
The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools?

Abstract
:1. Introduction
2. Polymorphism: The Awareness
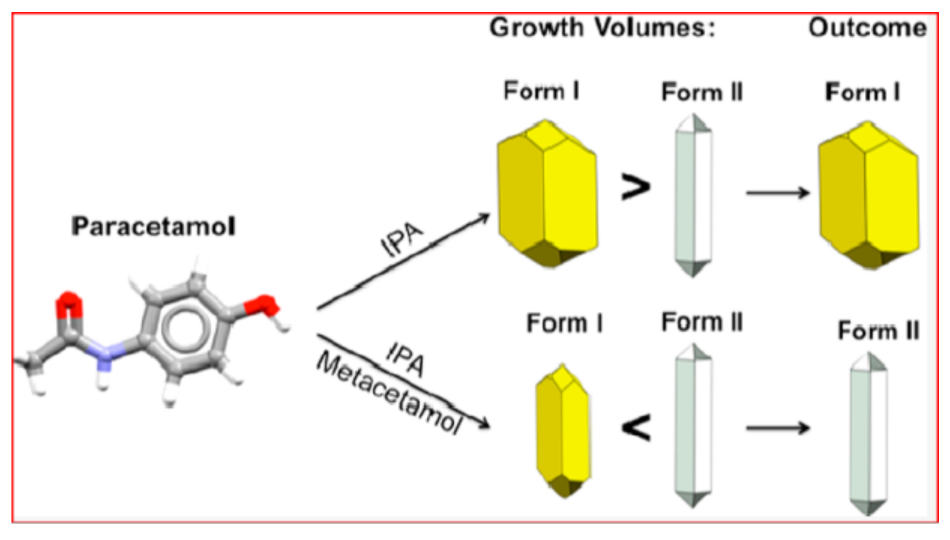
3. Polymorphism: The Implications. Different Crystals. Different Properties
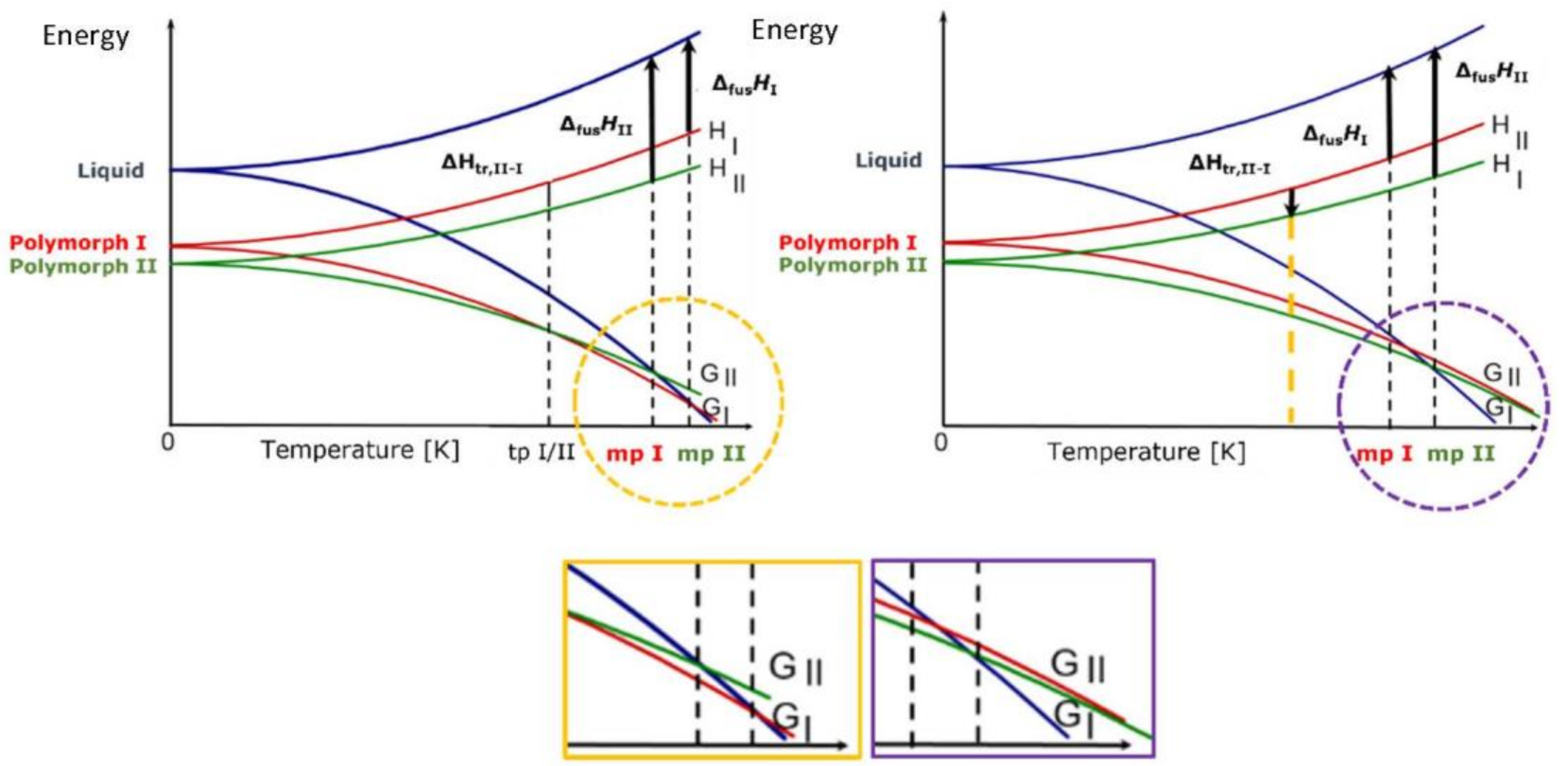
4. Polymorphism: The Rationale
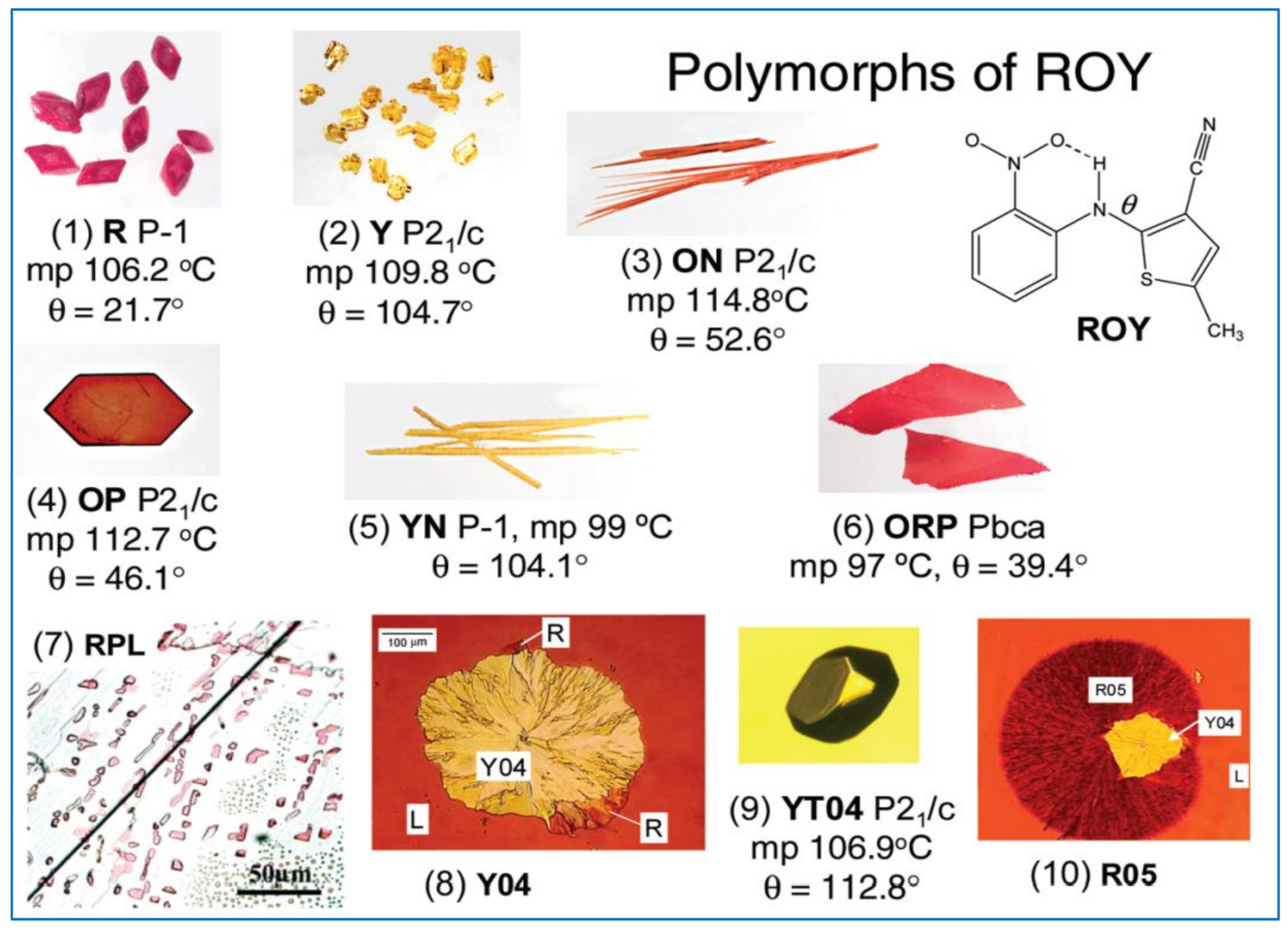
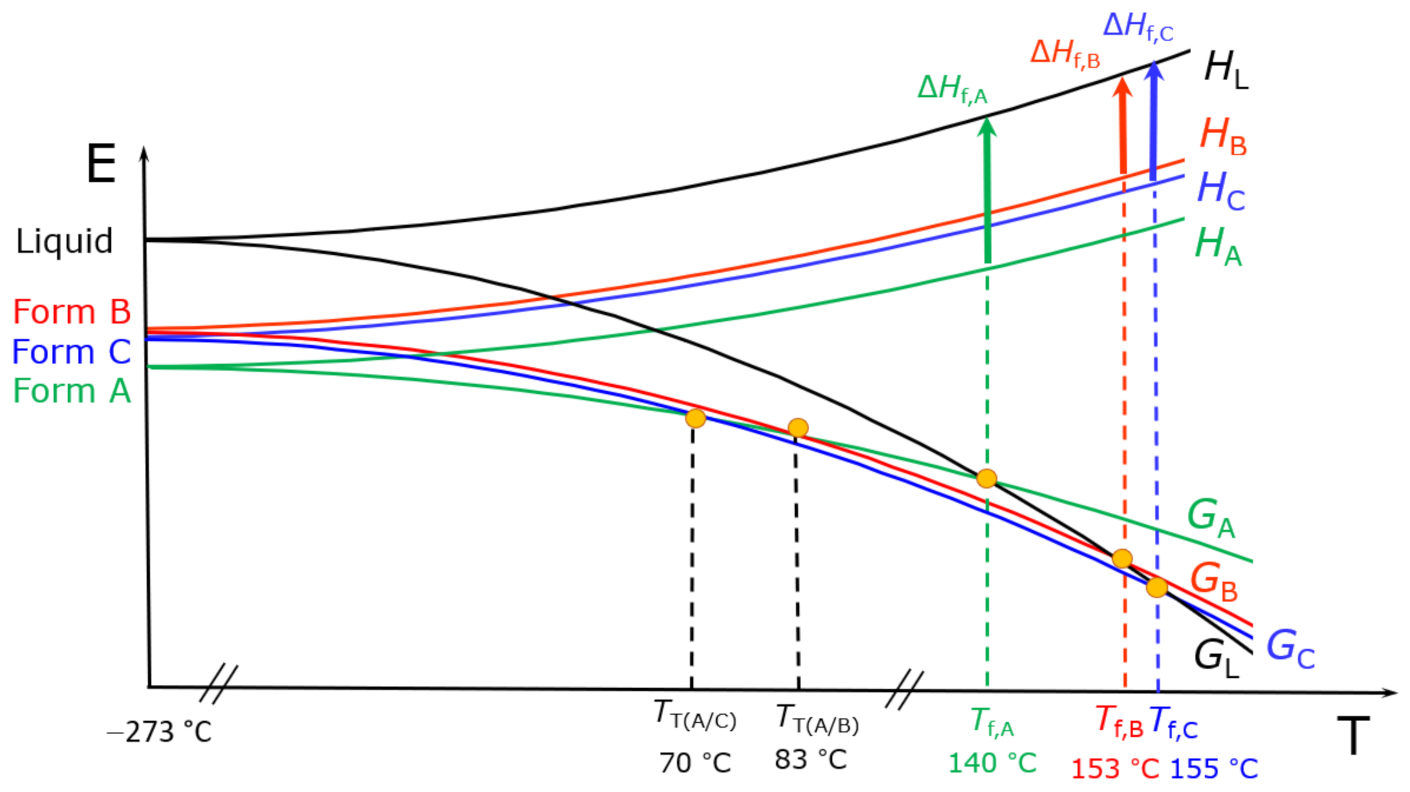
4.1. Examples of Polymorphism in Single Component (Unary) Systems
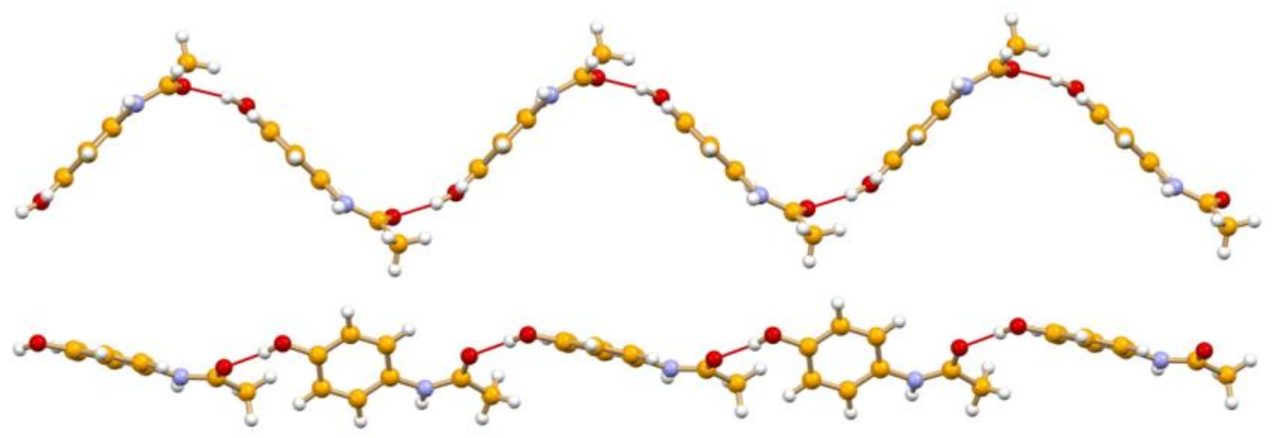
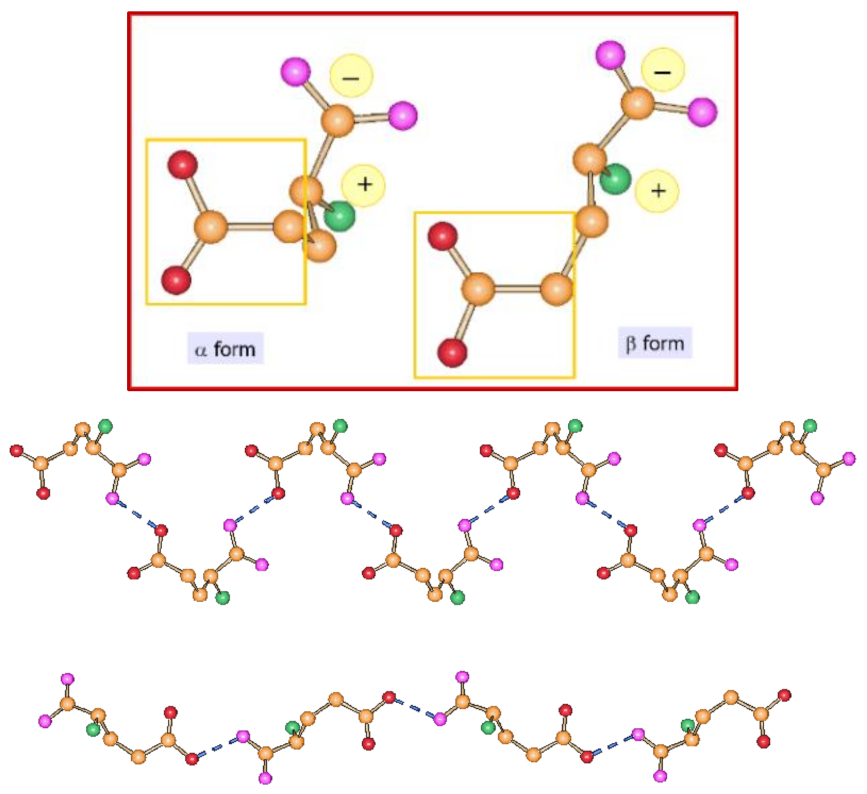
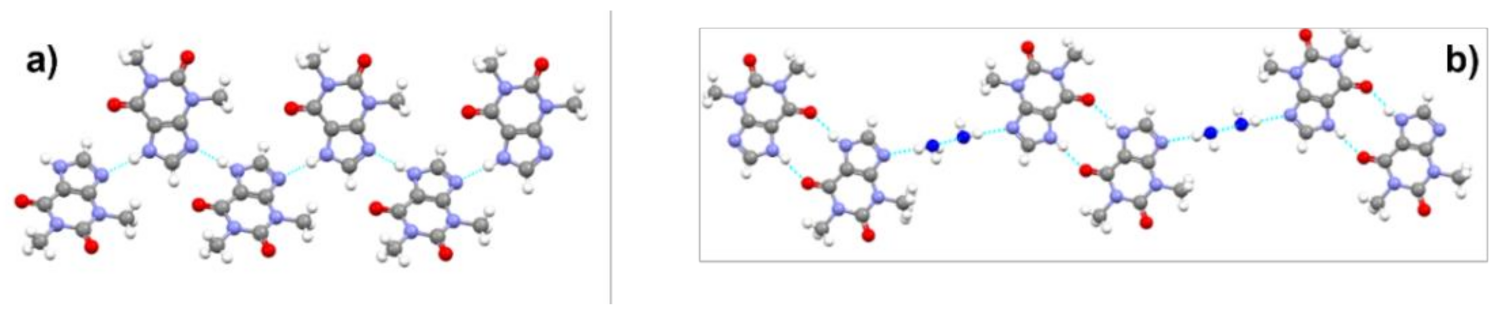
4.2. Conformational Polymorphism
4.3. Tautomeric Polymorphism
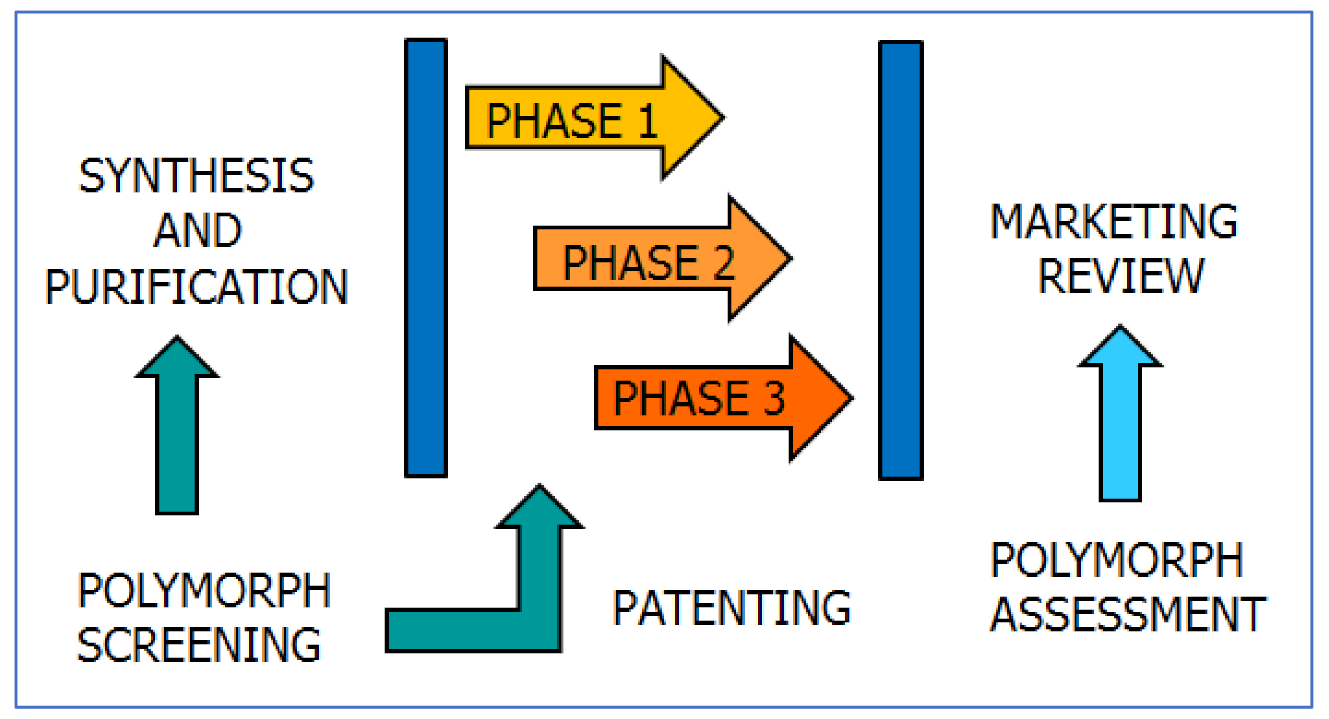
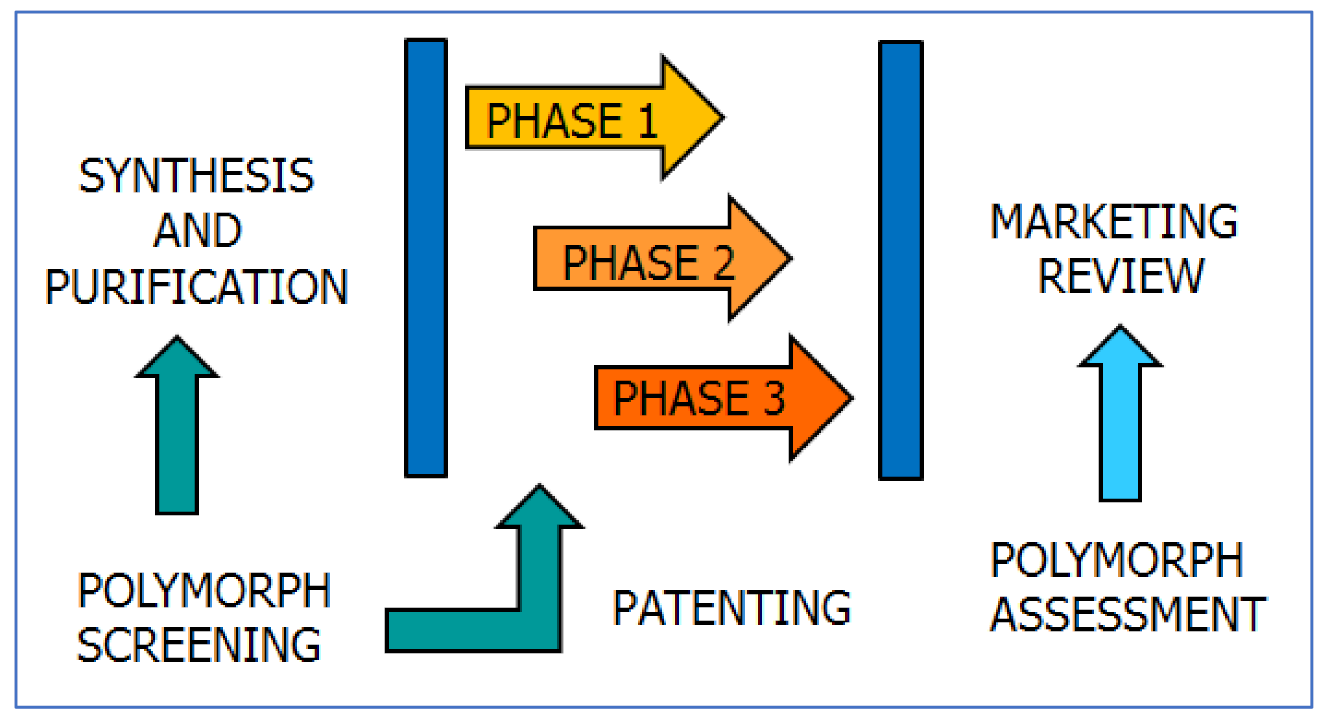
5. Polymorphism: The Impact
5.1. Polymorphism: The Reaction of the Scientific Community
5.2. Crystal Structure Prediction
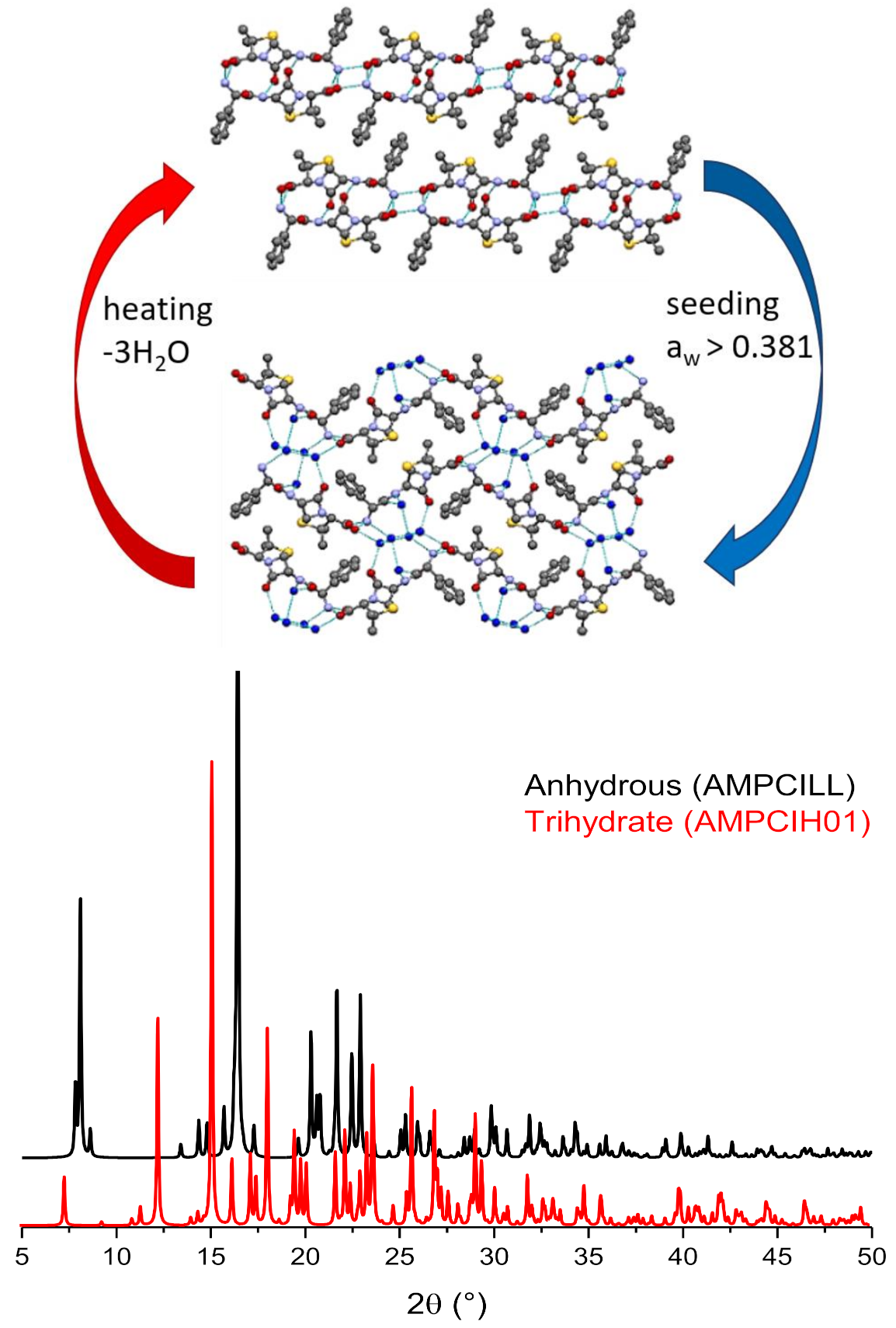
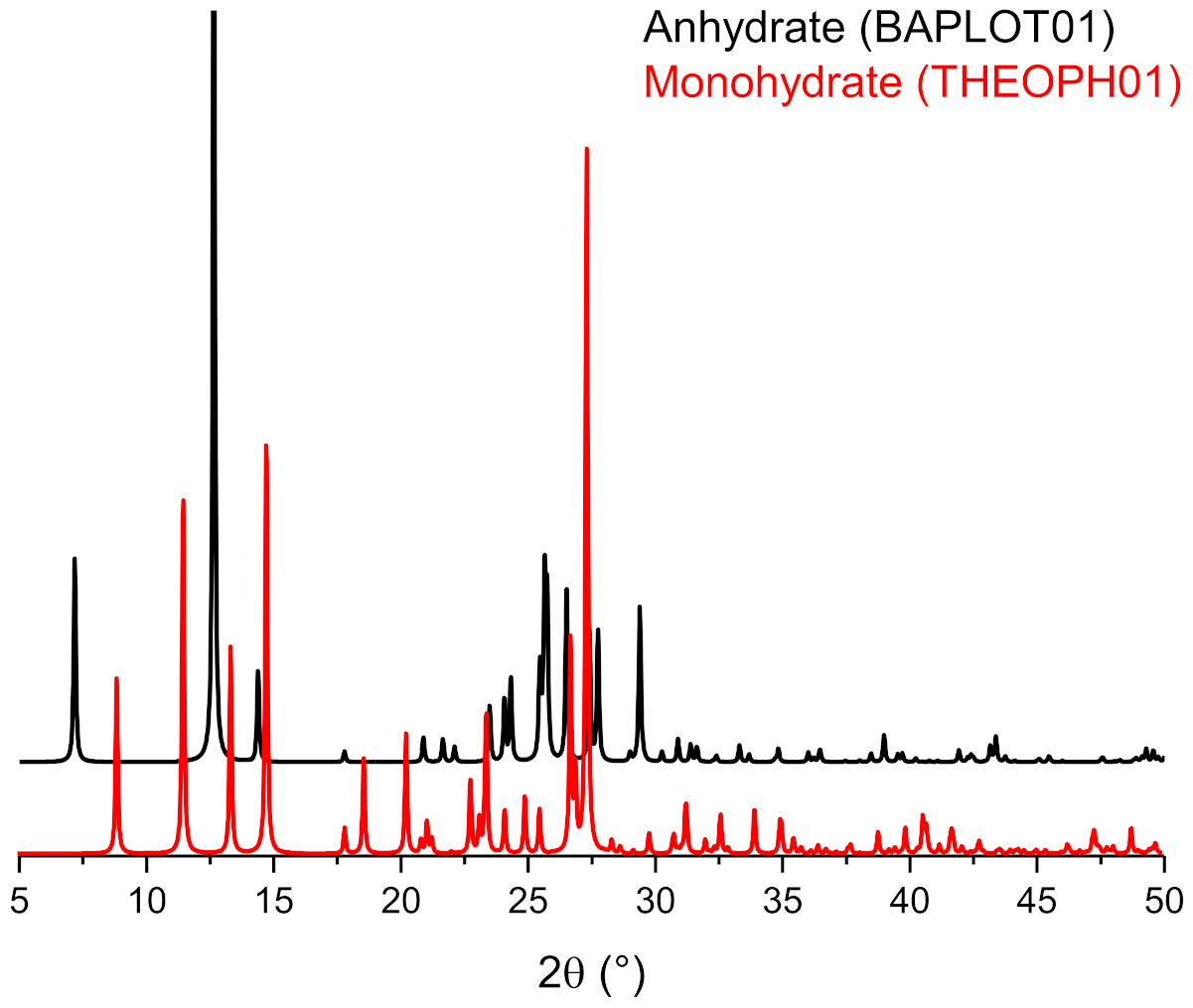
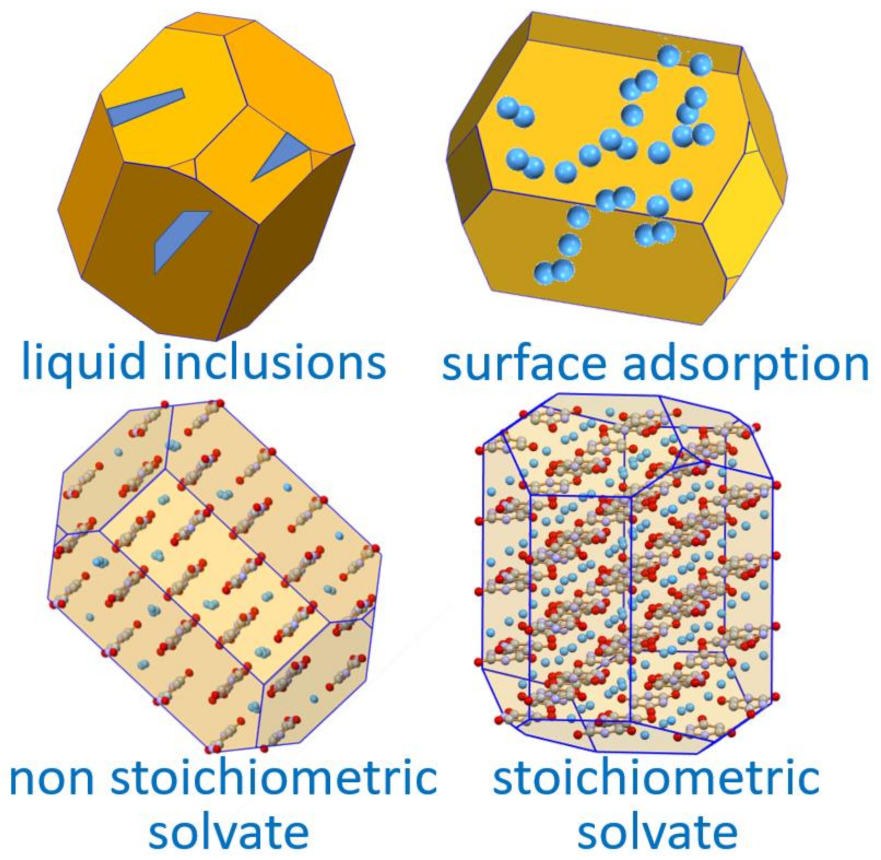
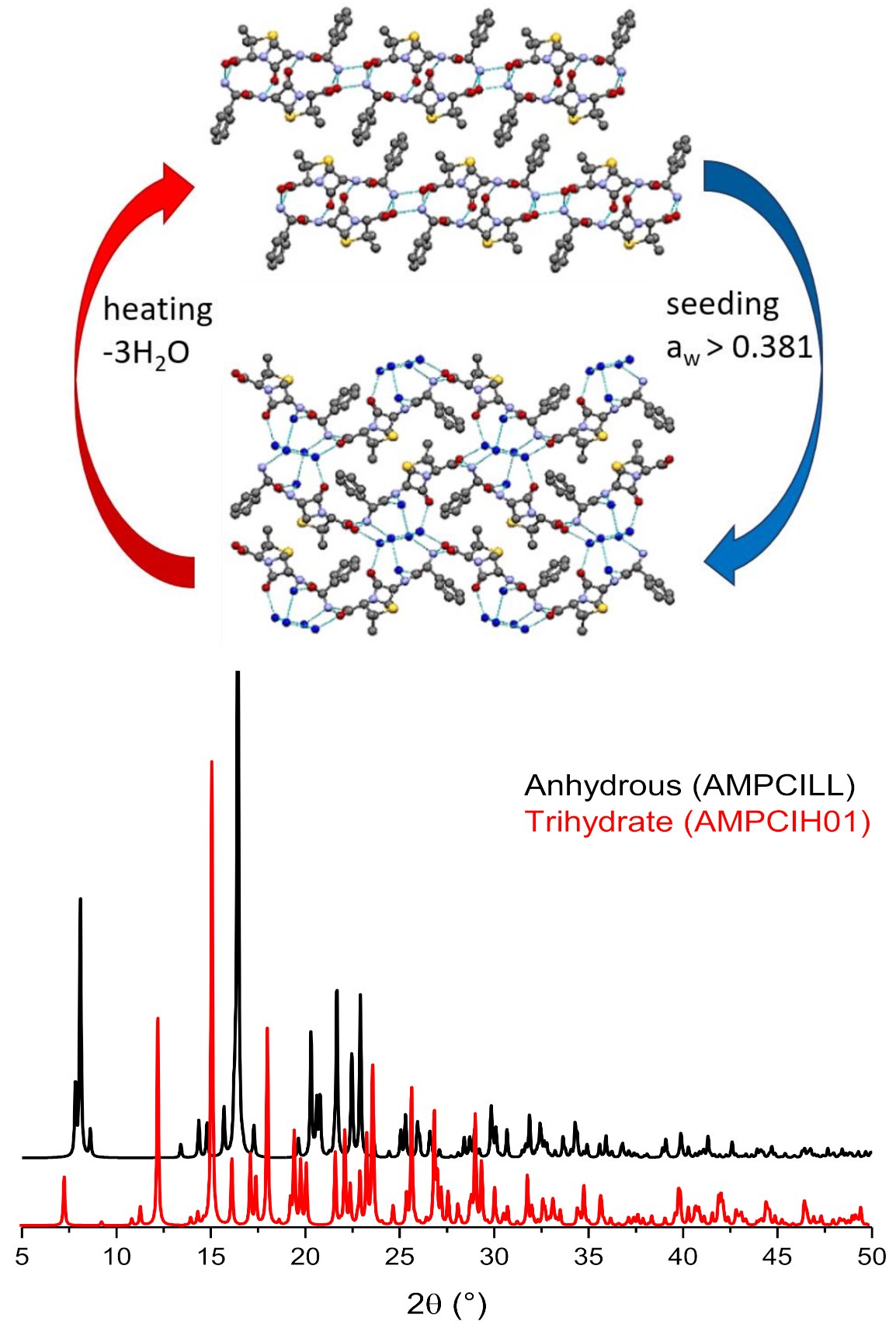
6. Solvates and, Especially, Hydrates
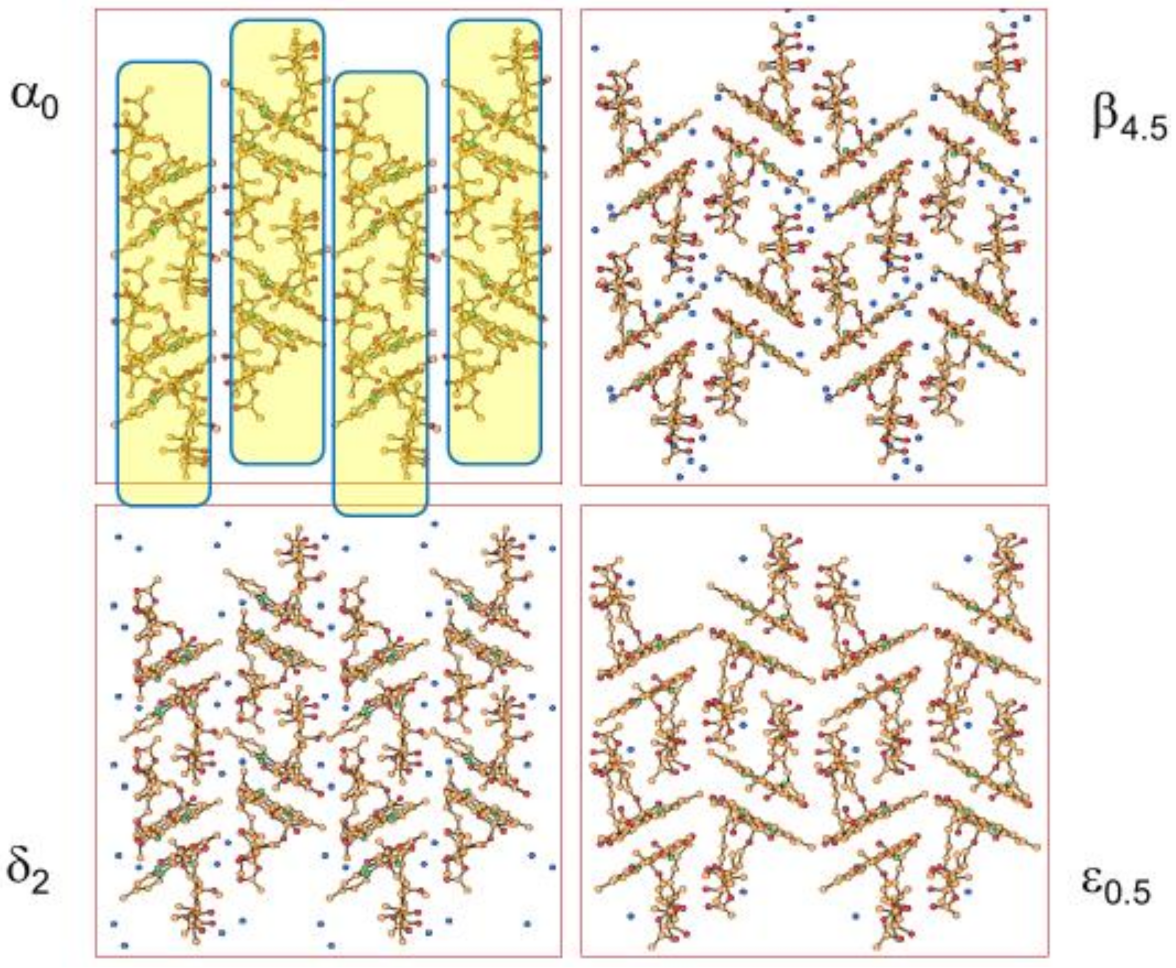
Stoichiometric versus Non-Stoichiometric Hydrates
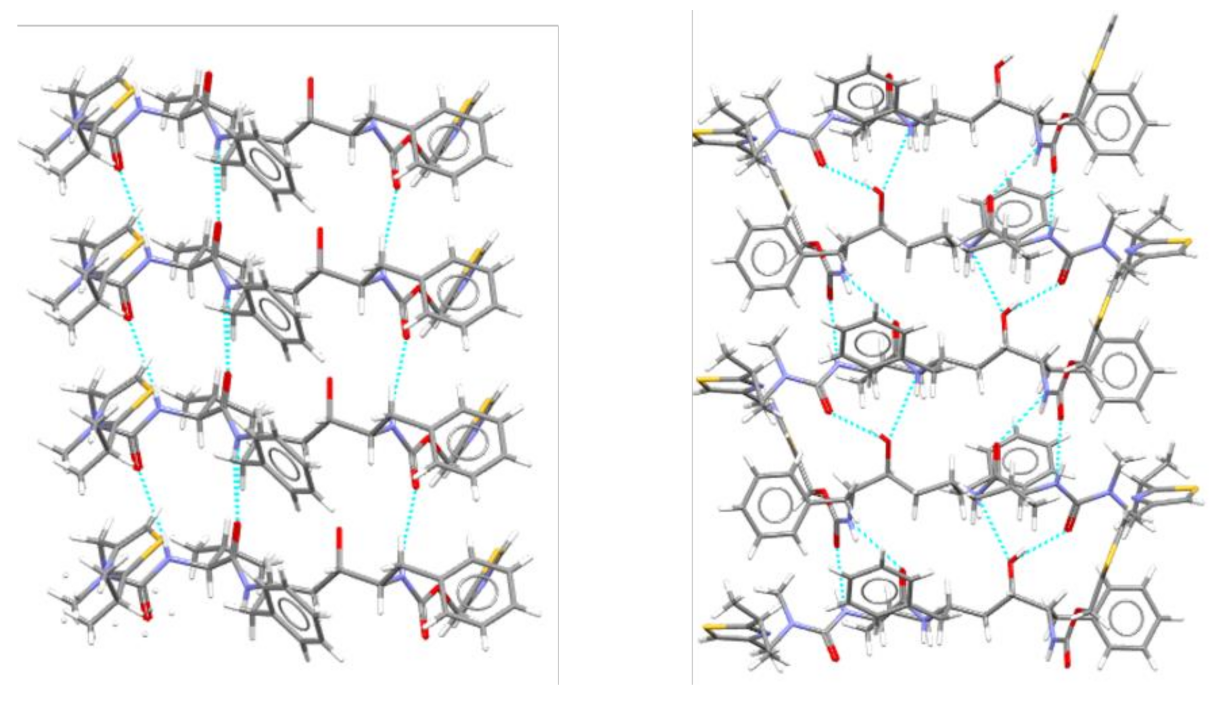
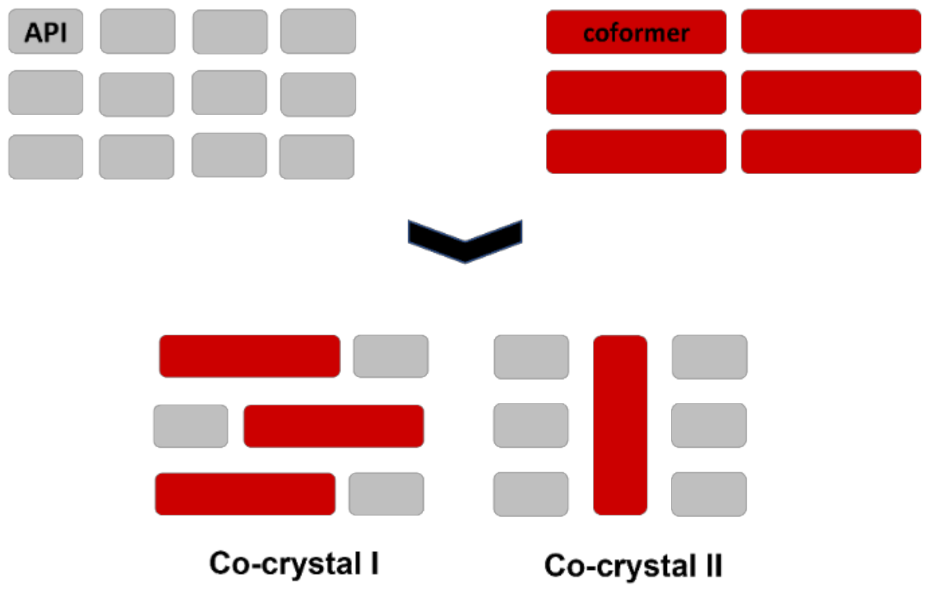
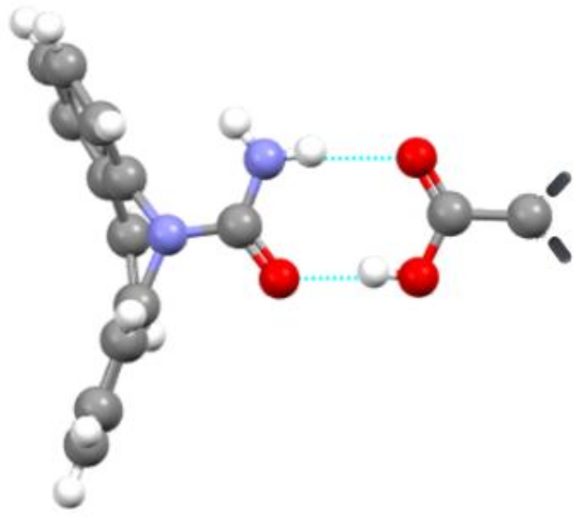
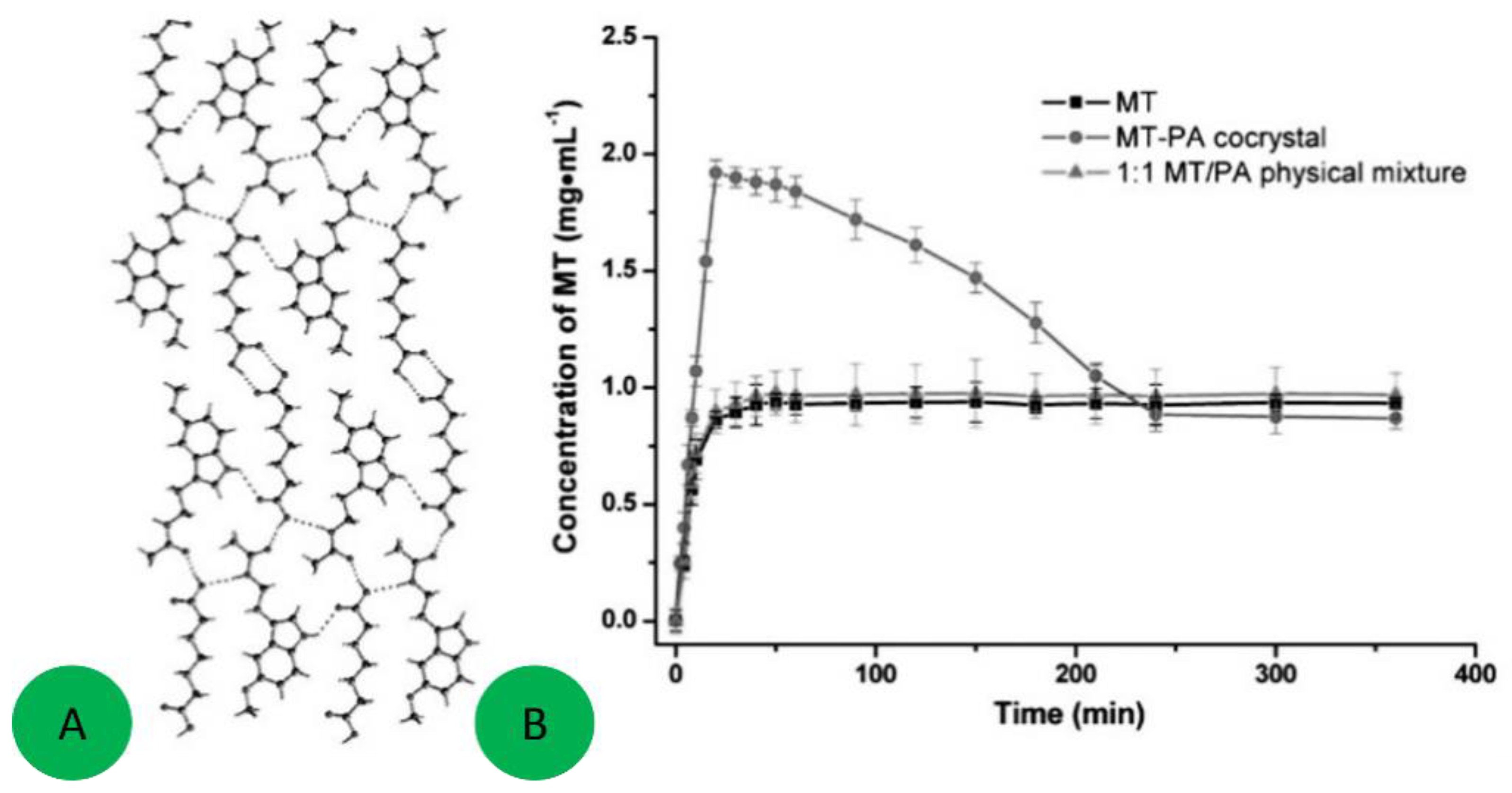
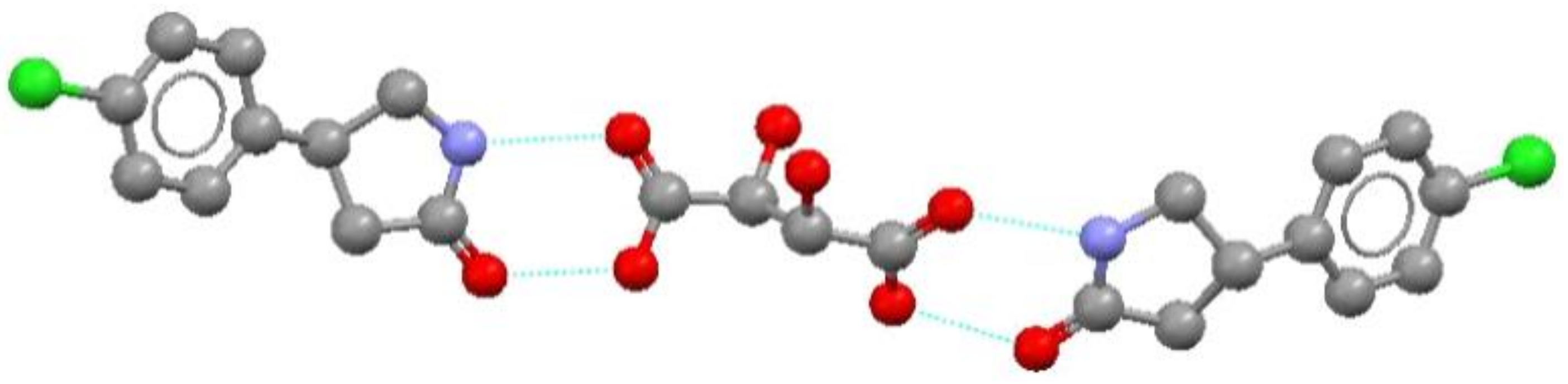
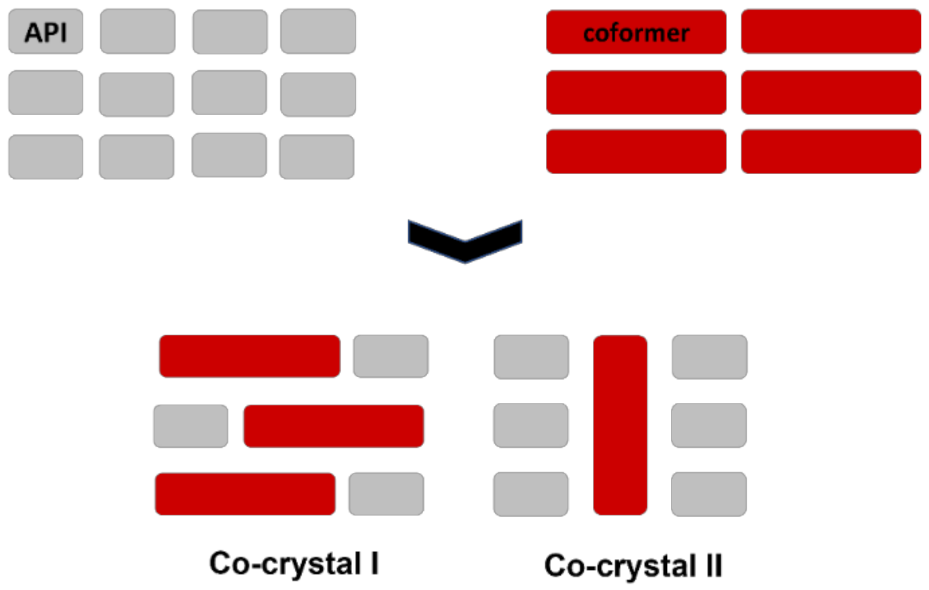
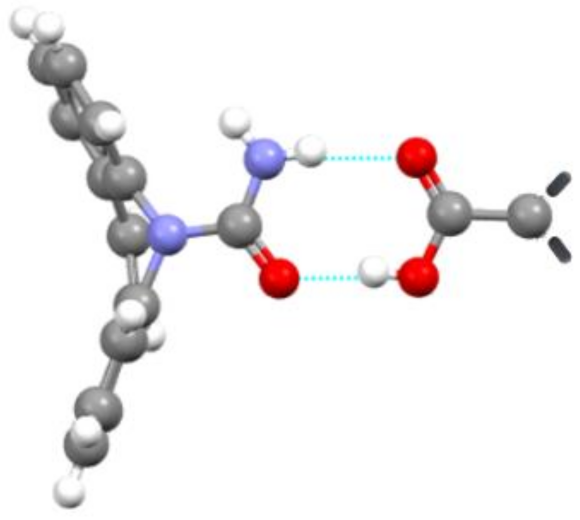
7. Molecular Co-Crystals
7.1. Co-Crystals in Pharmaceutics
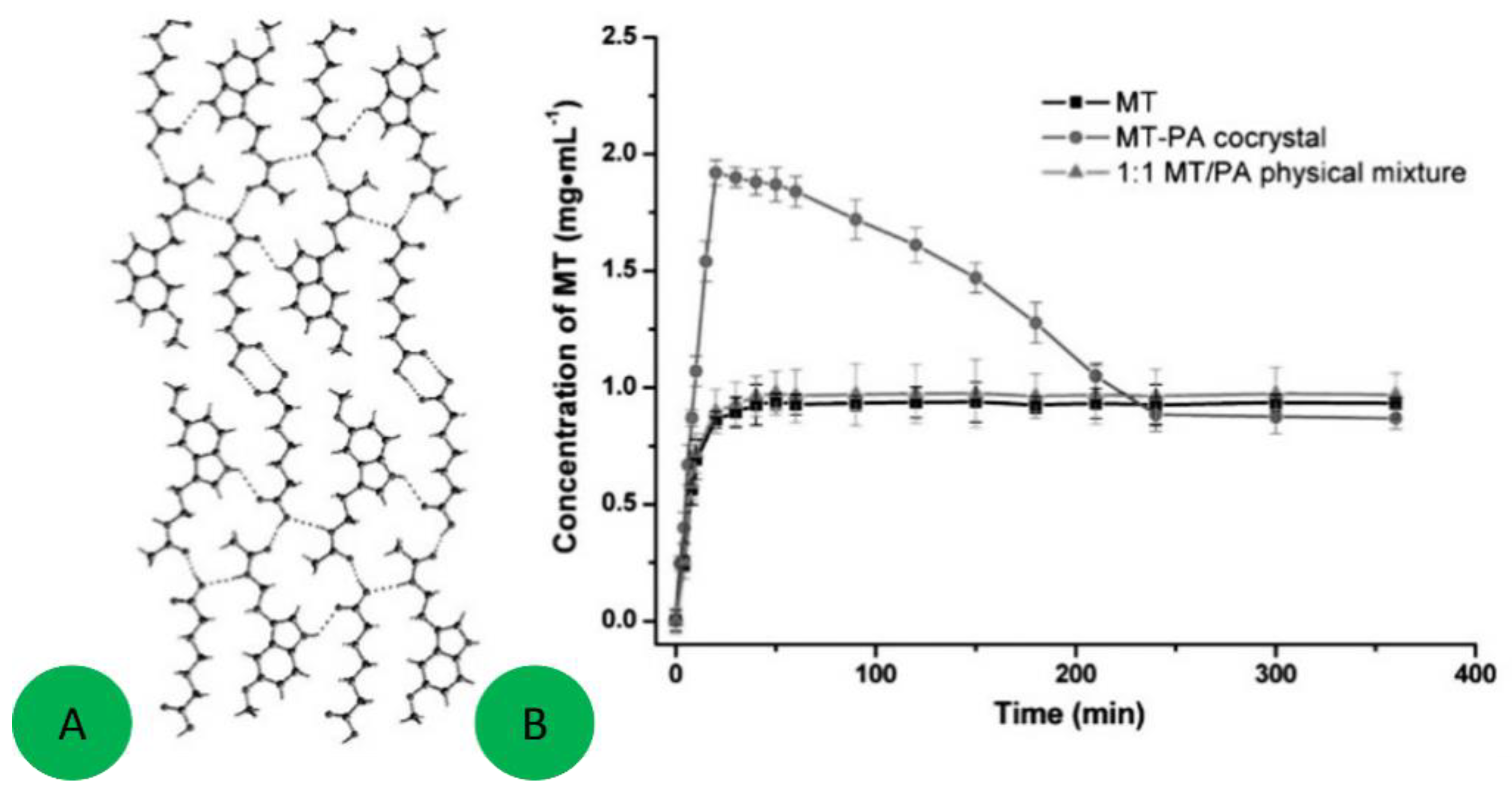
7.2. Co-Crystals Properties

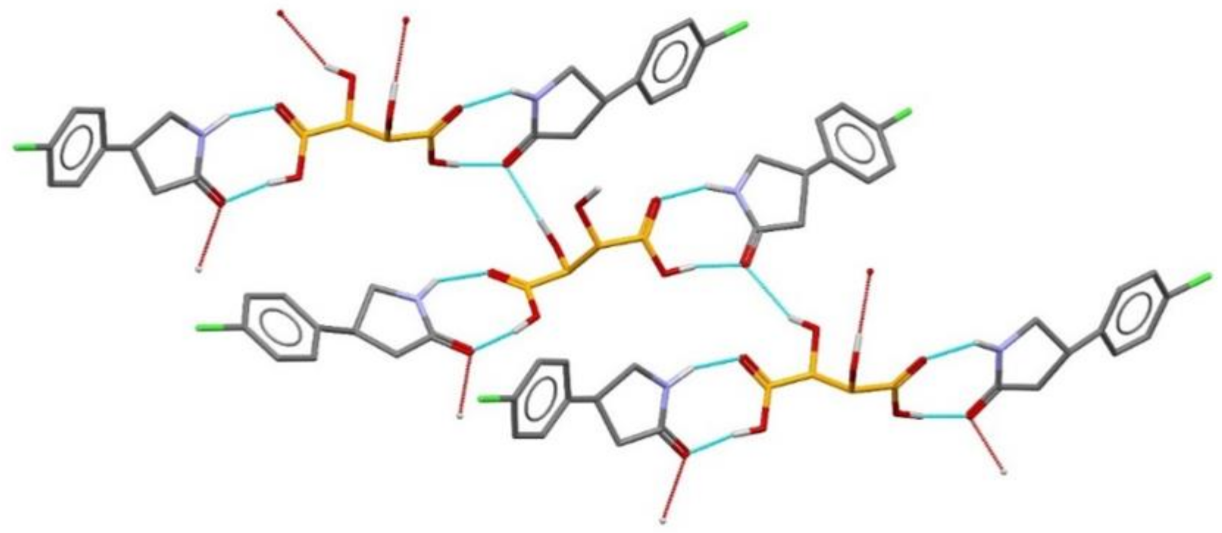
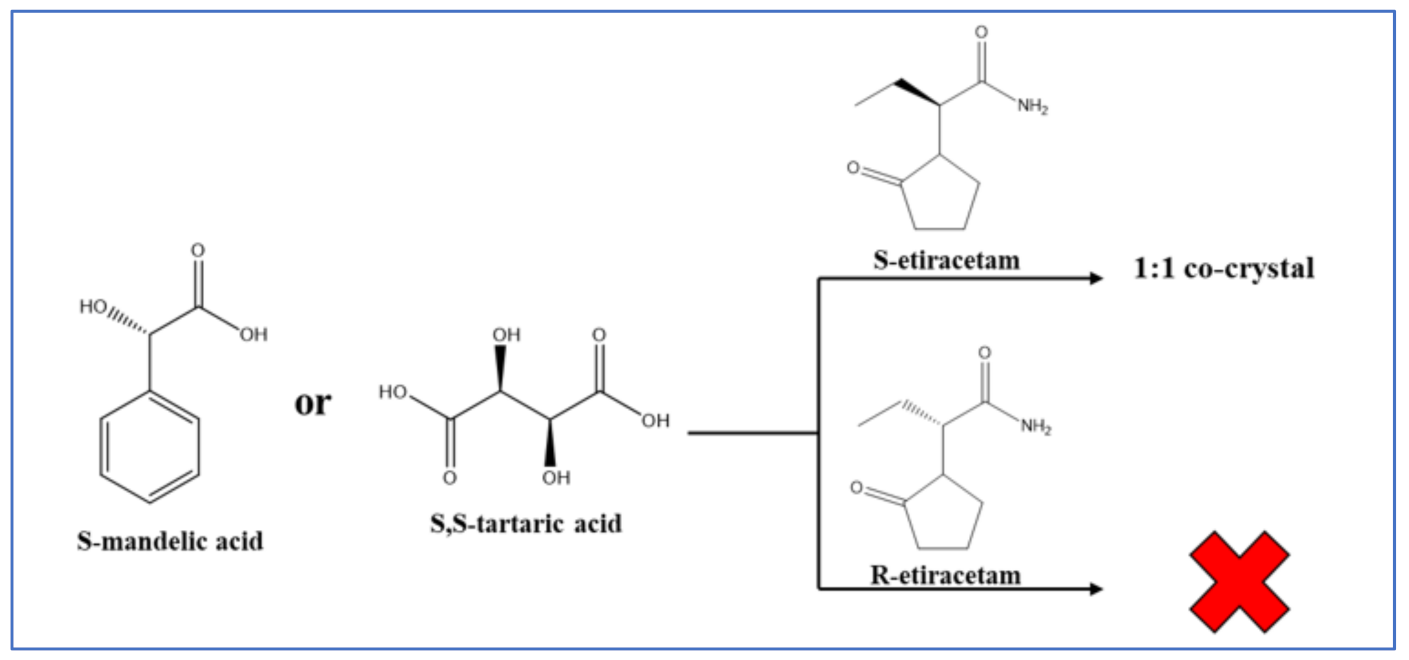
7.3. Chiral Resolution via Co-Crystallization
8. Conclusions
- (i)
- An API can take many solid forms (polymorphs, solvates, salts, co-crystals) all containing exactly the same active principle but in different structural arrangements and/or associated to different molecules (solvents, coformers, counterions, salts, etc.);
- (ii)
- Different solid forms will possess different properties, which might affect a drug’s processing, formulation, distribution, storage, administration to the patient and, ultimately, its therapeutic efficacy;
- (iii)
- Since there is no way to guarantee that a transformation to a more stable form will occur, or that a more stable form will appear, a preliminary, thorough exploration of the crystal domain is the only way to minimize the chances of the undesired appearance of a new and more stable form at a later stage of development;
- (iv)
- Besides maximising control on the crystal form of the API, the investigation of hydrates and co-crystals may afford alternative, often improved or innovative properties of the drug;
- (v)
- Polymorphs, hydrates, and co-crystals may meet the requisites of novelty, utility, and non-obviousness useful for extending the patenting life of a drug.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mitscherlich, E. Sur la relation qui existe entre la forme cristalline e le proportions chimiques, I. Memoires sur les arseniates et les phosphates. Ann. Chim. Phys. 1822, 19, 350–419. [Google Scholar]
