Abstract
Tumors are still one of the main causes of death; therefore, the search for new therapeutic agents that will enable the implementation of effective treatment is a significant challenge for modern pharmacy. One of the important factors contributing to the development of neoplastic diseases is the overexpression of enzymes responsible for the regulation of cell division processes such as cyclin-dependent kinases. Numerous studies and examples of already-developed drugs confirm that isatin is a convenient basis for the development of new groups of inhibitors for this class of enzyme. Therefore, in this work, a new group of potential inhibitors of the CDK2 enzyme, utilizing isatin derivatives and substituted benzoylhydrazines, has been designed based on the application of computational chemistry methods, such as docking and molecular dynamics, and their inhibiting ability was assessed. In the cases of the selected compounds, a synthesis method was developed, and the selected physicochemical properties of the newly synthesized derivatives were estimated. As part of the completed project, new compounds are developed which are potential inhibitors of the CDK2 enzyme.
1. Introduction
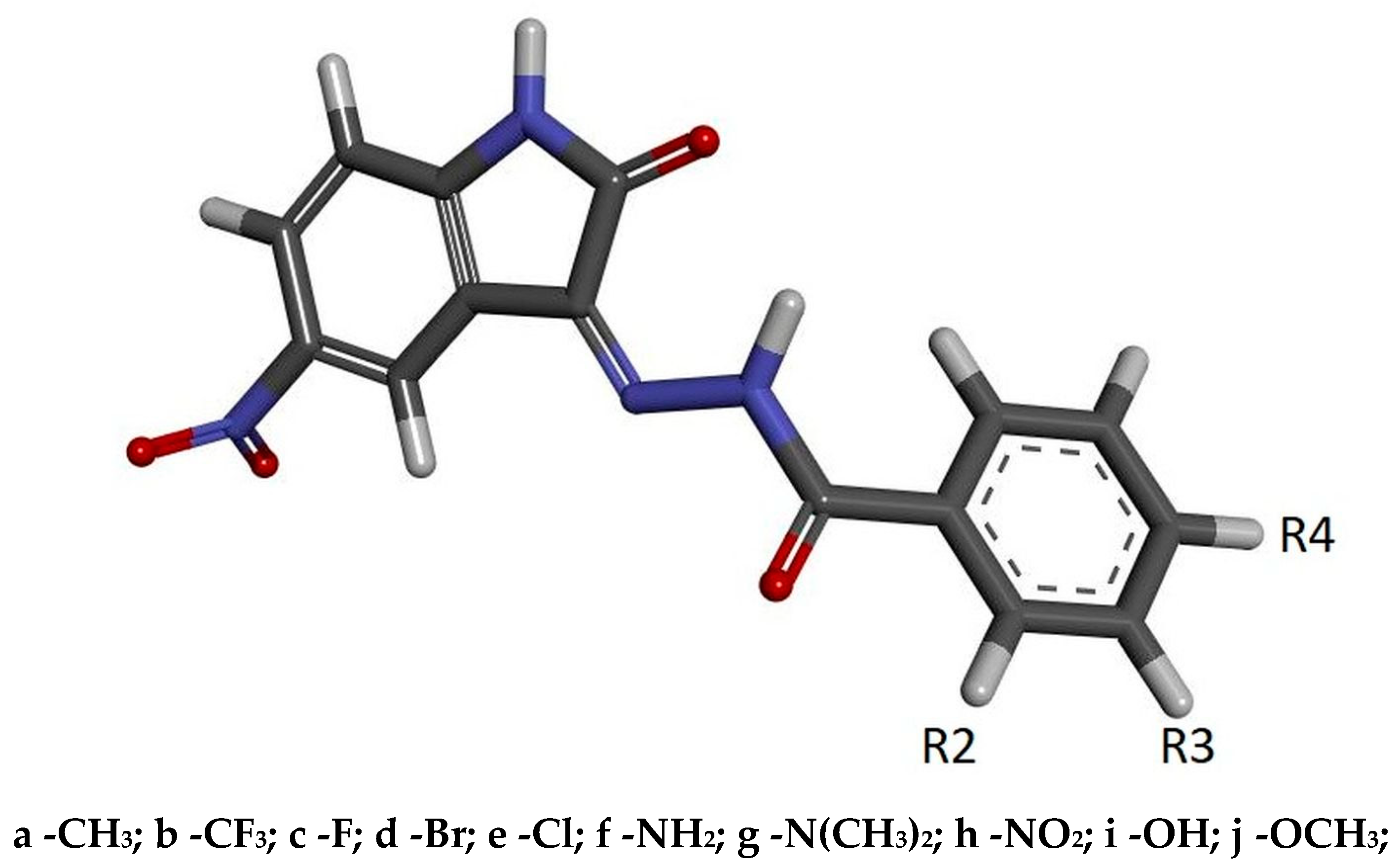
Isatin (1H-indol-2,3-dione) is a chemical compound which, as natural alkaloid, may be extracted from plants of the Isatis genus, occurring all over the world at various latitudes [1,2,3]. The significant chemical and biological activities of isatin meant that this compound and its numerous derivatives, obtained from natural products, have widely been used in medicine for many centuries [4,5]. In terms of the pharmaceutical activity of such compounds, they have anti-inflammatory, antiviral, antibacterial, antifungal, anticonvulsant, anxiogenic and anticancer properties [6,7,8,9,10]. The large spectra of pharmacological activities of isatin derivatives are associated with the significant chemical variety of these compounds, within which one can distinguish hydrazones, thiosemicarbazones, oximes, spiro-oxindoles, imines and many more. The common element of this broad group of compounds is the oxindole system, which is the core repeatedly used in the development of new groups of competitive inhibitors [11,12,13,14,15,16,17,18,19,20,21]. The anticancer activity of new drugs is mainly related to the competitive inhibition of biological targets, the overexpression of which drives the accelerated growth of cancer cells. The group of enzymes associated with the development of cancers includes the set of SER/THR kinases classified as cyclin-dependent kinases (CDKs), especially CDK2. This enzyme plays a crucial role in the regulation of the cell cycle. The CDK2 and other cyclin-dependent kinases are also crucially important in the regulation of the other enzymes involved in transcription and replication processes [22,23,24]. A significant group of CDK2 inhibitors is isatin derivatives and its analogs, which contain a characteristic oxindole core in their structure. Numerous studies show that this type of compound has a notable affinity for the active site of this enzyme [12,16,25]. The location of the donors and acceptors of hydrogen bonds in the oxindol core ensures the possibility of creating stable interactions in the hinge region with aminoacids such as LEU83 and GLU81 [16,25]. The presence of additional hydrogen bond acceptors in oxindolic systems may provide an opportunity to create interactions with LYS33 and ASP145 [15,26,27]. Based on these guidelines, this work will develop a new group of isatin derivatives based on the reaction of isatin derivatives with the set of substituted benzoylhydrazines. The schematic representation of the isatin-based benzoylhydrazine structure is presented in Figure 1. During the creation of new potential inhibitors, the impact of the presence of various substituents (positions R2–R7) on their binding activity with the CDK2 active site will be evaluated.
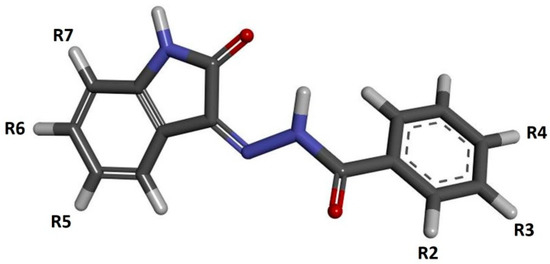
Figure 1.
The graphic representation of isatin derivatives. Symbols R2–R7 represent places of addition of new chemical residues modifying the chemical properties of the native compound.
2. Results and Discussion
2.1. Design and Computational Analysis of Binding Activity
The isatin derivatives were created in two stages. Firstly, the modification of the isatin molecule was chosen. Based on the commercially available isatine derivatives containing substituents localized in positions 5, 6 and 7, model structures of inhibitors obtained by reaction with non-substituted benzoylhydrazide were created. The binding affinity of such molecules towards cyclin dependent kinase was evaluated using docking methods.
