novas correlações cinéticas e termodinâmicas utilizando microcalorimetria isotérmica contínua. Quim. Nova 2010, 33, 880–884. [Google Scholar] [CrossRef]
- Aydin, C.M.; Abdel-Sadek, S.; Zheng, K.; Yahia, I.S.; Yakuphanoglu, F. Synthesis, diffused reflectance and electrical properties of nanocrystalline Fe-doped ZnO via sol-gel calcination technique. Opt. Laser Technol. 2013, 48, 447–452. [Google Scholar] [CrossRef]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S. Introduction to Spectroscopy, 3rd ed.; Thomson Learning: Boston, MA, USA, 2001. [Google Scholar]
- Kumirska, J.; Czerwicka, M.; Czerwicka, Z.; Bychowska, A.; Brzozowski, K.; Thöming, J.; Stepnowski, P. Application of spectroscopic methods for structural analysis of chitin and chitosan. Mar. Drugs 2010, 8, 1567–1636. [Google Scholar] [CrossRef] [PubMed]
- Fakayode, S.O. Purity analysis of the pharmaceuticals naproxen and propranolol: A guided-inquiry laboratory experiment in the analytical chemistry laboratory. J. Chem. Educ. 2015, 92, 157–162. [Google Scholar] [CrossRef]
- Focher, B.; Beltrame, P.L.; Torri, A.N.G. Alkaline N-deacetylation of chitin enhanced by flash treatments. Reaction kinetics and structure modifications. Carbohydr. Polym. 1990, 12, 405–418. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.; Ferrero, A.; Pizzoli, M. Light-scattering, X-ray diffraction, elemental analysis and infrared spectro-photometry characterization of chitosan, a chelating polymer. Talanta 1972, 19, 1222–1226. [Google Scholar] [CrossRef]
- Signini, R.; Campana Filho, S.P. Purificação e caraterização de quitosana comercial. Polímeros 1998, 8, 63–68. [Google Scholar] [CrossRef]
- Signini, R.; Campana Filho, S.P. Característica e propriedades de quitosanas purificadas nas formas neutras, acetato e cloridrato. Polímeros 2001, 11, 58–64. [Google Scholar] [CrossRef]
- Rasool, B.E.; Abu, G.; Fahmy, S.; Saad, H.; Khan, S. Development and evaluation of ibuprofen transdermal gel formulations. Trop. J. Pharm. Res. 2010, 9, 355–363. [Google Scholar] [CrossRef]
- Mohideen, S.; Shivakanth, M.; Sureshkumar, R.; Krishnan, S.N.; Surendranath, Y.; Satyanarayana, T. Development and validation of analytical method for naproxen and pantoprazole in capsule dosage form. Pharm. Sin. 2011, 2, 114–121. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Code | Ratio CTS:NAP (mol:mol) | Reaction Time (h) | Temperature (°C) |
|---|---|---|---|
| CN1 | 1.00:1.05 | 24 | 60 |
| CN2 | 1.00:1.05 | 24 | 40 |
| CN3 | 1.00:1.05 | 48 | 60 |
| CN4 | 1.00:1.50 | 24 | 60 |
| CN5 | 1.00:1.50 | 48 | 60 |
| CN6 | 1.00:1.50 | 48 | 40 |
| Chemical Shifts (δ) (ppm) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1′ | C=O | C2′ | Naphthalene Ring | C1 | C11′ | (c) | (b) | C4 | C3,C5 | C6 | (a) | C2 | C12′ | C13′ | CH3 | C14′ | |
| NAP | 178.9 | - | 158.1 | 115.0–150.0 | - | 104.2 | - | - | - | - | - | - | - | 53.1 | 46.8 | - | 17.0 |
| CTS | - | 174.7 | - | - | 105.2 | - | - | - | 81.6 | 75.5 | 61.1 | - | 57.5 | - | - | 23.7 | - |
| CN1 | 182.7 | 173.9 | 157.9 | 115.0–150.0 | 105.2 | 98.0 | - | - | 81.4 | 74.5 | 59.9 | - | 57.1 | - | 50.0 | 23.7 | 18.3 |
| CN2 | 183.1 | 174.7 | 158.1 | 115.0–150.0 | 105.2 | 97.6 | - | - | 81.5 | 75.6 | 59.9 | - | 57.1 | - | 49.4 | 23.7 | 20.4 |
| CN3 | 182.9 | 174.6 | 157.8 | 115.0–150.0 | 105.2 | 99.5 | - | - | 81.6 | 75.5 | 60.0 | - | 57.2 | - | 49.6 | 23.6 | 19.2 |
| CN4 | 182.6 | 174.6 | 157.9 | 115.0–150.0 | 105.2 | 98.0 | - | - | 81.5 | 75.5 | 60.0 | - | 57.2 | - | 49.9 | 23.8 | 21.1 |
| CN5 | 182.6 | 175.0 | 158.1 | 115.0–150.0 | 105.2 | 97.7 | - | - | 81.5 | 75.5 | 60.0 | - | 57.2 | - | 49.5 | 23.6 | 20.6 |