- McCrone, W.C. Physics and Chemistry of the Organic Solid State. In Physics Today, 2nd ed.; Fox, D., Labes, M.M., Weissenberg, A., Eds.; Interscience: New York, NY, USA, 1965; Volume 2, 725p. [Google Scholar]
- Bernstein, J. Polymorphism in Molecular Crystals, 2nd ed.; Oxford University Press: Oxford, UK, 2020. [Google Scholar]
- Brittain, H.G. (Ed.) Polymorphism in Pharmaceutical Solids, 2nd ed.; Informa Healthcare: London, UK, USA, 1999. [Google Scholar]
- Hilfiker, R.; Von Raumer, M. (Eds.) Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development, 2nd ed.; Hilfiker, R.; Von Raumer, M. (Eds.) Wiley-VCH: Weinheim, Germany, 2019. [Google Scholar]
- Gruss, M. (Ed.) Solid State Development and Processing of Pharmaceutical Molecules: Salts, Cocrystals, and Polymorphism; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Wouters, J.; Quéré, L. (Eds.) Pharmaceutical Salts and Co-Crystals, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 2011. [Google Scholar]
- Aakeröy, C.B.; Sinha, A.S. (Eds.) Co-Crystals: Preparation, Characterization and Applications, 1st ed.; Royal Society of Chemistry: Piccadilly, UK, 2018. [Google Scholar]
- Braga, D.; Grepioni, F.; Gavezzotti, A.; Bernstein, J. Re: “Crystal Engineering in the Regulatory and Patent Literature of Pharmaceutical Solid Forms”. Cryst. Growth Des. 2017, 17, 933–939. [Google Scholar] [CrossRef]
- Seddon, K.R.; Zaworotko, M.J. Crystal Engineering: The Design and Application of Functional Solids; Kluwer: Dordrecht, The Netherlands, 1999. [Google Scholar]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Cryst. B 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Görbitz, C.H.; Hersleth, H.P. On the Inclusion of Solvent Molecules in the Crystal Structures of Organic Compounds. Acta Cryst. B 2000, 56, 526–534. [Google Scholar] [CrossRef]
- Werner, J.E.; Swift, J.A. Data mining the Cambridge Structural Database for hydrate-anhydrate pairs with SMILES strings. CrystEngComm 2020, 22, 7290–7297. [Google Scholar] [CrossRef]
- ICH Official Website. Available online: https://www.ich.org/ (accessed on 14 June 2022).
- U.S. Food & Drug Administration. Guidance Document, Pharmaceutical Solid Polymorphism: Chemistry, Manufacturing, and Controls Information. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/andaspharmaceutical-solid-polymorphism-chemistry-manufacturing-and-controls-information (accessed on 14 June 2022).
- European Patent Office. Available online: http://www.european-patent-office.org/ (accessed on 14 June 2022).
- Dunitz, J.D. Phase Transitions in Molecular Crystals from a Chemical Viewpoint. Pure Appl. Chem. 1991, 63, 177–185. [Google Scholar] [CrossRef]
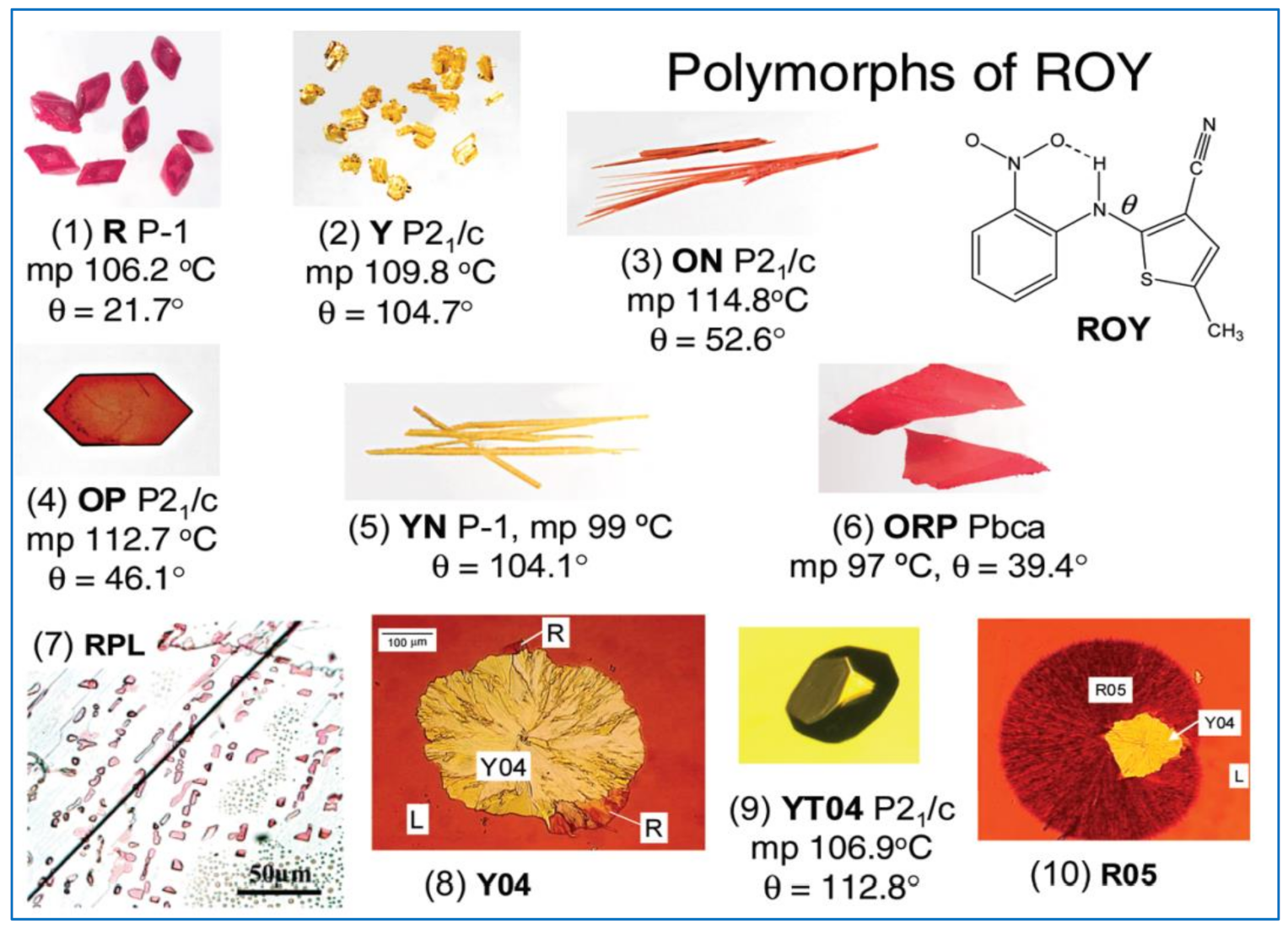
- Chen, S.; Guzei, I.A.; Yu, L. New Polymorphs of ROY and New Record for Coexisting Polymorphs of Solved Structures. J. Am. Chem. Soc. 2005, 127, 9881–9885. [Google Scholar] [CrossRef]
- Yu, L.; Stephenson, G.A.; Mitchell, C.A.; Bunnell, C.A.; Snorek, S.V.; Bowyer, J.J.; Borchardt, T.B.; Stowell, J.G.; Byrn, S.R. Thermochemistry and Conformational Polymorphism of a Hexamorphic Crystal System. J. Am. Chem. Soc. 2000, 122, 585–591. [Google Scholar] [CrossRef]