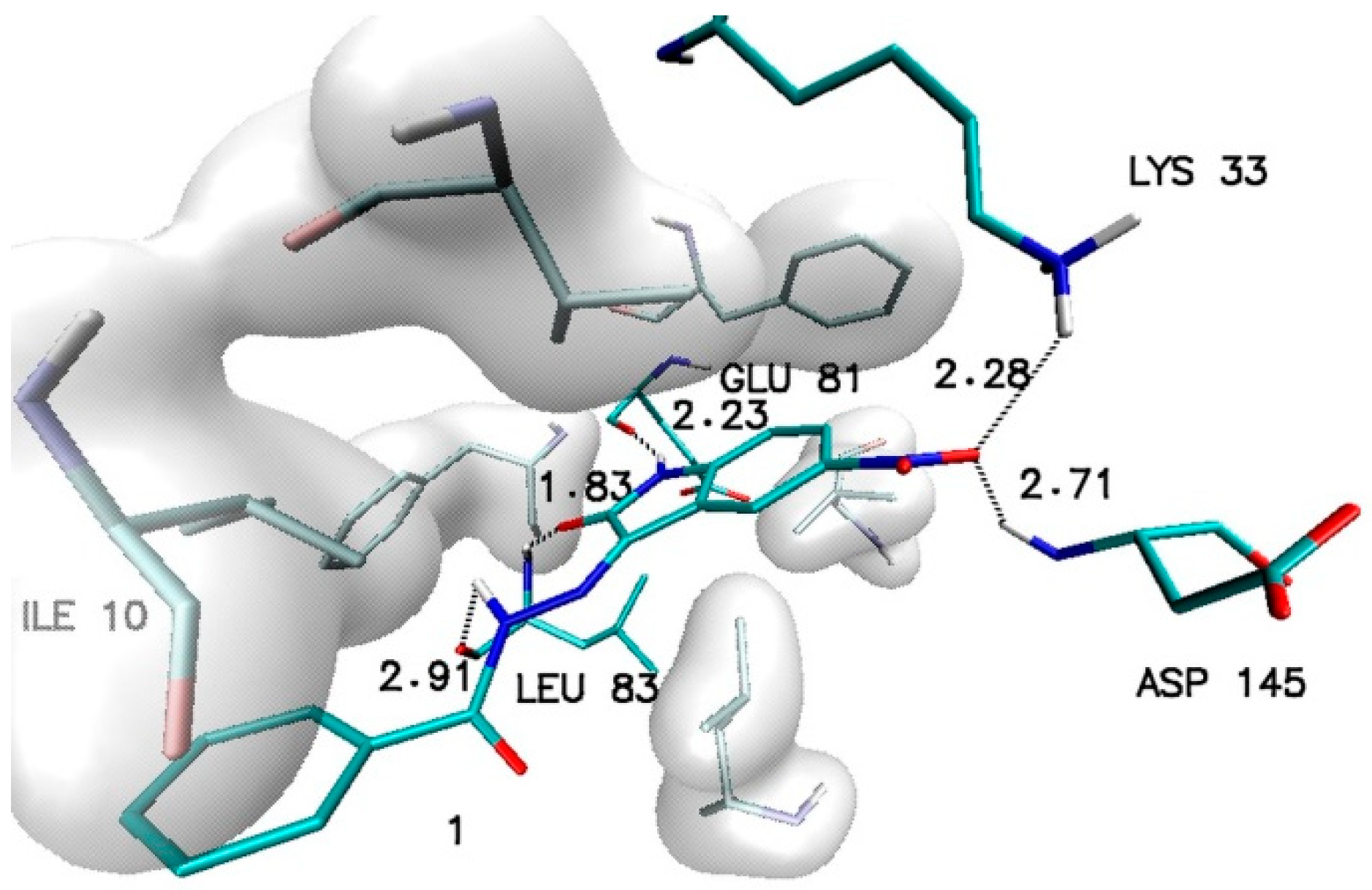
The outcome of these calculations is presented in Table 1. Considering all of the collected data, it can be observed that the least effective are modifications located in position 7, while the most favorable effects involving an increase in affinity are observed in the case of modifications located in positions 5 and 6. Among all of the considered structures, the highest value of affinity was recorded for the 5-nitroisatin derivative. The structure of the complex created by this molecule with the CDK2 active site is presented in Figure 2.

Table 1.
The values of binding affinity (∆G) of substituted isatin derivatives towards CDK2 active site. The reference value obtained for non-substituted isatin derivative is −8.9 (kcal/mol).
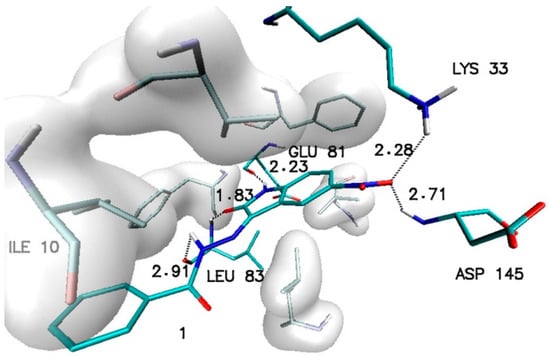
Figure 2.
The graphic representation of interactions involved in stabilization of CDK2 complex with N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (1).
The stability of such a system is maintained by a network of interactions including hydrogen bonds and hydrophobic interactions. Besides the hydrogen bonds created by atoms from a molecular core with GLU81 and LEU83, the activity of oxygen atoms from the nitro group was also observed, which participates in interactions with LYS33 and ASP145. Based on the values of binding affinity and the presence of additional binding factors, related with the existence of two additional hydrogen bond acceptors, 5-nitroisatin was chosen for the next stage of research. Based on commercially available substituted benzoylhydrazide derivatives, 30 new potential CDK2 inhibitors were created; the scheme of the foreseen modifications of the native molecule are presented in Figure 3. In the nomenclature adopted in this work, a native molecule based on unmodified benzoylhydrazide was marked with the number “1”, while in the case of subsequent derivatives, the designation consists of a number symbolizing the place of substitution and the letter assigned for a specific chemical group. The binding capabilities towards the CDK2 active site of each newly created molecule were evaluated using docking methods. Table 2 presents the values of binding affinity and the inhibition constants obtained for all 5-nitroisatin-based benzoylhydrazines.
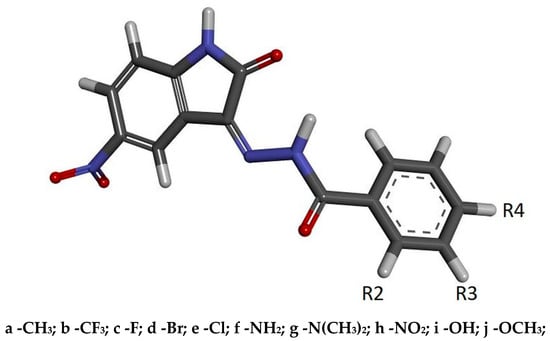
Figure 3.
The graphic representation of 5-nitroisatin-based benzoylhydrazines. The markings R2, R3 and R4 represent the places of chemical group substitution. The derivative markings consist of a number symbolizing the place of substitution and the letter assigned for a specific chemical group.
Table 2.
The values of biding affinity and inhibition constants (IC) of 5-nitroisatin-based benzoylhydrazines towards CDK2 active site.
The inhibition constant (IC) for ligand molecules was estimated based on the Van ’t Hoff isotherm equation.
Comparing the values obtained for new molecules with the native one, it can be observed that the introduction of modifications in the aromatic system of the considered derivatives does not always increase their binding capacities and, in some cases, even lowers them. It is especially visible in the case of modifications located in the para position (R4), since none of the newly generated molecules with such modification demonstrated a noticeable increase in binding activity.
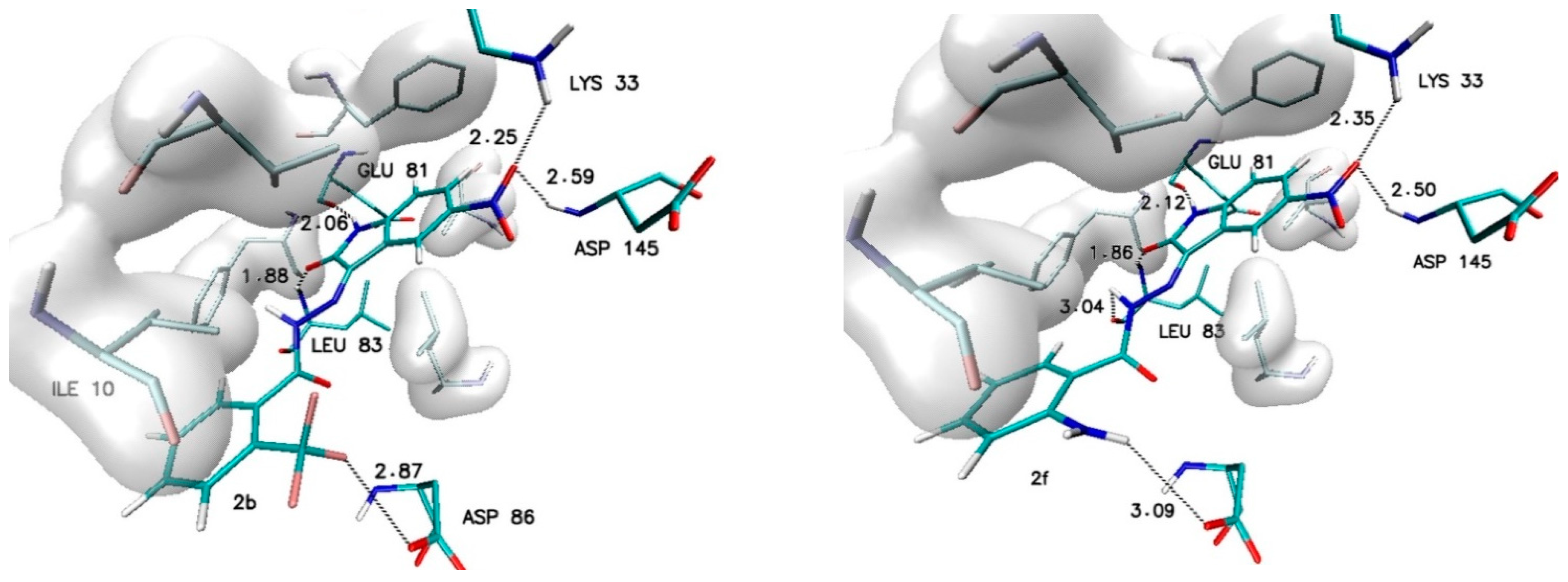
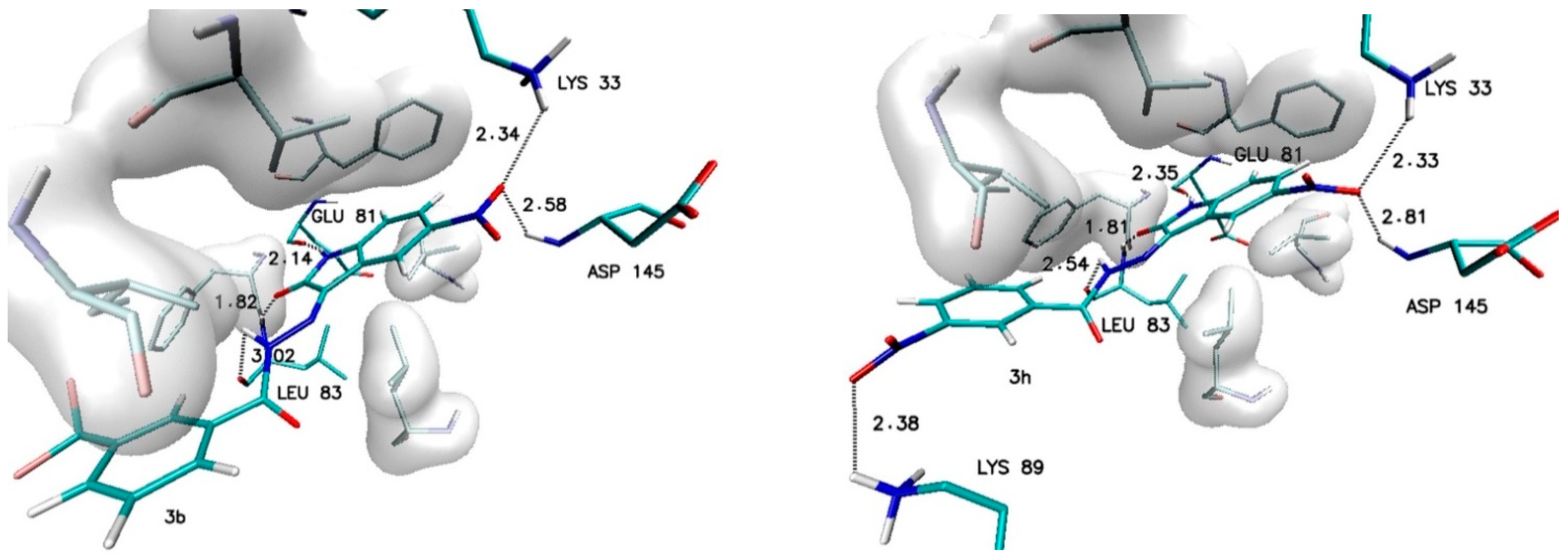
Different observations can be made in the case of modifications present in the ortho (R2) and meta (R3) positions. Among all considered substituents, the greatest influence on the binding capability was observed in the case of the trifluoromethyl group, the appearance of which caused the greatest increase in affinity value (−0.5 kcal/mol) and the most convenient value of the inhibition constant (46.8 nM). An analogous increase was also recorded for the methyl group located in the meta position (R3). In the case of the other considered substituents, the observed increases in affinity ranged from −0.2 to −0.3 kcal/mol. For all complexes formed by derivatives with an increase in affinity of at least −0.2 kcal/mol, structural stability was analyzed using molecular dynamics methods. The structures of the chosen complexes are presented in Figure 4 and Figure S1 (Supplementary Materials). Such systems are maintained by a network of numerous interactions of different types. The most important hydrogen bonds identified in all the analyzed systems are summarized in Table 3. Each considered molecule creates bindings with aminoacids such as LYS33, GLU81, LEU83 and ASP145. Based on the geometric hydrogen bond strength classification, interactions created with GLU81, LEU83 and LYS33 can be qualified as medium-strength hydrogen bonds, while interactions with ASP145 are classed weak bonds. In the case of molecules such as 2b, 2f and 3h, interactions created by atoms from chemical groups located in benzoylhydrazide part of the molecule were also observed.