| CN6 | 182.8 | 174.5 | 158.1 | 115.0–150.0 | 105.2 | 97.6 | - | - | 81.6 | 75.6 | 60.0 | - | 57.2 | - | 49.4 | 23.9 | 17.6 |
| CEPN | 182.4 | 173.8 | 153.8 | 115.0–150.0 | 105.1 | 98.1 | 100.2 | 86.1 | 81.4 | 75.42 | 60.9 | 59.8 | 56.9 | - | 49.3 | 26.3 | 22.6 |
| Sample * | CTS | CN1 | CN2 | CN3 | CN4 | CN5 | CN6 | CEPN | |
|---|---|---|---|---|---|---|---|---|---|
| Parmeter * | |||||||||
| C1 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |
| C1′ | - | 0.17 | 0.15 | 0.12 | 0.12 | 0.12 | 0.11 | 0.036 | |
| (%) | - | 17 | 15 | 12 | 12 | 12 | 11 | 3.6 | |
| Samples | νO-H/νN-H | νC-H asym./sym. | νC=O Carboxyl | δC=O Amide I | δN-H Amine | δC=C Naphth. Ring | δC-N Amide | δC-N Amide II | δCH3/δCOO− | CH3 Amide | δC-N Amide III | νC-O Ether | δC-H | νC-O-C β(1→4) | δC-OH | C-H Glucopy. Ring. | δC-H | δN-H Ammonium |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NAP | 3196 | 2976 2939 | 1728 | - | - | 1602 | - | - | 1394 | - | - | 1265 | 1176 | - | - | - | 819 | 484 |
| CTS | 3600–3100 | 2917 2877 | - | 1662 | 1604 | - | 1552 | 1435 | - | 1382 | 1325 | 1159 | 1089 1029 | 891 | ||||
| CN1 | 3600–3100 | 2937 2870 | - | 1664 | 1606 | 1606 | 1573 | 1425 | 1381 | 1381 | 1321 | 1269 | 1209 | 1157 | 1082 1022 | 887 | 819 | 484 |
| CEPN | 3400 | 2937 2873 | - | 1666 | 1602 | 1602 | 1589 | 1440 | 1384 | 1384 | 1330 | 1273 | 1163 | 1080 1033 | 896 | 810 | 480 |
| Sample | Process | TGA/DTG | DTA | ||
|---|---|---|---|---|---|
| Temperature Range (°C) | Mass Loss (%) | Ratio * | Temperature Peak (°C) | ||
| NAP | 1st step | 139.4–365.3 | 97 | - | 283.8 (endo) 303.4 (exo) |
| 2nd step | 365.3–491.0 | 1.50 | 430.3 (exo) | ||
| Residue | 491.0–1000.0 | 1.50 | - | ||
| CTS | Dehydration | 19.9–172.2 | 10.2 | 0.99 | 67.5 (endo) |
| 1st step | 172.2–390.1 | 42.4 | 304.4 (exo) | ||
| 2nd step | 390.1–609.9 | 42.0 | 544.2 (exo) | ||
| Residue | 609.9–1000.0 | 5.95 | - | ||
| CN1 | Dehydration | 19.6–151.4 | 9.52 | 1.13 | 64.8 (endo) |
| 1st step | 151.4–381.3 | 41.9 | 302.5 (exo) | ||
| 2nd step | 381.3–617.3 | 47.3 | 522.0 (exo) | ||
| Residue | 617.3–1000.0 | 1.69 | - | ||
| CEPN | Dehydration | 23.6–171.5 | 9.45 | 1.17 | 56.4 (endo) |
| 1st step | 171.5–399.0 | 38.7 | 301.2 (exo) | ||
| 2nd step | 399.0–637.9 | 45.3 | 553.5 (exo) | ||
| Residue | 637.9–1000.0 | 6.60 | - | ||
| Sample | pH | Kpart/mol L−1 * | mNAP Dissolved/mg | Equilibrium Time/min |
|---|---|---|---|---|
| CN1 | 2.0 | 5.82 ± 0.03 | 0.67 | 17.54 |
| 7.0 | 4.69 ± 0.02 | 0.54 | 25.48 | |
| CEPN | 2.0 | 6.58 ± 0.07 | 0.76 | 104.3 |
| 7.0 | 5.31 ± 0.03 | 0.61 | 36.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medeiros, R.S.; Ferreira, A.P.G.; Venâncio, T.; Cavalheiro, É.T.G. Preparation, Characterization and Study of the Dissociation of Naproxen from Its Chitosan Salt. Molecules 2022, 27, 5801. https://doi.org/10.3390/molecules27185801
Medeiros RS, Ferreira APG, Venâncio T, Cavalheiro ÉTG. Preparation, Characterization and Study of the Dissociation of Naproxen from Its Chitosan Salt. Molecules. 2022; 27(18):5801. https://doi.org/10.3390/molecules27185801
Chicago/Turabian StyleMedeiros, Ricardo S., Ana P. G. Ferreira, Tiago Venâncio, and Éder T. G. Cavalheiro. 2022. "Preparation, Characterization and Study of the Dissociation of Naproxen from Its Chitosan Salt" Molecules 27, no. 18: 5801. https://doi.org/10.3390/molecules27185801
APA StyleMedeiros, R. S., Ferreira, A. P. G., Venâncio, T., & Cavalheiro, É. T. G. (2022). Preparation, Characterization and Study of the Dissociation of Naproxen from Its Chitosan Salt. Molecules, 27(18), 5801. https://doi.org/10.3390/molecules27185801