- Leévesque, A.L.; Maris, T.; Wuest, J.D. ROY Reclaims Its Crown: New Ways to Increase Polymorphic Diversity. J. Am. Chem. Soc. 2020, 142, 11873–11883. [Google Scholar] [CrossRef]
- Yu, L. Polymorphism in Molecular Solids: An Extraordinary System of Red, Orange, and Yellow Crystals. Acc. Chem. Res. 2010, 43, 1257–1266. [Google Scholar] [CrossRef]
- Burger, A. Topics in Pharmaceutical Sciences; Breimer, D.D., Speiser, P., Eds.; Elsevier: Amsterdam, The Netherlands, 1983. [Google Scholar]
- Threlfall, T.L. Analysis of organic polymorphs. A review. Analyst 1995, 120, 2435–2460. [Google Scholar] [CrossRef]
- Bučar, D.K.; Lancaster, R.W.; Bernstein, J. Disappearing Polymorphs Revisited. Angew. Chem. 2015, 54, 6972–6993. [Google Scholar] [CrossRef]
- Haisa, M.; Kashino, S.; Kawai, R.; Maeda, H. The Monoclinic Form of P-Hydroxyacetanilide. Acta Cryst. B 1976, 32, 1283–1285. [Google Scholar] [CrossRef]
- Haisa, M.; Kashino, S.; Maeda, H. The Orthorhombic Form of P-Hydroxyacetanilide. Acta Crystallogr. Sect. B 1974, 30, 2510–2512. [Google Scholar] [CrossRef]
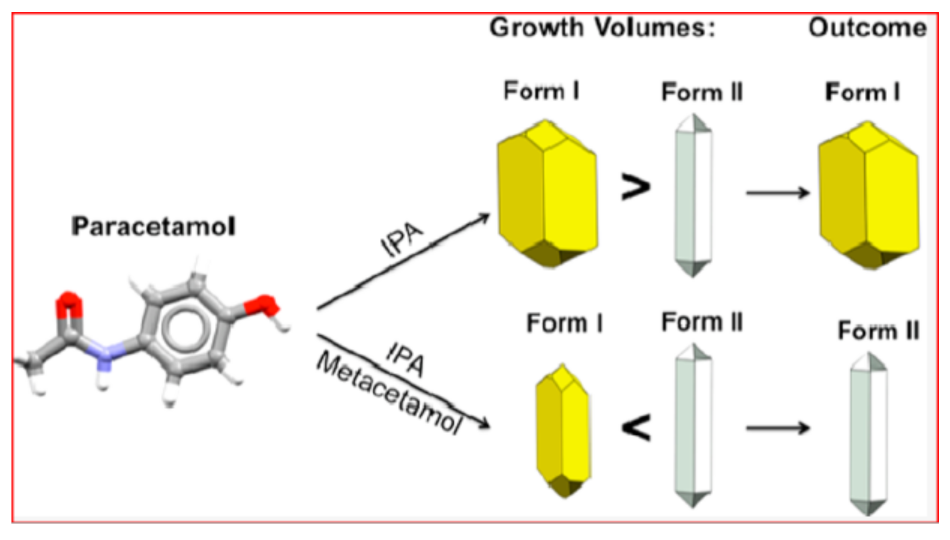
- Martino, P.D.; Conflant, P.; Drache, M.; Huvenne, J.P.; Guyot-Hermann, A.M. Preparation and Physical Characterization of Forms H and HI of Paracetamol. J. Therm. Anal. Cal. 1997, 48, 447–458. [Google Scholar] [CrossRef]
- Liu, Y.; Gabriele, B.; Davey, R.J.; Cruz-Cabeza, A.J. Concerning Elusive Crystal Forms: The Case of Paracetamol. J. Am. Chem. Soc. 2020, 142, 6682–6689. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.Y.Y.; Williams, D.R. Wettability of Paracetamol Polymorphic Forms I and II. Langmuir 2006, 22, 6905–6909. [Google Scholar] [CrossRef]
- Mishra, R.; Srivastava, A.; Sharma, A.; Tandon, P.; Baraldi, C.; Gamberini, M.C. Structural, Electronic, Thermodynamical and Charge Transfer Properties of Chloramphenicol Palmitate Using Vibrational Spectroscopy and DFT Calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 101, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, K.; Kulpe, S.; Schulz, B.; Kunath, D. The structure of the modification of chloramphenicol palmitate—A redetermination. Acta Crystallogr. Sect. B 1981, 37, 1673–1676. [Google Scholar] [CrossRef]
- Diodone, R.; Hidber, P.C.; Kammerer, M.; Meier, R.; Schwitter, U.; Thun, J. Industry Case Studies. In Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development, 2nd ed.; Hilfiker, R., Von Raumer, M., Eds.; Wiley-VCH: Weinheim, Germany, 2019; Chapter 15; pp. 453–456. [Google Scholar]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, M.T.; Sibik, J.; Zeitler, J.A.; Korter, T.M. Examination of L-Glutamic Acid Polymorphs by Solid-State Density Functional Theory and Terahertz Spectroscopy. Phys. Chem. A 2016, 120, 7490–7495. [Google Scholar] [CrossRef] [PubMed]
- Chierotti, M.R.; Gobetto, R.; Pellegrino, L.; Milone, L.; Venturello, P. Mechanically Induced Phase Change in Barbituric Acid. Cryst. Growth Des. 2008, 8, 1454–1457. [Google Scholar] [CrossRef]
- Schmidt, M.U.; Brüning, J.; Glinnemann, J.; Hützler, M.W.; Mörschel, P.; Ivashevskaya, S.N.; Van De Streek, J.; Braga, D.; Maini, L.; Chierotti, M.R.; et al. The Thermodynamically Stable Form of Solid Barbituric Acid: The Enol Tautomer. Angew. Chem. Int. Ed. 2011, 50, 7924–7926. [Google Scholar] [CrossRef] [PubMed]