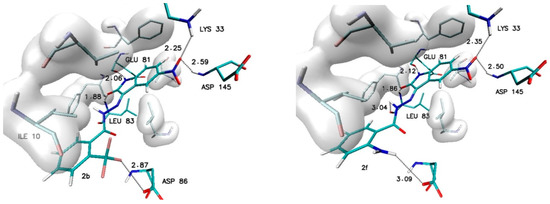
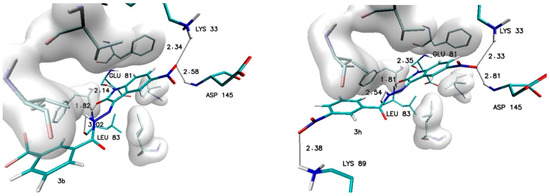
Figure 4.
The graphic representation of interactions involved in stabilization of CDK2 complex with chosen 5-nitroisatin-based benzoylhydrazines.
Table 3.
The values of hydrogen bond lengths [Å] involved in stabilization of 5-nitroisatin-based benzoylhydrazines complexes with the CDK2 active site.
The structural stability of considered complexes and the time evolution of such systems was evaluated based on an analysis of root mean square deviation (RMSD) values and structural descriptors, defining the interactions involved in complex stabilization such as distances and angles measured for hydrogen bond donors and acceptors identified for particular hydrogen bond. Figure S2 (Supplementary Materials) presents distributions of the RMSD values estimated for inhibitors and CDK2 protein. In each case, values were calculated with regard to the starting point, which was the geometry of the complex obtained during docking stage.
An analysis of such distributions and averaged values is presented in Table 4; this shows that not all proposed derivatives have the ability to create stable complexes with the active place of the considered protein. In a few cases, such as molecules 2c, 2e, 3c and 3d, large fluctuations in RMSD values are observed, indicating the noticeable conformational instability of such molecules in the space of a CDK2 active site. The inability to maintain the original conformation obtained during docking or to adopt a new one ensuring a better fit to the active place may indicate insufficient matching, which is caused by the presence of a possible steric hindrance in the considered molecules. Completely different characteristics of the discussed properties are shown by molecules such as 1, 2b, 2d, 3b, 3e, 3f and 3h, for which uniform distributions and low standard deviation for the analyzed populations of RMSD values indicate the conformational stability of the analyzed systems.
Table 4.
The averaged values of RMSD for ligands and CDK2 protein for all steps used during structural analysis.
An important factor for assessing the stability of the complexes formed by potential inhibitors with the active site of a biological target is the evaluation of the durability of the interactions responsible for its maintenance. During the docking stage, the primary structures of the complexes were created, in which interactions such as hydrogen and halogen bonds were identified. Table 5 and Table S1 (Supplementary Materials) contain a list of the most important impacts that showed sufficient durability to exclude their incidental occurrence. Among all of the considered interactions, the most important and the most stable in each of the complexes are hydrogen bonds formed by the oxygen and the hydrogen atoms of the isatin core with LEU83 and GLU81. The presence of these hydrogen bonds was confirmed in 100% of conformers accumulated during molecular dynamics for each of the analyzed complexes. The observed differences between particular systems are visible in the distribution of the bond lengths dominant within the compared populations. In the case of bonds created with GLU81, over 90% of tested conformers creates at least medium-strength hydrogen bonds (distance ≤ 2 Å), the exceptions are only the complexes created by 2c and 3c molecules. The distances measured for hydrogen bonds created with LEU83 exhibit much larger diversity—in this case, there is at least a 70% share of medium-strength hydrogen bonds observed only for conformers from complexes created by 1, 2d, 2e, 2f, 3b, 3e and 3f molecules. The activity of oxygen atoms from the nitro group localized in the isatin core and the significance of hydrogen bonds created by them is diametrically different for complexes created by individual derivatives. Among the analyzed systems, one can distinguish complexes where the bonds with ASP145 almost disappeared and those created by LYS33 have been significantly reduced, as in the case of 2c and 3c complexes. A group of complexes is also noticeable where, for at least 80% of the conformers, stable bonds with at least one of the amino acids are observed, for example, complexes formed by the molecules 1, 2d, 2f, 3b, 3e, and 3h. The highest binding activity for the nitro group was recorded in the case of derivative 2b, in which stable hydrogen bonds were observed both for the ASP145 and LYS33 in over 90% of the analyzed conformers. Based on the structures of complexes obtained during docking for many of the derivatives considered, the possibility of creating a second hydrogen bond with LEU83 was expected. An analysis of the results of molecular dynamics simulation showed the existence of stable and numerous interactions of this type only for complexes created by 2b (85% of conformers) and 3f (98.9% of conformers) derivatives. An analysis of the interactions in the complex formed by the derivative 2f shows the presence of a halogen bond between the fluorine atoms of the 3-fluoromethyl group and the oxygen atom of ILE10. The distance between the donor and acceptor of this interaction in the scope of the entire simulation ranges from 2.6 to 3.2 Å. The presence of such an interaction contributes to the overall stabilizing effect of the analyzed complex.
Table 5.
The cumulative analysis of the length of the interactions identified in CDK2 complexes with selected 5-nitroisatin-based benzoylhydrazines. The distances presented in the table represent the middle values of intervals with a width of 0.2 Å.
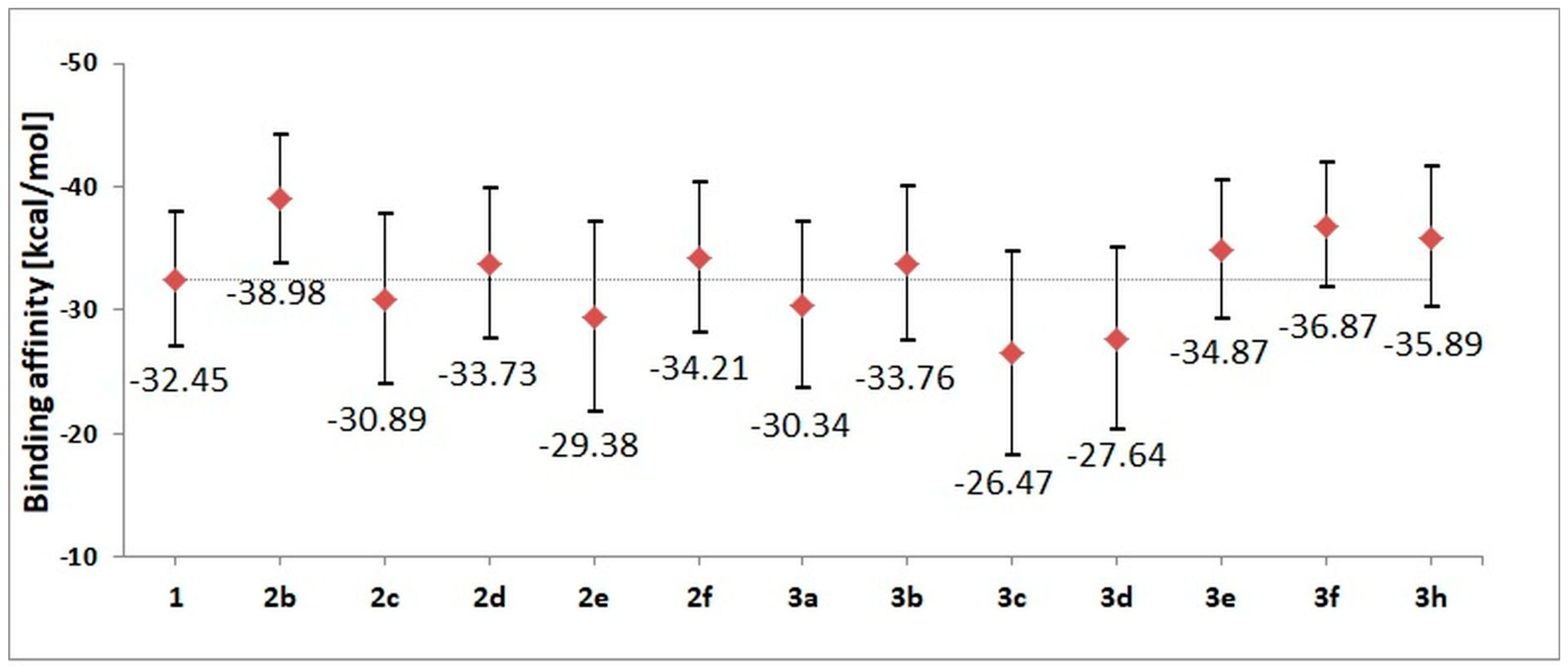
The stability of complexes was also evaluated using the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method, which helped to evaluate the values of binding affinity characterizing particular complexes. Based on the conformers from the last 60 ns of molecular dynamics simulation, enthalpic contributions to the binding affinity were estimated. The obtained values are presented in Figure 5; numeric labels represent accurate values of affinity, while the graphic markers correspond to a spread of values, taking into account the standard deviation. Based on the gathered values, the largest affinity toward the active site of CDK2 is shown by 2b, 3f and 3h derivatives. The energy characteristics of the systems corroborate the previous structural observations based on an analysis of the conformational properties of ligands and the stability of interactions involved in the maintenance of the complexes. Based on the summary characteristics of the analyzed systems, for the experimental phase, including the synthesis and characterization of molecular and spectroscopic properties, derivatives 1, 2b, 2d, 2f, 3b, 3e, 3f and 3h were selected.
