Molecular Genetics of Renal Cell Carcinoma: A Narrative Review Focused on Clinical Relevance


 ,
,
Simple Summary
Abstract
1. Introduction
2. Genetic Landscape of Kidney Cancer
2.1. Genetic Variants in Sporadic Kidney Cancer
2.2. Hereditary Kidney Cancer Syndromes
2.2.1. Von Hippel–Lindau (VHL) Syndrome
2.2.2. Hereditary Papillary Renal Carcinoma (HPRC)
2.2.3. Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)
2.2.4. Birt–Hogg–Dubé (BHD) Syndrome
2.2.5. Tuberous Sclerosis Complex (TSC)
2.2.6. Succinate-Dehydrogenase-Deficient RCC
2.2.7. BRCA1-Associated Protein (BAP1)
2.2.8. Translocation RCC
2.2.9. ELOC-Mutated RCC
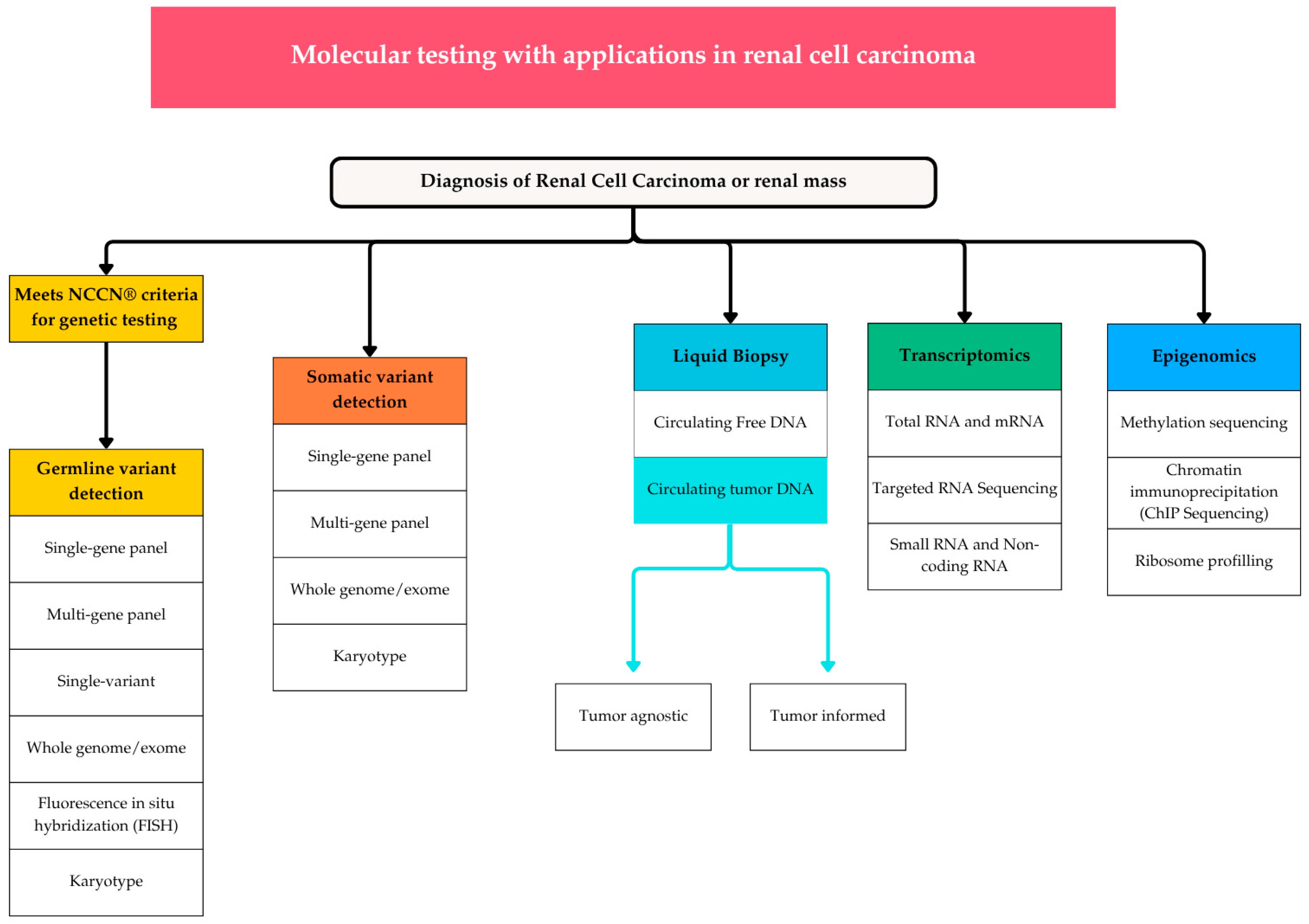
3. Molecular Testing Modalities
3.1. Germline Genetic Testing
3.2. Somatic Genetic Testing
3.3. Cell-Free DNA (cfDNA) and Circulating Tumor DNA (ctDNA)
3.4. RNA Sequencing and Methylation Profiling
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, N.; Bai, H.; Zhang, Z.; Yu, B.; Zhao, H.; Li, J.; Zheng, G. Global, regional, and national burden of kidney cancer and attributable risk factors in adults aged 65 years and older from 1990 to 2021 and projections to 2040. BMC Cancer 2025, 25, 481. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Linehan, W.M.; Pinto, P.A.; Bratslavsky, G.; Pfaffenroth, E.; Merino, M.; Vocke, C.D.; Toro, J.R.; Bottaro, D.; Neckers, L.; Schmidt, L.S.; et al. Hereditary kidney cancer: Unique opportunity for disease-based therapy. Cancer 2009, 115, 2252–2261. [Google Scholar] [CrossRef]
- Bratslavsky, G.; Mendhiratta, N.; Daneshvar, M.; Brugarolas, J.; Ball, M.W.; Metwalli, A.; Nathanson, K.L.; Pierorazio, P.M.; Boris, R.S.; Singer, E.A.; et al. Genetic risk assessment for hereditary renal cell carcinoma: Clinical consensus statement. Cancer 2021, 127, 3957–3966. [Google Scholar] [CrossRef]
- Ball, M.W.; Shuch, B.M. Inherited kidney cancer syndromes. Curr. Opin. Urol. 2019, 29, 334–343. [Google Scholar] [CrossRef]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; Ricketts, C.J. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef]
- Bories, C.; Lejour, T.; Adolphe, F.; Kermasson, L.; Couve, S.; Tanguy, L.; Luszczewska, G.; Watzky, M.; Poillerat, V.; Garnier, P.; et al. DCLRE1B/Apollo germline mutations associated with renal cell carcinoma impair telomere protection. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167107. [Google Scholar] [CrossRef]
- Adolphe, F.; Ferlicot, S.; Verkarre, V.; Posseme, K.; Couve, S.; Garnier, P.; Droin, N.; Deloger, M.; Job, B.; Giraud, S.; et al. Germline mutation in the NBR1 gene involved in autophagy detected in a family with renal tumors. Cancer Genet. 2021, 258–259, 51–56. [Google Scholar] [CrossRef]