- Chierotti, M.R.; Ferrero, L.; Garino, N.; Gobetto, R.; Pellegrino, L.; Braga, D.; Grepioni, F.; Maini, L.; Ferrero, L.; Garini, N.; et al. The Richest Collection of Tautomeric Polymorphs: The Case of 2-Thiobarbituric. Acid. Chem. Eur. J. 2010, 16, 4347–4358. [Google Scholar]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. J. Pharm. Res. 2001, 6, 59–66. [Google Scholar]
- Morissette, S.L.; Soukasene, S.; Levinson, D.; Cima, M.J.; Almarsson, Ö. Elucidation of crystal form diversity of the HIV protease inhibitor ritonavir by high-throughput crystallization Polymorphism and Crystalforms. Proc. Natl. Acad. Sci. USA 2003, 100, 2180–2184. [Google Scholar] [CrossRef]
- Georgi, C. European Patent Specification. Supply Manag. 2006, 1, 1–11. [Google Scholar]
- Wolff, H.-M.; Queré, L.; Riedner, J. Polymorphic Form of Rotigotine. EP2215072B1, 25 November 2008. [Google Scholar]
- Rietveld, I.B.; Céolin, R. Rotigotine: Unexpected Polymorphism with Predictable Overall Monotropic Behavior. J. Pharm. Sci. 2015, 104, 4117–4122. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Bernstein, J. Disappearing Polymorphs. Acc. Chem. Res. 1995, 28, 193–200. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism and Patents from a Chemist’s Point of View. In Polymorphism in the Pharmaceutical Industry; Elsevier: Amsterdam, The Netherlands, 2006; pp. 365–384. [Google Scholar]
- Price, S.L. Predicting Crystal Structures of Organic Compounds. Chem. Soc. Rev. 2014, 43, 2098–2111. [Google Scholar] [CrossRef] [PubMed]
- Price, L.S.; Price, S.L. Packing Preferences of Chalcones: A Model Conjugated Pharmaceutical Scaffold. Cryst. Growth Des. 2022, 22, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Price, S.L. Is Zeroth Order Crystal Structure Prediction (CSP_0) Coming to Maturity? What Should We Aim for in an Ideal Crystal Structure Prediction Code? Faraday Discuss. 2018, 211, 9–30. [Google Scholar] [CrossRef]
- CSP Blind Tests—The Cambridge Crystallographic Data Centre (CCDC). Available online: https://www.ccdc.cam.ac.uk/Community/initiatives/cspblindtests/past-csp-blind-tests (accessed on 14 June 2022).
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the Sixth Blind Test of Organic Crystal Structure Prediction Methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 439–459. [Google Scholar] [CrossRef]
- Available online: https://www.ccdc.cam.ac.uk/Community/initiatives/cspblindtests/csp-blind-test-7/ (accessed on 14 June 2022).
- Griesser, U.J. Importance of Solvates. In Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development; Hilfiker, R., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 211–234. [Google Scholar]
- Generally Recognized as Safe (GRAS). Available online: https://www.fda.gov/food/food-ingredients-packaging/generally-recognized-safe-gras (accessed on 15 June 2022).
- Braun, D.E.; Koztecki, L.H.; Mcmahon, J.A.; Price, S.L.; Reutzel-Edens, S.M. Navigating the Waters of Unconventional Crystalline Hydrates. Mol. Pharm. 2015, 12, 3069–3088. [Google Scholar] [CrossRef]