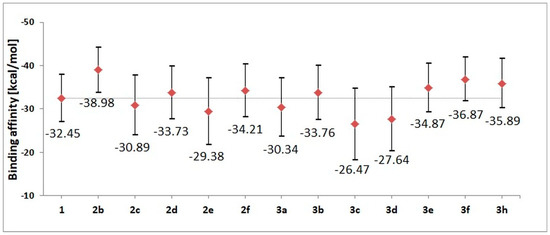
Figure 5.
The values of binding enthalpy (kcal/mol) estimated for all complexes considered during molecular dynamics stage.
2.2. Synthesis and NMR Data
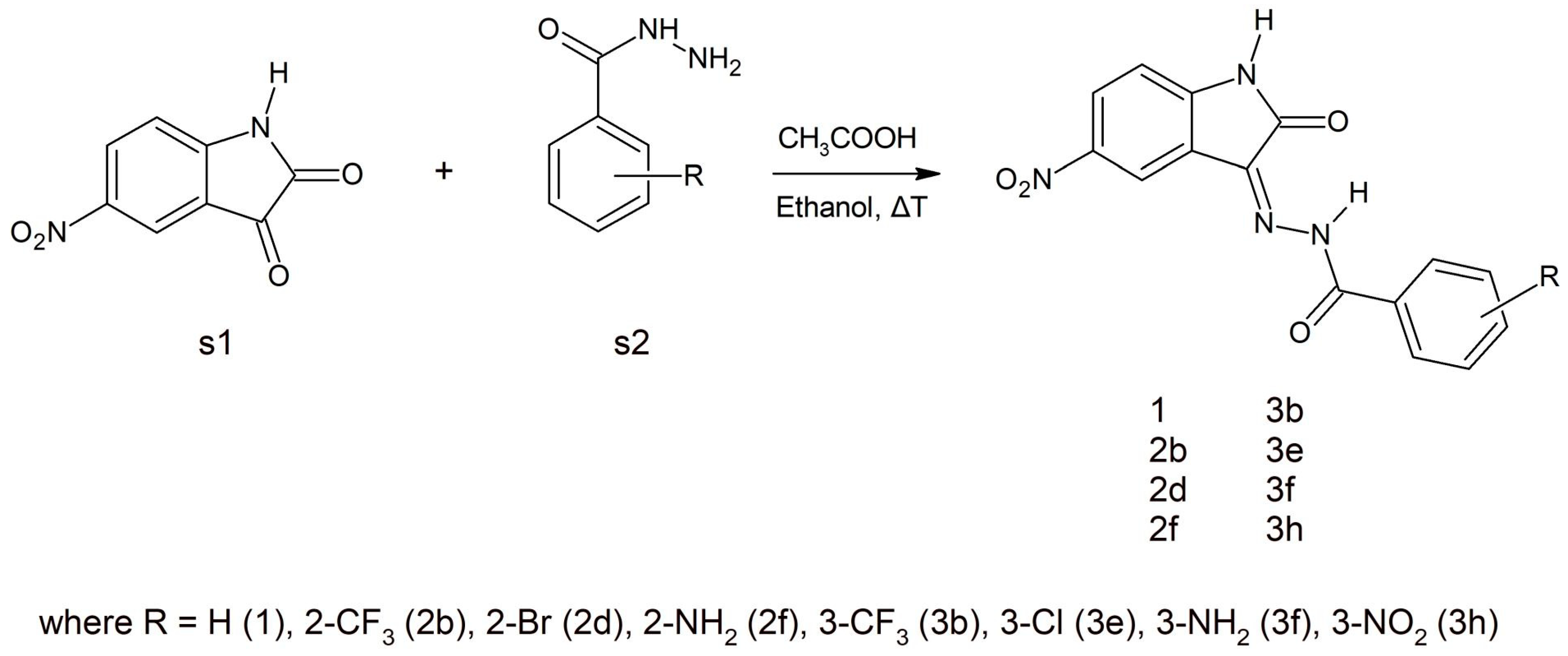
Various synthetic procedures for 5-nitroisatin-based benzoylhydrazines can be found in the literature [28,29,30]. The simplest indicates that 5-nitroisatin (s1) is heated with substituted benzoylhydrazine (s2) in ethanol in the presence of acetic acid (Scheme 1). All the derivatives were analyzed using IR, NMR and also elemental analysis.
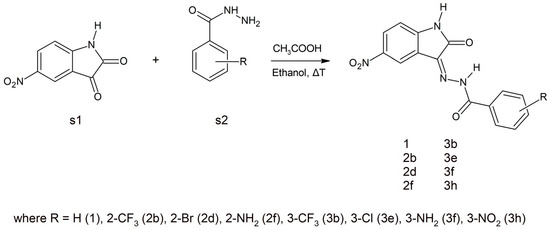
Scheme 1.
A schematic representation of the synthesis of the selected 5-nitroisatin-based benzoylhydrazines.
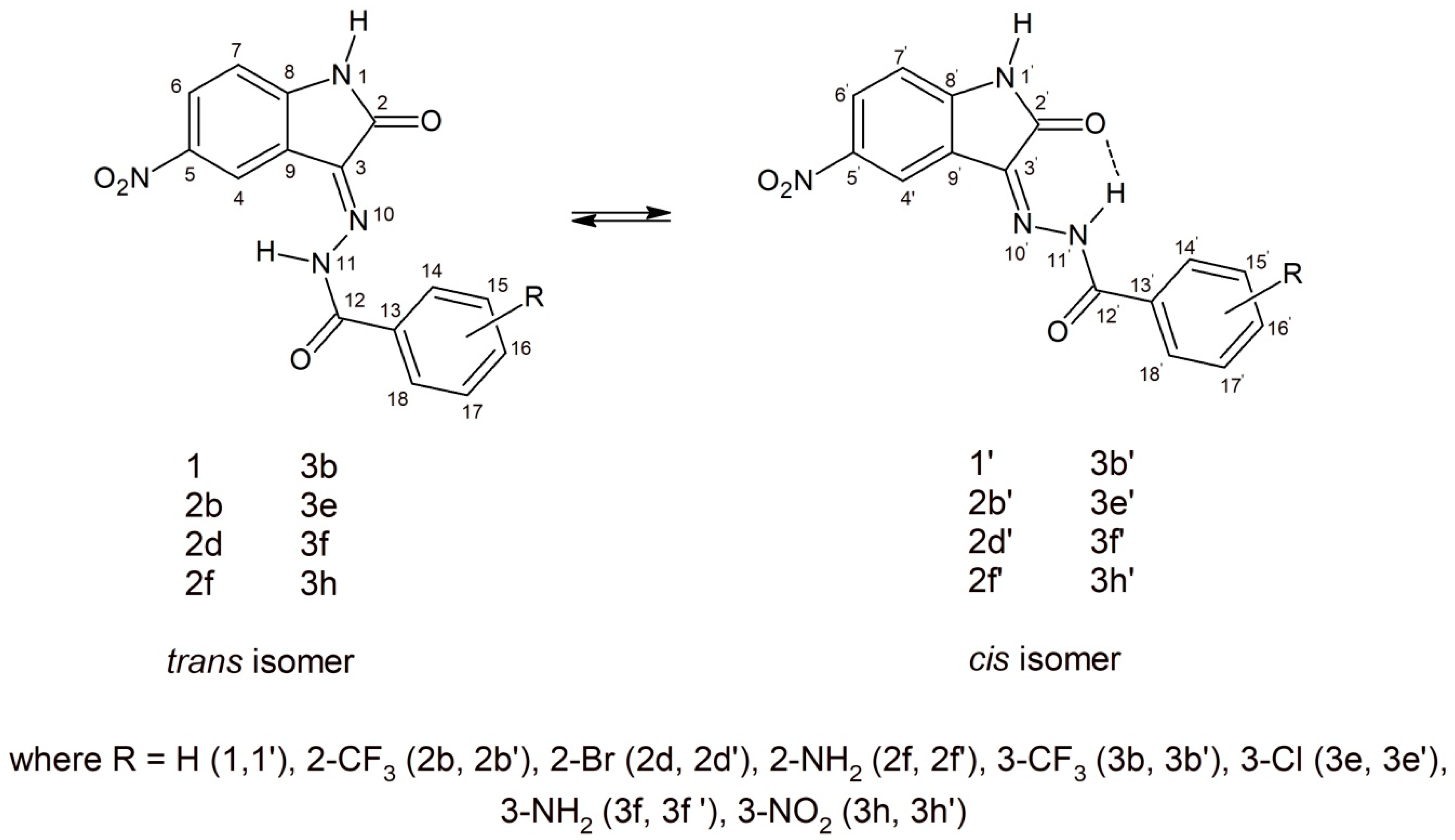
Structurally, the synthesized 5-nitroisatin-based benzoylhydrazines can exist in cis- and trans-isoforms. Although all of the derivatives demonstrated the presence of both isomers in the solution at room temperature, with the cis-isomer being the predominant one. It should be noted that in all cases, their 1H NMR spectra showed a more intense signals for hydrogen atoms of cis-isomer. Moreover, the calculated energies showed that more stable are cis-isomers (Table 6).