- Moch, H.; Amin, M.B.; Berney, D.M.; Comperat, E.M.; Gill, A.J.; Hartmann, A.; Menon, S.; Raspollini, M.R.; Rubin, M.A.; Srigley, J.R.; et al. The 2022 World Health Organization Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2022, 82, 458–468. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Mori, K.; Matsukawa, A.; Kawada, T.; Katayama, S.; Bekku, K.; Laukhtina, E.; Rajwa, P.; Quhal, F.; Pradere, B. Updated systematic review and network meta-analysis of first-line treatments for metastatic renal cell carcinoma with extended follow-up data. Cancer Immunol. Immunother. 2024, 73, 38. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Gurram, S.; Harthy, M.A.; Singer, E.A.; Sidana, A.; Shuch, B.M.; Ball, M.W.; Friend, J.C.; Mac, L.; Purcell, E.; et al. Results from a phase II study of bevacizumab and erlotinib in subjects with advanced hereditary leiomyomatosis and renal cell cancer (HLRCC) or sporadic papillary renal cell cancer. J. Clin. Oncol. 2020, 38, 5004. [Google Scholar] [CrossRef]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Alva, A.; Bagshaw, H.; Baine, M.; Beckermann, K.; Carlo, M.I.; Choueiri, T.K.; Costello, B.A.; et al. NCCN Guidelines(R) Insights: Kidney Cancer, Version 2.2024. J. Natl. Compr. Cancer Netw. 2024, 22, 4–16. [Google Scholar] [CrossRef]
- Mendhiratta, N.; Hauver, H., Jr.; Hatton, W.; Ostrusky, A.; Sathe, D.S.; Gurram, S.; Rice, P.; Chalfin, H. Outcomes of a universal germline screening program in a community urology practice. Clin. Genet. 2024, 106, 277–283. [Google Scholar] [CrossRef]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L.; for a Guideline Development Group of the American College of Medical Genetics and Genomics Professional Practice and Guidelines Committee and of the National Society of Genetic Counselors Practice Guidelines Committee. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Gomella, P.T.; Linehan, W.M.; Ball, M.W. Precision Surgery and Kidney Cancer: Knowledge of Genetic Alterations Influences Surgical Management. Genes 2021, 12, 261. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef]
- Shuch, B.; Amin, A.; Armstrong, A.J.; Eble, J.N.; Ficarra, V.; Lopez-Beltran, A.; Martignoni, G.; Rini, B.I.; Kutikov, A. Understanding pathologic variants of renal cell carcinoma: Distilling therapeutic opportunities from biologic complexity. Eur. Urol. 2015, 67, 85–97. [Google Scholar] [CrossRef]
- Walther, M.M.; Lubensky, I.A.; Venzon, D.; Zbar, B.; Linehan, W.M. Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: Clinical implications. J. Urol. 1995, 154, 2010–2014; discussion 2014–2015. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Klco, J.; Nakamura, E.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002, 1, 237–246. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Duffey, B.G.; Choyke, P.L.; Glenn, G.; Grubb, R.L.; Venzon, D.; Linehan, W.M.; Walther, M.M. The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J. Urol. 2004, 172, 63–65. [Google Scholar] [CrossRef]