- Jurczak, E.; Mazurek, A.H.; Szeleszczuk, L.; Pisklak, D.M.; Zielinska-Pisklak, M. Pharmaceutical Hydrates Analysis-Overview of Methods and Recent Advances. Pharmaceutics 2020, 12, 959. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Zhu, H.; Yuen, C.; Grant, D.J.W. Influence of Water Activity in Organic Solvent + Water Mixtures on the Nature of the Crystallizing Drug Phase. 1. Theophylline. Int. J. Pharm. 1996, 135, 151–160. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F. Crystal Engineering: State of the Art and Open Challenges in Intermolecular Interactions in Crystals. In Intermolecular Interactions in Crystals: Fundamentals of Crystal Engineering; Novoa, J.J., Ed.; The Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Gift, A.D.; Luner, P.E.; Luedeman, L.; Taylor, L.S. Influence of Polymeric Excipients on Crystal Hydrate Formation Kinetics in Aqueous Slurries. J. Pharm. Sci. 2008, 97, 5198–5211. [Google Scholar] [CrossRef]
- Fang, T.; Haiyan, Q.; Zimmermann, A.; Mtunk, T.; Joergensen, A.C.; Rantanen, J. Factors affecting crystallization of hydrates. J. Pharm. Pharmac. 2010, 62, 1534–1546. [Google Scholar]
- Marchi, E.; Montecchi, L. Imidazo-Rifamycin Derivatives with Antimicrobial Utility. U.S. Patent 4341785A, 27 July 1982. [Google Scholar]
- Gillis, J.C.; Brogden, R.N. Rifaximin: A Review of Its Antibacterial Activity, Pharmacokinetic Properties and Therapeutic Potential in Conditions Mediated by Gastrointestinal Bacteria. Drugs 1995, 49, 467–484. [Google Scholar] [CrossRef] [PubMed]
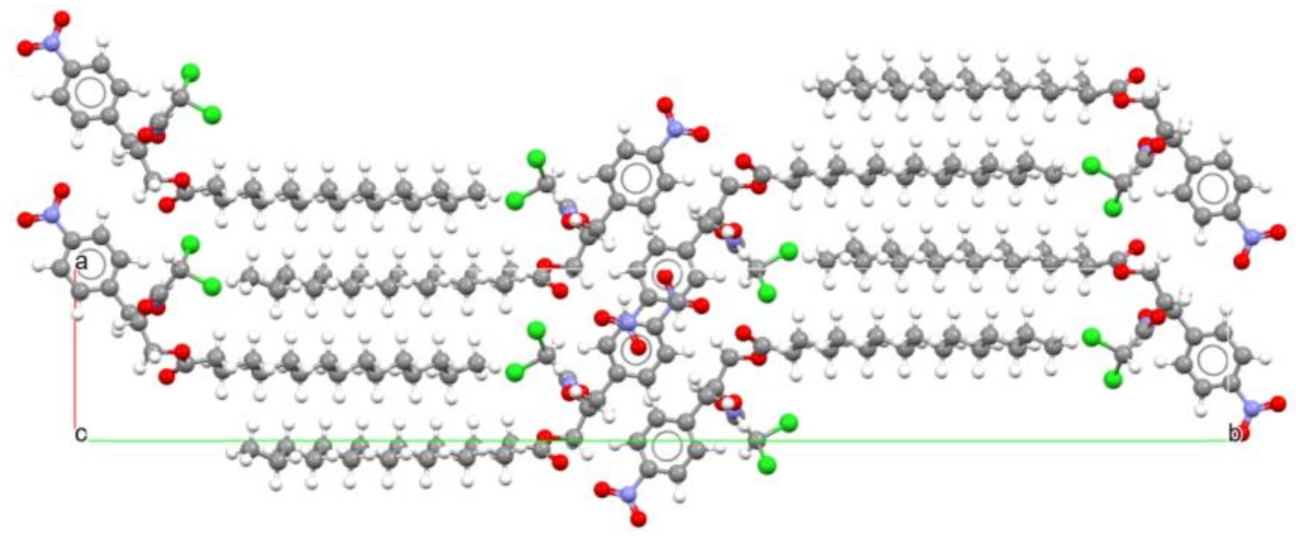
- Braga, D.; Grepioni, F.; Chelazzi, L.; Campana, M.; Confortini, D.; Viscomi, G.C. The Structure-Property Relationship of Four Crystal Forms of Rifaximin. CrystEngComm 2012, 14, 6404–6411. [Google Scholar] [CrossRef]
- Viscomi, G.C.; Campana, M.; Barbanti, M.; Grepioni, F.; Polito, M.; Confortini, D.; Rosini, G.; Righi, P.; Cannata, V.; Braga, D. Crystal Forms of Rifaximin and Their Effect on Pharmaceutical Properties. CrystEngComm. 2008, 10, 1074–1081. [Google Scholar] [CrossRef]
- Viscomi, G.C.; Campana, M.; Braga, D.; Confortini, D.; Cannata, V.; Severini, D.; Righi, P.; Rosini, G. Polymorphous Forms of Rifaximin, Processes for Their Production and Use thereof in Medicinal Preparations. U.S. Patent US7045620, 5 June 2006. [Google Scholar]
- Viscomi, G.C.; Campana, M.; Confortini, D.; Barbanti, M.M.; Braga, D. New polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations. PCT Patent WO2006/094662, 14 September 2006. [Google Scholar]
- Grepioni, F.; Braga, D.; Chelazzi, L.; Shemchuk, O.; Maffei, P.; Sforzini, A.; Viscomi, G.C. Improving solubility and storage stability of Rifaximin via solid-state solvation with Transcutol®. CrystEngComm. 2019, 21, 5278–5283. [Google Scholar] [CrossRef]
- Viscomi, G.C.; Maffei, P.; Sforzini, A.; Grepioni, F.; Chelazzi, L. Solvated Crystal Form of Rifaximin, Production, Compositions and Uses. U.S. Patent 9,938,298 B2 (45), 10 April 2018. [Google Scholar]
- Hoogsteen, K. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Crystallogr. 1963, 16, 907–916. [Google Scholar] [CrossRef]
- Etter, M.C. Hydrogen bonds as design elements in organic chemistry. J. Phys. Chem. 1991, 95, 4601–4610. [Google Scholar] [CrossRef]