Table 6.
Energies of cis- and trans-isomers of 5-nitroisatin-based benzoylhydrazines estimated in DMSO at the B3LYP/6-311 ++ G(d,p) level of theory. ∆ECIS-TRANS represents the difference between cis- and trans-isomers (∆ECIS-TRANS = ECIS − ETRANS).
As shown in Figure 6, in the chemical shift structure of the cis form, there is a possibility of intramolecular hydrogen bonding between the hydrogen atom of the hydrazone section and the oxygen atom of the adjacent carbonyl group [31]. In addition, in derivatives substituted in position 2 (2b, 2d and 2f), the presence of an intramolecular hydrogen bond between the substituent and the carbonyl oxygen of benzoylhydrazine was observed. According to R.S. Hunoor et al. [32], the N–H group (hydrazine part) is sandwiched between carbonyl (C=O) and N=C azometine nitrogen and intramolecular hydrogen bonding with carbonyl oxygen of 5-nitroisatin. All these interactions make it difficult to unambiguously assign aromatic hydrogens and carbons to individual isomers of 2-substitued 5-nitroisatin-based benzoylhydrazines.
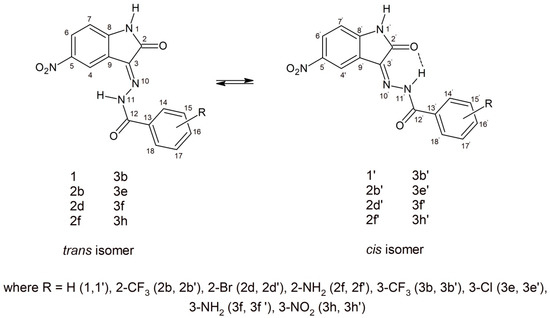
Figure 6.
Possible geometry of the synthesized 5-nitroisatin-based benzoylhydrazines.
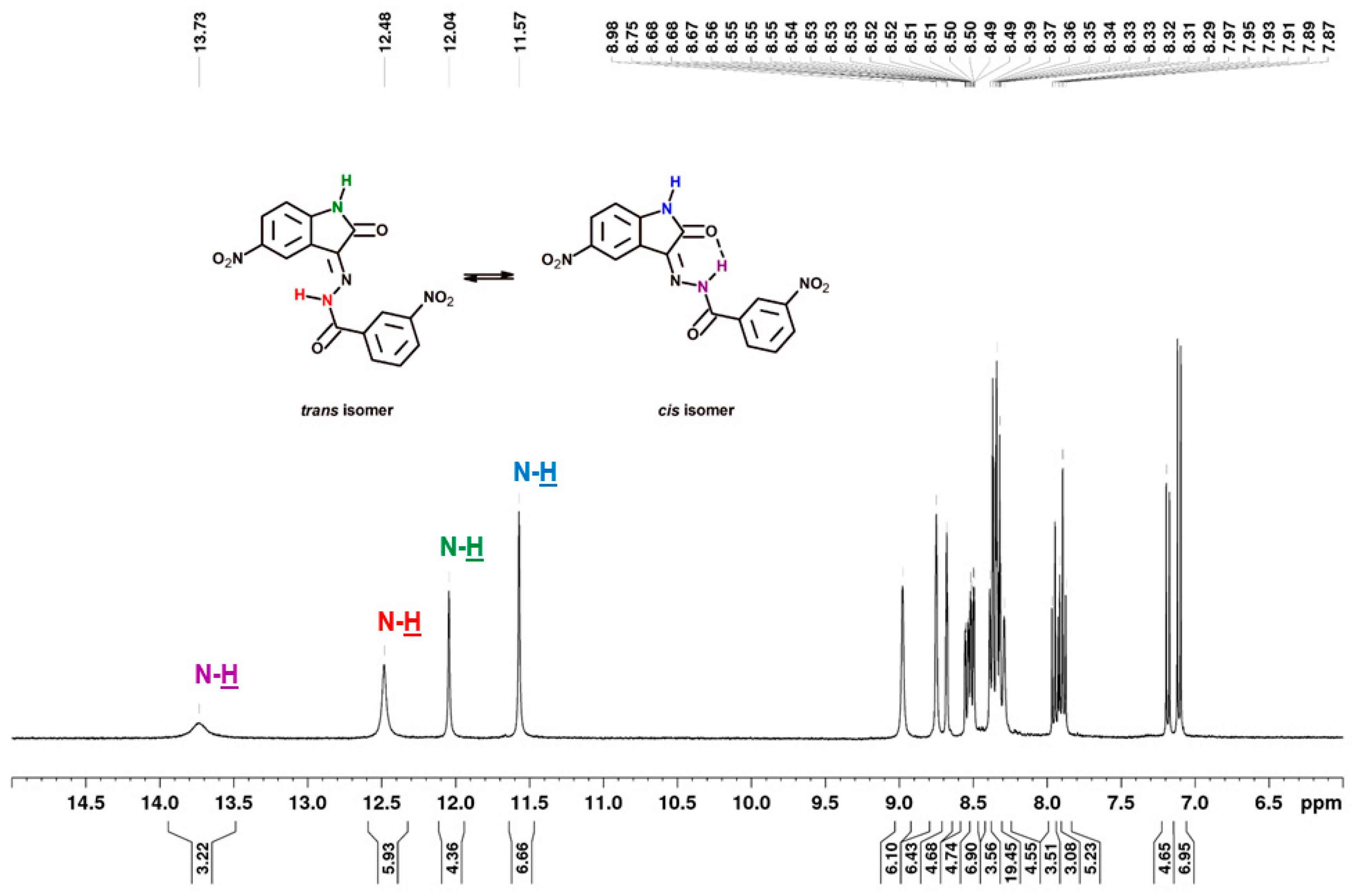
As previously mentioned, the compounds showed in Figure 6 were present in solution as trans- and cis-isomers. In all cases, their 1H NMR spectra showed a signal with chemical shift in the range of 13.16–13.37 ppm, corresponding to the N–H of benzoylhydrazine involved in the relevant intramolecular hydrogen bond for cis-isomers. For the same proton the trans-isomer (without intramolecular hydrogen bond), the signal was observed in the range of 12.02–12.88 ppm. The 1H-NMR signal of N–H of isatin can be seen at δ = 11.51–11.57 ppm and δ = 11.96–12.04 ppm in DMSO, which is a singlet, for cis- and trans-isomers, respectively (Figure 7). The first signal observed with the least chemical shift in aromatic protons relates to the H-7 and H-7′ of isatin, which appears as a doublet signal in the region of 6.85 to 7.18 ppm (J = 8.56–8.86 Hz). The next protons of isatin (H-4, H-4′ and H-6, H-6′) appeared in the region of 8.23 to 9.06 ppm. The aroyl (hydrazone) part of the target compounds showed appropriate signals in the range of 6.67–8.18 ppm, depending on the type of substituent. The carbon signals of the most characteristic carbon atoms, such as carbonyl carbon C-12/C-12′ and lactonyl carbon C-2/C-2′, were observed at ca. 167 and 164 ppm, respectively. The azomethine carbon C-3/C-3′ at ca. 137 ppm was not always recorded.
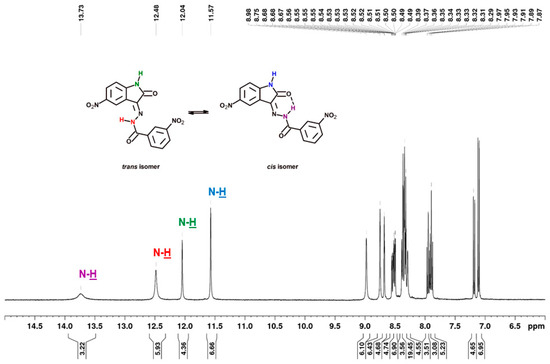
Figure 7.
1H NMR spectrum (400 MHz) of 3-nitro-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (3h) in DMSO-d6.
2.3. Infrared Spectral Studies
Vibrational spectroscopy is used extensively in the study of molecular conformations, the identification of functional groups and reaction kinetics, etc. Infrared spectral data for the synthesized 5-nitroisatin-based benzoylhydrazines are presented in Table 7. Sharp bands of medium intensity at 3167–3303 cm−1 in the analyzed compounds spectrums were due to stretching vibrations of the –NH group. The absorption band characteristic of ν(C=O) vibrations of isatin and benzoylhydrazine fragments appeared in the range 1697–1751 cm−1 and 1622–1698 cm−1, respectively, and the absorption band at 1513–1528 cm−1 was assigned to azomethine ν(C=N) stretching.
Table 7.
Characteristic band-strength values of the synthesized 5-nitroisatin-based benzoylhydrazines.