- Srinivasan, R.; Iliopoulos, O.; Beckermann, K.E.; Narayan, V.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Iversen, A.B.; Cornell, J.; et al. Belzutifan for von Hippel-Lindau disease-associated renal cell carcinoma and other neoplasms (LITESPARK-004): 50 months follow-up from a single-arm, phase 2 study. Lancet Oncol. 2025, 26, 571–582. [Google Scholar] [CrossRef]
- Zbar, B.; Tory, K.; Merino, M.; Schmidt, L.; Glenn, G.; Choyke, P.; Walther, M.M.; Lerman, M.; Linehan, W.M. Hereditary papillary renal cell carcinoma. J. Urol. 1994, 151, 561–566. [Google Scholar] [CrossRef]
- Sebai, M.; Tulasne, D.; Caputo, S.M.; Verkarre, V.; Fernandes, M.; Guerin, C.; Reinhart, F.; Adams, S.; Maugard, C.; Caron, O.; et al. Novel germline MET pathogenic variants in French patients with papillary renal cell carcinomas type I. Hum. Mutat. 2022, 43, 316–327. [Google Scholar] [CrossRef]
- Marona, P.; Gorka, J.; Kotlinowski, J.; Majka, M.; Jura, J.; Miekus, K. C-Met as a Key Factor Responsible for Sustaining Undifferentiated Phenotype and Therapy Resistance in Renal Carcinomas. Cells 2019, 8, 272. [Google Scholar] [CrossRef]
- Sweeney, P.; El-Naggar, A.K.; Lin, S.H.; Pisters, L.L. Biological significance of c-met over expression in papillary renal cell carcinoma. J. Urol. 2002, 168, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Tangen, C.; Plets, M.; Thompson, I.M., Jr.; Narayan, V.; George, D.J.; Heng, D.Y.C.; Shuch, B.; Stein, M.; Gulati, S.; et al. Final Overall Survival Analysis of S1500: A Randomized, Phase II Study Comparing Sunitinib With Cabozantinib, Crizotinib, and Savolitinib in Advanced Papillary Renal Cell Carcinoma. J. Clin. Oncol. 2024, 42, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.H.; Toure, O.; Glenn, G.M.; Pithukpakorn, M.; Neckers, L.; Stolle, C.; Choyke, P.; Grubb, R.; Middelton, L.; Turner, M.L.; et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J. Med. Genet. 2006, 43, 18–27. [Google Scholar] [CrossRef]
- Yang, Y.; Lane, A.N.; Ricketts, C.J.; Sourbier, C.; Wei, M.H.; Shuch, B.; Pike, L.; Wu, M.; Rouault, T.A.; Boros, L.G.; et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS ONE 2013, 8, e72179. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef]
- Tong, W.H.; Sourbier, C.; Kovtunovych, G.; Jeong, S.Y.; Vira, M.; Ghosh, M.; Romero, V.V.; Sougrat, R.; Vaulont, S.; Viollet, B.; et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell 2011, 20, 315–327. [Google Scholar] [CrossRef]
- Yang, Y.; Valera, V.A.; Padilla-Nash, H.M.; Sourbier, C.; Vocke, C.D.; Vira, M.A.; Abu-Asab, M.S.; Bratslavsky, G.; Tsokos, M.; Merino, M.J.; et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: In vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet. Cytogenet. 2010, 196, 45–55. [Google Scholar] [CrossRef]