- Dunitz, J.D. Crystal and cocrystal: A second opinion. CrystEngComm 2003, 5, 506. [Google Scholar] [CrossRef]
- Almarsson, Ö.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. Do pharmaceutical cocrystals represent a new path to improved medicines? Chem. Comm. 2004, 17, 1889–1896. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Salmon, D.J. Building cocrystals with molecular sense and supramolecular sensibility. CrystEngComm 2005, 7, 439–448. [Google Scholar] [CrossRef]
- Duggirala, N.K.; Perry, M.L.; Almarsson, O.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640. [Google Scholar] [CrossRef]
- Bolla, G.; Sarma, B.; Nangia, A.K. Crystal Engineering of Pharmaceutical Cocrystals in the Discovery and Development of Improved Drugs. Chem. Rev. 2022, 122, 11514–11603. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and fictions about polymorphism. Chem. Soc. Rev 2015, 44, 8619. [Google Scholar] [CrossRef]
- Childs, S.L.; Rodriguez-Hornedo, N.; Reddy, L.S.; Jayasankar, A.; Maheshwari, C.; McCausland, L.; Shipplett, R.; Stahly, B.C. Screening strategies based on solubility and solution composition generate pharmaceutically acceptable cocrystals of carbamazepine. CrystEngComm 2008, 10, 856–864. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Maini, L.; Prosperi, S.; Gobetto, R.; Chierotti, M.R. From unexpected reactions to a new family of ionic co-crystals: The case of barbituric acid with alkali bromides and caesium iodide. Chem. Commun 2010, 46, 7715–7717. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Grepioni, F.; Maini, L.; Capucci, D.; Nanna, S.; Wouters, J.; Aerts, L.; Quéré, L. Combining piracetam and lithium salts: Ionic cocrystals and co-drugs? Chem. Commun. 2012, 48, 8219–8221. [Google Scholar] [CrossRef] [PubMed]
- Grepioni, F.; Wouters, J.; Braga, D.; Nanna, S.; Fours, B.; Coquerel, G.; Geraldine Longfils, G.; Rome, S.; Aerts, L.; Quéré, L. Ionic cocrystals of racetams: Solid-state properties enhancement of neutral active pharmaceutical ingredients via addition of Mg2+ and Ca2+ chlorides. CrystEngComm 2014, 16, 5887–5896. [Google Scholar] [CrossRef]
- Ong, T.T.; Kavuru, P.; Nguyen, T.; Cantwell, R.; Wojtas, Ł.; Zaworotko, M.J. 2:1 cocrystals of homochiral and achiral amino acid zwitterions with Li+ salts: Water-stable zeolitic and diamondoid metal-organic materials. J. Am. Chem. Soc. 2011, 133, 9224–9227. [Google Scholar] [CrossRef]
- Oertling, H. Interactions of alkali- and alkaline earth-halides with carbohydrates in the crystalline state—The overlooked salt and sugar cocrystals. CrystEngComm 2016, 18, 1676–1692. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Forbes, S.; Desper, J. Using cocrystals to systematically modulate aqueous solubility and melting behavior of an anticancer drug. J. Am. Chem. Soc. 2009, 131, 17048–17049. [Google Scholar] [CrossRef]
- Shiraki, K.; Takata, N.; Takano, R.; Hayashi, Y.; Terada, K. Dissolution Improvement and the Mechanism of the Improvement from Cocrystallization of Poorly Water-soluble Compounds. Pharm. Res. 2008, 25, 2581–2592. [Google Scholar] [CrossRef] [PubMed]