2.4. Spectroscopic Properties
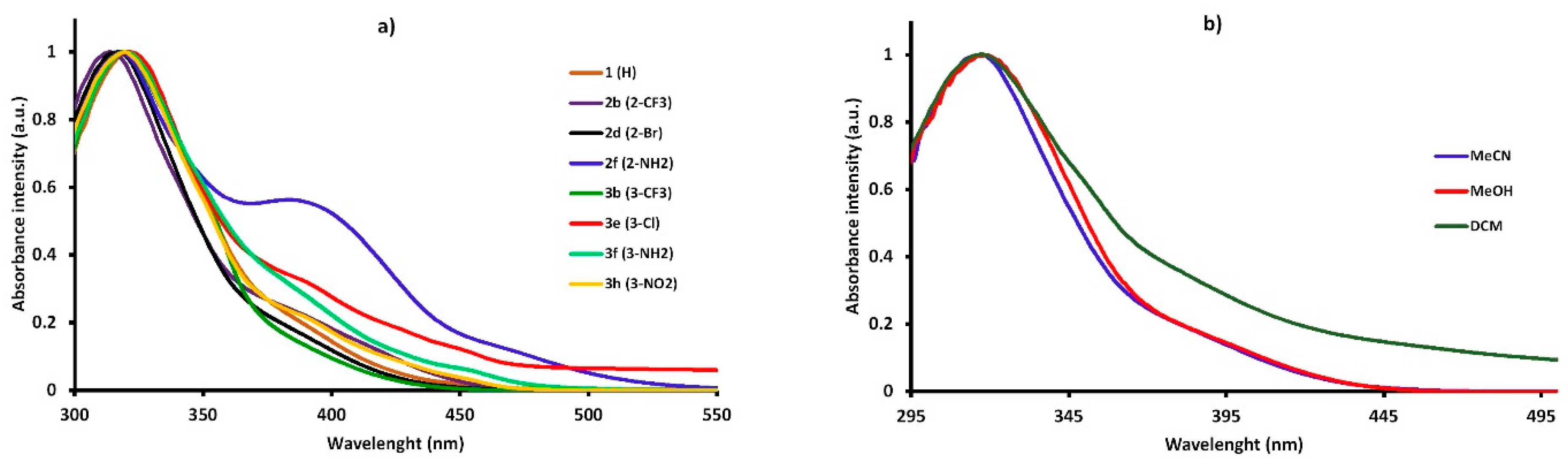
The spectral data of the synthesized compounds in different solvents are shown in Table 8. The study showed that new 5-nitroisatin-based benzoylhydrazines absorbed at around 320 nm and emitted (between 388 nm and 579 nm) over a wide spectral range with the extinction coefficients ranging from 1.92 × 104 M−1 cm−1 to 2.80 × 104 M−1 cm−1. The extinction coefficients did not achieve relatively high values. Additionally, no clear correlations between the solvent polarities and the molar extinction coefficients were found. The shape of the absorption spectra remained very similar to that of the parent compound (1) (R=H), and it was not significantly dependent on the electron-withdrawing or electron-releasing group in the different positions of the phenyl ring (Figure 8a). The position of the band of absorption did not depend on the polarity of the solvent (Figure 8b). The emission spectra were broad, with the single maximum of fluorescence that slightly shifted towards higher-wavelength values as the polarity of solvent increased.
Table 8.
Spectroscopic properties of the synthesized 5-nitroisatin-based benzoylhydrazines in solvents of different polarity.
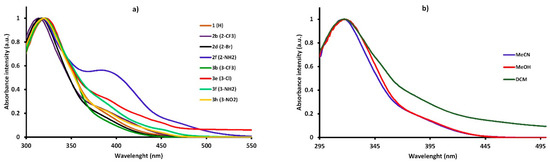
Figure 8.
The normalized UV–Vis absorption of the synthesized 5-nitroisatin-based benzoylhydrazines (a) and the normalized UV–Vis absorption of 2-bromo-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (2d) in solvents of different polarity (b).
3. Materials and Methods
3.1. Computational Methods
3.1.1. The Docking Procedure
The structure of cyclin dependent kinase 2 (CDK2) was obtained from Brookhaven Protein Database PDB (CDK2—PDB ID 1E9H) [25]. The AutodockTools package [33] was used in all preliminary steps, including the identification and localization of the active site and the preparation of all structures of considered ligands and protein. In the case of each structure used during the molecular docking stage, all non-polar hydrogen atoms were removed. The localization of the center (x = 2.5, y = 35.0, z = 63.0) and the dimensions of the grid box have been adjusted to the size of the active site (16 × 14 × 22 Å). The docking procedure for all considered ligands was realized utilizing Autodock Vina package [34]. The value of exhaustiveness parameter was set at 20 for all realized simulations. In the case of each considered molecule, the docking procedure was repeated five times with a different value of the random seed.
3.1.2. The Molecular Dynamics Simulations
The structure of the CDK2 protein was described using ff14sb [35] force-field parameters, while in the case of the inhibitor molecules, gaff force field parameters were used. In the case of isatin derivatives, the values of charges were estimated based on the Merz–Kollmann scheme using the RESP procedure at the HF/6-31G* level [36]. The complexes of the CDK2 protein with considered inhibitors were neutralized and immersed in the periodic cube box of TIP3P water molecules. Each system was heated to 300 K in the preliminary stage; the control of the temperature was realized utilizing Langevin thermostat [37]. The 80-nanosecond molecular dynamics simulations were realized with an applied shake algorithm and periodic boundary conditions. The structural analysis of considered systems, including the stability of interactions involved in the maintenance of complexes, were realized utilizing a VMD package [38]. The definition of hydrogen bonds was based on the following boundary criteria: distance D (donor)–A (acceptor) < 3.5 Å, distance H–A < 3 Å and angle D–H–A > 90°. The affinity of isatin derivatives towards the CDK2 active site was evaluated using the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) [39]. The molecular dynamics simulations were realized using the AMBER 14 package [40].
3.1.3. The Quantum Mechanics Calculations
The geometries of all the isatin derivatives were calculated using density functional theory (DFT) implemented in the Gaussian 09 package [41]. All calculations were realized using B3LYP functional at 6-311 + G (d,p) level of theory. The solvatation effects during calculations were included by applying a self-consistent reaction field (SCRF) approach [42] based on accurate numerical solutions of the Poisson–Boltzmann equation [43,44].
3.2. Materials
All reagents and solvents were purchased from Sigma-Aldrich (Poznań, Poland) and used without further purification. The highest (≥99%) purity of all used chemicals was required for spectroscopic studies.
3.3. Synthesis
The general procedure for the synthesis of the N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide.
Equimolar amounts of 5-nitroisatin (s1) (0.002 mol) and substituted benzolhydrazine (s2) (0.002 mol) were added to 96% ethanol (50 mL) containing 3 drops of glacial acetic acid. The mixture was heated under reflux for 5 h and then cooled to room temperature. The resulting solid was collected by filtration, washed with cold ethanol and recrystallized from ethanol to give the following compounds (1, 2b, 2d, 2f, 3b, 3e, 3f, and 3h).
The elemental analysis is as follows:
N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (1) [30] Yellow solid, yield 85%, d.t. 338.4 °C, IR (ATR), cm−1: 3167, 1744, 1625, 1525, 1340. Trans-isomer (1): 1H NMR (DMSO-d6 from TMS) δ (ppm): 12.22 (s, 1H), 11.56 (s, 1H), 8.89 (s, 1H), 8.34 (dd,1H), 7.96 (m, 2H), 7.68 (m, 6H), 7.10 (d, J = 8.76, 1H). 13C NMR δ (ppm): 167.55, 165.44, 148.02, 142.31, 139.06, 132.96, 129.73, 129.18, 129.04, 128.06, 122.76, 115.78, 111.21. Cis-isomer (1′): 1H NMR (DMSO-d6 from TMS) δ (ppm): 13.74 (s, 1H), 12.03 (s, 1H), 8.31 (m, 2H), 7.92 (m, 2H), 7.68 (m, 6H), 7.18 (d, J = 8.24 Hz, 1H). 13C NMR δ (ppm): 163.87, 149.86, 143.41, 133.63, 132.18, 121.08, 116.29, 112.03. C15H10N4O4, Calcd. C, 58.07, H, 3.25, N, 18.06. Found C, 58.19, H, 3.30, N, 21.48.
2-trifluoromethyl-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (2b). Yellow solid, yield 87%, m.p. 267.4 °C, d.t. 310.3 °C, IR (ATR), cm−1: 3232, 1740, 1690, 1513, 1315. 1H NMR (DMSO-d6 from TMS) δ (ppm): 13.18 (s, 1H, 2b’), 12.88 (s, 1H, 2b), 11.96 (s, 1H, 2b’), 11.51 (s, 1H, 2b), 9.17 (bs, 1H), 8.85 (bs, 1H), 8.32 (m, 2H, 2b and 2b’), 7,87 (m, 8H, 2b and 2b’), 7.14 (d, J = 8.56 Hz, 1H, 2b’), 7.07 (d, J = 8.86 Hz, 1H, 2b). 13C NMR δ (ppm): 165.14, 163.46, 150.03, 148.30, 143.31, 142.42, 134.20, 132.92, 131.32, 129.38, 128.26, 126.91, 125.54, 122.82, 122.46, 120.75, 115.38, 112.05, 111.21. C16H9F3N4O4, Calcd. C, 50.80, H, 2.40, N, 14.81. Found C, 50.74, H, 2.34, N, 14,93.
2-bromo-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (2d) Yellow solid, yield 85%, m.p. 268.4 °C, d.t. 305.9 °C, IR (ATR), cm−1: 3303, 1749, 1698, 1495, 1339. 1H NMR (DMSO-d6 from TMS) δ (ppm): 13.16 (s, 1H, 2d’), 12.43 (s, 1H, 2d), 11.97 (s, 1H, 2d’), 11.52 (s, 1H, 2d), 9.06 (s, 1H, 2d), 8.34 (dd, 2H, 2d and 2d’), 7,79 (m, 2H, 2d and 2d’), 7.55 (m, 6H, 2d and 2d’), 7.15 (d, J = 8.72 Hz, 1H, 2d’), 7.07 (d, J = 8.72 Hz, 1H, 2d). 13C NMR δ (ppm): 165.20, 149.96, 148.24, 143.32, 142.43, 137.26, 135.64, 133.20, 132.24, 130.05, 129.31, 128.23, 122.49, 120.85, 119.68, 115.43, 112.06, 111.20. C15H9BrN4O4, Calcd. C, 46.29, H, 2.33, N, 14.40. Found C, 46.00, H, 2.43, N, 14.59.