- Bateman, N.W.; Tarney, C.M.; Abulez, T.; Soltis, A.R.; Zhou, M.; Conrads, K.; Litzi, T.; Oliver, J.; Hood, B.; Driggers, P.; et al. Proteogenomic landscape of uterine leiomyomas from hereditary leiomyomatosis and renal cell cancer patients. Sci. Rep. 2021, 11, 9371. [Google Scholar] [CrossRef]
- Menko, F.H.; Maher, E.R.; Schmidt, L.S.; Middelton, L.A.; Aittomaki, K.; Tomlinson, I.; Richard, S.; Linehan, W.M. Hereditary leiomyomatosis and renal cell cancer (HLRCC): Renal cancer risk, surveillance and treatment. Fam. Cancer 2014, 13, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Zbar, B.; Alvord, W.G.; Glenn, G.; Turner, M.; Pavlovich, C.P.; Schmidt, L.; Walther, M.; Choyke, P.; Weirich, G.; Hewitt, S.M.; et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dube syndrome. Cancer Epidemiol. Biomark. Prev. 2002, 11, 393–400. [Google Scholar]
- Pavlovich, C.P.; Grubb, R.L., 3rd; Hurley, K.; Glenn, G.M.; Toro, J.; Schmidt, L.S.; Torres-Cabala, C.; Merino, M.J.; Zbar, B.; Choyke, P.; et al. Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J. Urol. 2005, 173, 1482–1486. [Google Scholar] [CrossRef]
- Di Malta, C.; Zampelli, A.; Granieri, L.; Vilardo, C.; De Cegli, R.; Cinque, L.; Nusco, E.; Pece, S.; Tosoni, D.; Sanguedolce, F.; et al. TFEB and TFE3 drive kidney cystogenesis and tumorigenesis. EMBO Mol. Med. 2023, 15, e16877. [Google Scholar] [CrossRef]
- Schmidt, L.S.; Nickerson, M.L.; Warren, M.B.; Glenn, G.M.; Toro, J.R.; Merino, M.J.; Turner, M.L.; Choyke, P.L.; Sharma, N.; Peterson, J.; et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dube syndrome. Am. J. Hum. Genet. 2005, 76, 1023–1033. [Google Scholar] [CrossRef]
- Toro, J.R.; Wei, M.H.; Glenn, G.M.; Weinreich, M.; Toure, O.; Vocke, C.; Turner, M.; Choyke, P.; Merino, M.J.; Pinto, P.A.; et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: A new series of 50 families and a review of published reports. J. Med. Genet. 2008, 45, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Pavlovich, C.P.; Walther, M.M.; Eyler, R.A.; Hewitt, S.M.; Zbar, B.; Linehan, W.M.; Merino, M.J. Renal tumors in the Birt-Hogg-Dube syndrome. Am. J. Surg. Pathol. 2002, 26, 1542–1552. [Google Scholar] [CrossRef]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N. Engl. J. Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef]
- Rakowski, S.K.; Winterkorn, E.B.; Paul, E.; Steele, D.J.; Halpern, E.F.; Thiele, E.A. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006, 70, 1777–1782. [Google Scholar] [CrossRef]