- Good, D.J.; Naır, N.; Rodrıguez-Hornedo, R. Cocrystal Eutectic Constants and Prediction of Solubility Behavior. Cryst. Growth Des. 2010, 10, 1028–1032. [Google Scholar] [CrossRef]
- McNamara, D.P.; Childs, S.L.; Giordano, J.; Iarriccio, A.; Cassidy, J.; Shet, M.S.; Mannion, R.; O’Donnell, E.; Park, A. Use of a Glutaric Acid Cocrystal to Improve Oral Bioavailability of a Low Solubility API. Pharm. Res. 2006, 23, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
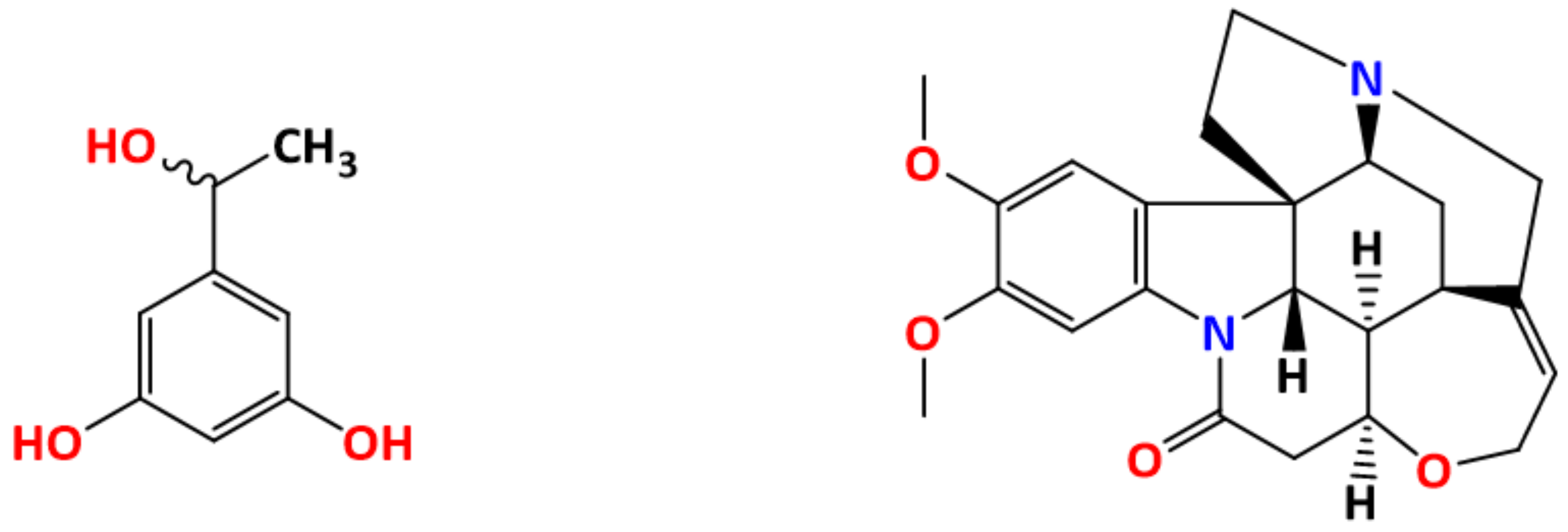
- Yan, Y.; Chen, J.M.; Geng, N.; Lu, T.B. Thermodynamics and preliminary pharmaceutical characterization of a melatonin–pimelic acid cocrystal prepared by a melt crystallization method. CrystEngComm 2015, 17, 612–620. [Google Scholar] [CrossRef]
- Grifasi, F.; Chierotti, M.R.; Gaglioti, K.; Gobetto, R.; Maini, L.; Braga, D.; Dichiarante, E.; Curzi, M. Using Salt Cocrystals to Improve the Solubility of Niclosamide. Cryst. Growth Des. 2015, 15, 1939–1948. [Google Scholar] [CrossRef]
- Chadha, K.; Karan, M.; Bhalla, Y.; Chadha, R.; Khullar, S.; Mandal, S.; Vasisht, K. Cocrystals of Hesperetin: Structural, Pharmacokinetic, and Pharmacodynamic Evaluation. Cryst. Growth Des. 2017, 17, 2386–2405. [Google Scholar] [CrossRef]
- Eisenlohr, F.; Meier, G. Die Aufspaltung von Racematen mit Hilfe von Molekülverbindungen. Ber. Der Dtsch. Chem. Ges. A B Ser. 1938, 71, 1005–1013. [Google Scholar] [CrossRef]
- Caira, M.R.; Nassimbeni, L.R.; Scott, J.L.; Wildervanck, A.F. Resolution of optical isomers of 4-amino-p-chlorobutyric acid lactam by cocrystallization. J. Chem. Cryst. 1996, 26, 117–122. [Google Scholar] [CrossRef]
- Springuel, G.; Leyssens, T. Innovative chiral resolution using enantiospecific cocrystallization in solution. Cryst. Growth Des. 2012, 12, 3374–3378. [Google Scholar] [CrossRef]
- George, F.; Norberg, B.; Robeyns, K.; Wouters, J.; Leyssens, T. Peculiar Case of Levetiracetam and Etiracetam α-Ketoglutaric Acid Cocrystals: Obtaining a Stable Conglomerate of Etiracetam. Cryst. Growth Des. 2016, 16, 5273–5282. [Google Scholar] [CrossRef]
- Toda, F. Isolation and optical resolution of materials utilizing inclusion crystallization. In Molecular Inclusion and Molecular Recognition—Clathrates I; Springer: Berlin/Heidelberg, Germany, 2005; pp. 43–69. [Google Scholar]
- Weber, E.; Wimmer, C.; Llamas-Saiz, A.L.; Foces-Foces, C. New chiral selectors derived from lactic acid. Cocrystalline and sorptive optical resolutions, and the crystal structure of an inclusion complex with 3-methylcyclohexanone. J. Chem. Soc. Chem. Commun. 1992, 733–735. [Google Scholar] [CrossRef]
- Nishikawa, K.; Tsukada, H.; Abe, S.; Kishimoto, M.; Yasuoka, N. Mode of molecular recognition during optical resolution: A structural study of the molecular complex involving both 3-hydroxypyrrolidines and (R,R)-(-)-trans-bis(hydroxydiphenylrnethyl)-1,4-dioxaspiro[4,5]decane. Chirality 1999, 11, 166–171. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physical and Thermodynamic Properties | Density and refractive index, thermal and electrical conductivity, hygroscopicity, melting points, free energy and chemical potential, heat capacity, vapor pressure, solubility, thermal stability, and color and shape of crystals. |