2-Amino-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (2f) Dark orange solid, yield 91%, d.t. 320.1 °C, IR (ATR), cm−1: 3490, 3378, 3197, 1749, 1623, 1517, 1334. Trans-isomer (2f): 1H NMR (DMSO-d6 from TMS) δ (ppm): 11.52 (s, 1H), 8.83 (d, J = 2.16 Hz, 1H), 8.32 (m, 1H), 7.67 (m, 1H), 7.31 (m, 2H), 7.09 (d, J = 8.67 Hz, 1H), 6.87 (m, 2H), 6.67 (m, 2H). 13C NMR δ (ppm): 167.92, 165.56, 150.91, 149.56, 142.25, 138.28, 133.83, 130.01, 128.88, 122.36, 117.84, 115.92, 113.01, 111.11. Cis-isomer (2f’): 1H NMR (DMSO-d6 from TMS) δ (ppm): 13.64 (s, 1H), 11.97 (s, 1H), 8.30 (s, 1H), 8.27 (m, 1H), 7.45 (dd, 1H), 7.31 (m, 2H), 7.15 (d, J = 8.67 Hz, 1H), 6.87 (m, 2H), 6.67 (m, 2H). 13C NMR δ (ppm):165.21, 163.86, 151.90, 147.66, 143.33, 135.69, 134.37, 127.65, 127.60, 117.42, 115.66, 111.90, 111.60. C15H11N5O4, Calcd. C, 55.39, H, 3.41, N, 21.53. Found C, 55.20, H, 3.51, N, 21.62.
3-trifluoromethyl-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (3b) Yellow solid, yield 86%, m.p. 324.5 °C, d.t. 328.7 °C, IR (ATR), cm−1: 3197, 1713, 1685, 1526, 1341. Cis-isomer (3b’): 1H NMR (DMSO-d6 from TMS) δ (ppm): 13.68 (s, 1H), 12.01 (s, 1H), 8.30 (dd, 1H), 8.23 (s, 1H), 8.18 (s, 1H), 8.17 (d, J = 8.12 Hz, 1H), 8.09 (d, J = 7.80 Hz, 1H), 7.89 (t, 1H), 7.15 (d, J = 8.68 Hz, 1H). 13C NMR δ (ppm): 163.67, 148.12, 143.40, 137.28, 133.25, 132.10, 131.00, 130.37, 129.72, 128.22, 125.52, 124.99, 122.811, 120.84, 116.32, 112.09. Only the cis-isomer was observed. C16H9F3N4O4, Calcd. C, 50.80, H, 2.40, N, 14.81. Found C, 50.92, H, 2.36, N, 14,73.
3-chloro-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (3e). Yellow solid, yield 85%, d.t. 358.5 °C, IR (ATR), cm−1: 3189, 1679, 1676, 1527, 1340. Trans-isomer (3e): 1H NMR (DMSO-d6 from TMS) δ (ppm): 12.29 (s, 1H), 11.57 (s, 1H), 8.92 (s, 1H), 8.35 (dd, 1H), 8.00 (s, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.76 (m, 1H), 7.64 (t, 1H), 7.11 (d, J = 8.75, 1H). 13C NMR δ (ppm): 166.49, 165.34, 150.00, 142.33, 135.38, 133.71, 132.62, 130.99, 129.31, 129.00, 127.95, 122.90, 115.68, 111.25. Cis-isomer (3e’): 1H NMR (DMSO-d6 from TMS) δ (ppm): 13.67 (s, 1H), 12.04 (s, 1H), 8.31 (m, 1H), 8.30 (s, 1H), 7.93 (m, 1H), 7.86 (d, J = 7.84 Hz, 1H), 7.80 (m, 1H), 7.69 (t, 1H), 7.18 (d, J = 8.6 Hz, 1H). 13C NMR δ (ppm): 148.14, 134.39, 133.33, 131.70, 129.31, 128.24, 116.36, 112.10. C15H9ClN4O4, Calcd. C, 52.26, H, 2.63, N, 16.25. Found C, 52.16, H, 2.72, N, 16.30.
3-Amino-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (3f) Yellow solid, yield 92%, d.t. 327.4 °C, IR (ATR), cm−1: 3481, 3387, 3204, 1752, 1622, 1514, 1339. Trans-isomer (3f): 1H NMR (DMSO-d6 from TMS) δ (ppm): 12.02 (s, 1H), 11.53 (s, 1H), 8.80 (d, J = 1.96 Hz, 1H), 8.33 (dd, 1H), 7.24 (m, 1H), 7.18 (s, 1H), 7.09 (m, 2H), 6.85 (m, 1H), 5.48 (bs, 4H). 13C NMR δ (ppm): 163.86, 150.04, 149.54, 143.37, 139.06, 134.02, 129.51, 129.06, 122.64, 118.26, 115.89, 114.07, 111.76. Cis-isomer (3f’): 1H NMR (DMSO-d6 from TMS) δ (ppm): 13.65 (s, 1H), 11.99 (s, 1H), 8.30 (m, 2H), 7.24 (m, 1H), 7.21 (m, 3H), 6.98 (d, J = 8.24 Hz, 1H), 5.48 (bs, 4H). 13C NMR δ (ppm): 167.54, 164.07, 149.77, 147.51, 142.27, 136.55, 132.83, 130.12, 127.87, 121.19, 118.78, 116.17, 114.37, 112.99, 111.96. C15H11N5O4, Calcd. C, 55.39, H, 3.41, N, 21.53. Found C, 55.43, H, 3.40, N, 21.50.
3-nitro-N′-[5-nitro-2-oxo-1,2-dihydro-3H-indol-3-ylidene]benzohydrazide (3h). Yellow solid, yield 92%, m.p. 275.2 °C, d.t. 345.5 °C, IR (ATR), cm−1: 3197, 1747, 1684, 1528, 1341. Trans-isomer (3h): 1H NMR (DMSO-d6 from TMS) δ (ppm): 12.48 (s, 1H), 11.57 (s, 1H), 8.98 (s, 1H), 8.75 (s, 1H), 8.51 (m, 1H), 8.37 (m, 2H), 7.89 (t, 1H), 7.11 (d, J = 8.72 Hz, 1H). 13C NMR δ (ppm): 165.24, 150.06, 148.05, 142.37, 135.73, 134.81, 134.02, 130.65, 129.37, 127.17, 122.85, 124.13, 115.61, 111.27. Cis-isomer (3h’): 1H NMR (DMSO-d6 from TMS) δ (ppm): 13.73 (s, 1H), 12.04 (s, 1H), 8.68 (s, 1H), 8.54 (m, 1H), 8.33 (m, 2H), 8.29 (s, 1H), 7.18 (d, J = 8.72 Hz, 1H). 13C NMR δ (ppm): 163.65, 148.45, 148.20, 143.41, 134.81, 134.20, 133.63, 131.44, 129.37, 128.30, 123.12, 120.80, 116.42, 112.10. C15H9N5O6, Calcd. C, 50.71, H, 2.55, N, 19.71. Found C, 50.86, H, 2.52, N, 19,59.
3.4. Experimental Measurements
3.4.1. NMR Measurements
The 1H NMR spectra were recorded using an Ascend III spectrometer operating at 400 MHz, Bruker. Chloroform was used as solvent and tetramethylsilane (TMS) as internal standard. Chemical shifts (δ) are reported in ppm relative to TMS and coupling constants (J) in Hz.
3.4.2. Elemental Analysis Measurements
The elemental analysis was conducted with a Vario MACRO 11.45–0000, Elemental Analyser System GmbH, operating with the VARIOEL software (version 5.14.4.22).
3.4.3. UV–VIS Measurements
The absorption and emission spectra were measured at room temperature in quartz cuvette (1 cm) using an Agilent Technology UV–Vis Cary 60 Spectrophotometer and a Hitachi F-7000 Spectrofluorometer, respectively.
3.4.4. FTIR Measurements
The infrared spectra were recorded using reflectance spectroscopy measurements realized using PerkinElmer’s FTIR Spectrum Two, spectrophotometer equipped with diamond ATR in the range of 4000–400 cm–1.
3.4.5. The Calorimetric Measurements
The thermal stability of 5-nitroisatin derivatives, considering the determination of the melting point (m.p.) and thermal decomposition temperature (d.t.), was realized using the DSC 6000 from PerkinElmer, Waltham, MA, USA. The calorimetric measurements were conducted with a heating rate of 10 K/min and 20 mL/min nitrogen flow to provide an inert atmosphere. The initial calibration the calorimeter was realized with indium and zinc standards; during measurements, standard aluminum pans were used.
4. Conclusions
The conducted research allowed for the development of a new group of 5-nitroisatin-based benzoylhydrazines, which show a significant affinity for the active site of CDK2. Based on the conducted analyses, it was possible to select groups of chemical substituents—the substitution of which, both in the isatin core and in the aromatic benzoylhydrazine ring, contributed to the improvement of the binding capacity of the considered derivatives. The conducted research allowed for the accurate characterization of the interactions responsible for maintaining the complexes of the considered inhibitors with the CDK2 enzyme. The most promising potential inhibitors, with the highest affinity values and forming the most stable complexes with the biological target, were directed to the experimental phase. A procedure for the synthesis of 5-nitroisatin derivatives was developed, and eight new compounds were obtained with a good yield. The structures of the newly synthesized compounds were confirmed using IR and NMR spectroscopy. It needs to be highlighted that the synthesized 5-nitroisatin-based benzoylhydrazines were present in the solution as trans- and cis-isomers. The absorption and fluorescence maxima were determined. It was found that the position of the bands of absorption and emission did not depend on the polarity of the solvent.
Based on the collected data—considering the binding capacity, the structural stability of the complexes and the molecular properties of the studied group of compounds—it can be concluded that the newly developed isatin derivatives can be used in the development of new anti-cancer therapies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23148046/s1.
Author Contributions
Conceptualization, P.C.; methodology, P.C. and A.S.; software, P.C.; docking and molecular dynamics P.C.; ab initio calculations P.C. and B.S.; validation, P.C. and A.S.; formal analysis, P.C. and A.S.; investigation, P.C.; resources, P.C.; synthesis A.S. and P.C.; UV-VIS, NMR measurements A.S.; IR and DSC measurements P.C., A.S. and B.S.; data curation, P.C. and A.S.; writing—original draft preparation, P.C. and A.S.; review and editing P.C., A.S. and B.S.; visualization, P.C. and A.S.; supervision, P.C.; project administration, P.C.; funding acquisition, P.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
This research was supported by PL-Grid Infrastructure (http://www.plgrid.pl/en).
Conflicts of Interest
The authors declare that there are no conflict of interest.
References
- Zhou, J.; Qu, F. Analysis of the Extracts of Isatis tinctoria by New Analytical Approaches of HPLC, MS and NMR. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Bergman, J.; Lindström, J.-O.; Tilstam, U. The structure and properties of some indolic constituents in Couroupita guianensis aubl. Tetrahedron 1985, 41, 2879–2881. [Google Scholar] [CrossRef]
- Bayly, M.J.; Duretto, M.F.; Holmes, G.D.; Forster, P.I.; Cantrill, D.J.; Ladiges, P.Y.; Bayly, M.J.; Duretto, M.F.; Holmes, G.D.; Forster, P.I.; et al. Transfer of the New Caledonian genus Boronella to Boronia (Rutaceae) based on analyses of cpDNA and nrDNA. Aust. Syst. Bot. 2015, 28, 111. [Google Scholar] [CrossRef]
- Xiao, Z.; Hao, Y.; Liu, B.; Qian, L. Indirubin and Meisoindigo in the Treatment of Chronic Myelogenous Leukemia in China. Leuk. Lymphoma 2002, 43, 1763–1768. [Google Scholar] [CrossRef]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.M.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef]
- Cheke, R.S.; Firke, S.D.; Patil, R.R.; Bari, S.B. ISATIN: New Hope Against Convulsion. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 76–101. [Google Scholar] [CrossRef]
- Bharathi Dileepan, A.G.; Daniel Prakash, T.; Ganesh Kumar, A.; Shameela Rajam, P.; Violet Dhayabaran, V.; Rajaram, R. Isatin based macrocyclic Schiff base ligands as novel candidates for antimicrobial and antioxidant drug design: In vitro DNA binding and biological studies. J. Photochem. Photobiol. B Biol. 2018, 183, 191–200. [Google Scholar] [CrossRef]
- Guo, H. Isatin derivatives and their anti-bacterial activities. Eur. J. Med. Chem. 2019, 164, 678–688. [Google Scholar] [CrossRef]
- Zhang, M.-Z.; Chen, Q.; Yang, G.-F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef]
- Prakash, C.R.; Theivendren, P.; Raja, S.; Prakash, C.R.; Theivendren, P.; Raja, S. Indolin-2-Ones in Clinical Trials as Potential Kinase Inhibitors: A Review. Pharmacol. Pharm. 2012, 3, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Carter, L.; Johnson, K.; Henry, R.R. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes 2002, 51, 2190–2198. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-A.; Kwon, S.-M.; Kim, J.-A.; Kang, K.W.; Yoon, J.-H.; Ahn, S.-G. 5′-Nitro-indirubinoxime, an indirubin derivative, suppresses metastatic ability of human head and neck cancer cells through the inhibition of Integrin β1/FAK/Akt signaling. Cancer Lett. 2011, 306, 197–204. [Google Scholar] [CrossRef]
- Martin, L.; Magnaudeix, A.; Wilson, C.M.; Yardin, C.; Terro, F. The new indirubin derivative inhibitors of glycogen synthase kinase-3, 6-BIDECO and 6-BIMYEO, prevent tau phosphorylation and apoptosis induced by the inhibition of protein phosphatase-2A by okadaic acid in cultured neurons. J. Neurosci. Res. 2011, 89, 1802–1811. [Google Scholar] [CrossRef]
- Avila, J.; Hernández, F. GSK-3 inhibitors for Alzheimer’s disease. Expert Rev. Neurother. 2007, 7, 1527–1533. [Google Scholar] [CrossRef]
- Czeleń, P. Investigation of the Inhibition Potential of New Oxindole Derivatives and Assessment of Their Usefulness for Targeted Therapy. Symmetry 2019, 11, 974. [Google Scholar] [CrossRef] [Green Version]
- Bramson, H.N.; Holmes, W.D.; Hunter, R.N.; Lackey, K.E.; Lovejoy, B.; Luzzio, M.J.; Montana, V.; Rocque, W.J.; Rusnak, D.; Shewchuk, L.; et al. Oxindole-based inhibitors of cyclin-dependent kinase 2 (CDK2): Design, synthesis, enzymatic activities, and X-ray crystallographic analysis. J. Med. Chem. 2001, 44, 4339–4358. [Google Scholar] [CrossRef]
- Czeleń, P.; Szefler, B. The oxindole derivatives, new promising gsk-3β inhibitors as one of the potential treatments for alzheimer’s disease—A molecular dynamics approach. Biology 2021, 10, 332. [Google Scholar] [CrossRef]
- Shin, E.-K.; Kim, J.-K. Indirubin derivative E804 inhibits angiogenesis. BMC Cancer 2012, 12, 164. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-Y.; Liu, Y.-W.; Chen, M.-H.; Ww, J.-Y.; Ho, H.-Y.; Wang, Q.-F.; Chuang, J.-J. Indirubin-3′-monoxime promotes autophagic and apoptotic death in JM1 human acute lymphoblastic leukemia cells and K562 human chronic myelogenous leukemia cells. Oncol. Rep. 2013, 29, 2072–2078. [Google Scholar] [CrossRef] [Green Version]
- Lozinskaya, N.A.; Babkov, D.A.; Zaryanova, E.V.; Bezsonova, E.N.; Efremov, A.M.; Tsymlyakov, M.D.; Anikina, L.V.; Zakharyascheva, O.Y.; Borisov, A.V.; Perfilova, V.N.; et al. Synthesis and biological evaluation of 3-substituted 2-oxindole derivatives as new glycogen synthase kinase 3β inhibitors. Bioorg. Med. Chem. 2019, 27, 1804–1817. [Google Scholar] [CrossRef]
- de Paiva, R.E.F.; Vieira, E.G.; da Silva, D.R.; Wegermann, C.A.; Ferreira, A.M.C. Anticancer Compounds Based on Isatin-Derivatives: Strategies to Ameliorate Selectivity and Efficiency. Front. Mol. Biosci. 2021, 7, 511. [Google Scholar]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Morgan, D.O. CYCLIN-DEPENDENT KINASES: Engines, Clocks, and Microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Davies, T.G.; Tunnah, P.; Meijer, L.; Marko, D.; Eisenbrand, G.; Endicott, J.A.; Noble, M.E. Inhibitor binding to active and inactive CDK2: The crystal structure of CDK2-cyclin A/indirubin-5-sulphonate. Structure 2001, 9, 389–397. [Google Scholar] [CrossRef]
- Czeleń, P.; Szefler, B. Molecular dynamics study of the inhibitory effects of ChEMBL474807 on the enzymes GSK-3β and CDK-2. J. Mol. Model. 2015, 21, 74. [Google Scholar] [CrossRef] [Green Version]
- Czeleń, P. Molecular dynamics study on inhibition mechanism of CDK-2 and GSK-3β by CHEMBL272026 molecule. Struct. Chem. 2016, 27, 1807–1818. [Google Scholar] [CrossRef] [Green Version]
- Katiyar, A.; Hegde, M.; Kumar, S.; Gopalakrishnan, V.; Bhatelia, K.D.; Ananthaswamy, K.; Ramareddy, S.A.; de Clercq, E.; Choudhary, B.; Schols, D.; et al. Synthesis and evaluation of the biological activity of N′-[2-oxo-1,2 dihydro-3H-indol-3-ylidene] benzohydrazides as potential anticancer agents. RSC Adv. 2015, 5, 45492–45501. [Google Scholar] [CrossRef]
- Debnath, K.; Pathak, S.; Pramanik, A. Facile synthesis of ninhydrin and isatin based hydrazones in water using PEG-OSO3H as a highly efficient and homogeneous polymeric acid-surfactant combined catalyst. Tetrahedron Lett. 2013, 54, 4110–4115. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Ibrahim, H.S.; Bua, S.; Abou-Seri, S.M.; Supuran, C.T. Novel hydrazido benzenesulfonamides-isatin conjugates: Synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur. J. Med. Chem. 2018, 157, 28–36. [Google Scholar] [CrossRef]
- Emami, S.; Valipour, M.; Komishani, F.K.; Sadati-Ashrafi, F.; Rasoulian, M.; Ghasemian, M.; Tajbakhsh, M.; Masihi, P.H.; Shakiba, A.; Irannejad, H.; et al. Synthesis, in silico, in vitro and in vivo evaluations of isatin aroylhydrazones as highly potent anticonvulsant agents. Bioorg. Chem. 2021, 112, 104943. [Google Scholar] [CrossRef]
- Hunoor, R.S.; Patil, B.R.; Badiger, D.S.; Chandrashekhar, V.M.; Muchchandi, I.S.; Gudasi, K.B. Co(II), Ni(II), Cu(II) and Zn(II) complexes of isatinyl-2-aminobenzoylhydrazone: Synthesis, characterization and anticancer activity. Appl. Organomet. Chem. 2015, 29, 101–108. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Potemkin, V.A.; Grishina, M.A.; Belik, A.V. A Method for Multiconformational Modeling of the Three-Dimensional Shape of a Molecule. J. Struct. Chem. 2002, 43, 1033–1039. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Adelman, S.A. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A., III; Duke, R.E.; Gohlke, H.; et al. AMBER 14 2014. Available online: https://www.researchgate.net/publication/270588529_Amber_2014 (accessed on 27 June 2022).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Carnegie Mellon University: Pittsburgh, PA, USA, 2016. [Google Scholar]
- Cramer, C.J.; Truhlar, D.G. Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics. Chem. Rev. 1999, 99, 2161–2200. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New model for calculation of solvation free energies: Correction of self-consistent reaction field continuum dielectric theory for short-range hydrogen-bonding effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).