| Spectroscopic Properties | Electronic, vibrational, and rotational properties, and nuclear magnetic resonance spectral features. |
| Kinetic Properties | Rate of dissolution, kinetics of solid-state reactions, and kinetic inertness. |
| Surface Properties | Surface free energy, crystal habit, surface area, and particle size distribution. |
| Mechanical Properties | Hardness, compression, and thermal expansion. |
| Chemical Properties | Chemical and photochemical reactivity. |
| Target | Chemical Diagram | Crystallization Conditions, Remarks, and Clarifications |
|---|---|---|
| (XXII) |  | Crystallized from an acetone/water mixture; chiral-like character due to potential flexibility of the six-membered ring, but no chiral precursors used in synthesis. |
| (XXIII) | 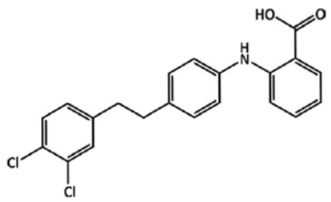 | Five known polymorphs (A–E). The most stable polymorphs at 257 and 293 K. Crystallization conditions include slow evaporation of acetone solution and of an ethyl acetate:water mixture. |
| (XXIV) |  | Crystallized from 1 M HCl solution. The substituents of the C=C double bond are in the cis configuration. |
| (XXV) |  | Slow evaporation of a methanol solution, which contained a racemic mixture of the enantiomers of Tröger’s base. |
| (XXVI) | 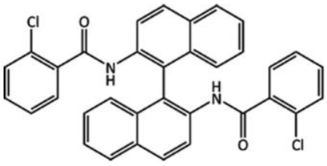 | Slow evaporation from 1:1 mixture of hexane and dichloromethane. No chiral precursors used in synthesis. |
| CRYSTAL FORMS | % of All Organic Structures |
|---|---|
| Organic crystal structures | 100 |
| Single component molecular organic structures | 72.1 |
| Single component polymorphic structures | 1.4 |
| Hydrates | 7.4 |
| Molecular organic hydrates | 2.7 |
| Polymorphic molecular organic hydrates | 1.0 |
| Cocrystals | 1.1 |
| Polymorphic cocrystals | 1.9 |
| Material | Molecular Structure | Solid State Form | Solubility (mg/mL) |
|---|---|---|---|
| Caffeine (CAF) | 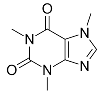 | Anh = triclinic β-phase form Hyd = monoclinic 4/5 hydrate | Anh = 49.7 Hyd = 21.8 |
| Carbamazepine (CBZ) | 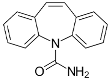 | Anh = monoclinic low temp. form Hyd = orthorhombic dihydrate | Anh = 0.424 Hyd = 0.139 |
| Sulfaguanidine (SFG) | 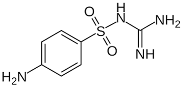 | Anh = monoclinic form II Hyd = monoclinic monohydrate | Anh = 1.38 Hyd = 1.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braga, D.; Casali, L.; Grepioni, F. The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools? Int. J. Mol. Sci. 2022, 23, 9013. https://doi.org/10.3390/ijms23169013
Braga D, Casali L, Grepioni F. The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools? International Journal of Molecular Sciences. 2022; 23(16):9013. https://doi.org/10.3390/ijms23169013
Chicago/Turabian StyleBraga, Dario, Lucia Casali, and Fabrizia Grepioni. 2022. "The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools?" International Journal of Molecular Sciences 23, no. 16: 9013. https://doi.org/10.3390/ijms23169013
APA StyleBraga, D., Casali, L., & Grepioni, F. (2022). The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools? International Journal of Molecular Sciences, 23(16), 9013. https://doi.org/10.3390/ijms23169013