- Yang, P.; Cornejo, K.M.; Sadow, P.M.; Cheng, L.; Wang, M.; Xiao, Y.; Jiang, Z.; Oliva, E.; Jozwiak, S.; Nussbaum, R.L.; et al. Renal cell carcinoma in tuberous sclerosis complex. Am. J. Surg. Pathol. 2014, 38, 895–909. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tretiakova, M.S.; Troxell, M.L.; Osunkoya, A.O.; Fadare, O.; Sangoi, A.R.; Shen, S.S.; Lopez-Beltran, A.; Mehra, R.; Heider, A.; et al. Tuberous sclerosis-associated renal cell carcinoma: A clinicopathologic study of 57 separate carcinomas in 18 patients. Am. J. Surg. Pathol. 2014, 38, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Muller, R.U.; Marlais, M.; Wlodkowski, T.; Haeberle, S.; de Argumedo, M.L.; Bergmann, C.; Breysem, L.; Fladrowski, C.; Henske, E.P.; et al. Clinical practice recommendations for kidney involvement in tuberous sclerosis complex: A consensus statement by the ERKNet Working Group for Autosomal Dominant Structural Kidney Disorders and the ERA Genes & Kidney Working Group. Nat. Rev. Nephrol. 2024, 20, 402–420. [Google Scholar] [CrossRef] [PubMed]
- Ramon, J.; Rimon, U.; Garniek, A.; Golan, G.; Bensaid, P.; Kitrey, N.D.; Nadu, A.; Dotan, Z.A. Renal angiomyolipoma: Long-term results following selective arterial embolization. Eur. Urol. 2009, 55, 1155–1161. [Google Scholar] [CrossRef]
- Bissler, J.J.; Kingswood, J.C.; Radzikowska, E.; Zonnenberg, B.A.; Frost, M.; Belousova, E.; Sauter, M.; Nonomura, N.; Brakemeier, S.; de Vries, P.J.; et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013, 381, 817–824. [Google Scholar] [CrossRef]
- Bissler, J.J.; Nonomura, N.; Budde, K.; Zonnenberg, B.A.; Fischereder, M.; Voi, M.; Louveau, A.L.; Herbst, F.; Bebin, E.M.; Curatolo, P.; et al. Angiomyolipoma rebound tumor growth after discontinuation of everolimus in patients with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis. PLoS ONE 2018, 13, e0201005. [Google Scholar] [CrossRef]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Shuch, B.; Vocke, C.D.; Metwalli, A.R.; Bratslavsky, G.; Middelton, L.; Yang, Y.; Wei, M.H.; Pautler, S.E.; Peterson, J.; et al. Succinate dehydrogenase kidney cancer: An aggressive example of the Warburg effect in cancer. J. Urol. 2012, 188, 2063–2071. [Google Scholar] [CrossRef]
- Kluckova, K.; Tennant, D.A. Metabolic implications of hypoxia and pseudohypoxia in pheochromocytoma and paraganglioma. Cell Tissue Res. 2018, 372, 367–378. [Google Scholar] [CrossRef]
- Gill, A.J. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology 2012, 44, 285–292. [Google Scholar] [CrossRef]
- Gill, A.J.; Hes, O.; Papathomas, T.; Sedivcova, M.; Tan, P.H.; Agaimy, A.; Andresen, P.A.; Kedziora, A.; Clarkson, A.; Toon, C.W.; et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: A morphologically distinct entity: A clinicopathologic series of 36 tumors from 27 patients. Am. J. Surg. Pathol. 2014, 38, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavia-Jimenez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Ferris, L.K.; Baumann, F.; Napolitano, A.; Lum, C.A.; Flores, E.G.; Gaudino, G.; Powers, A.; Bryant-Greenwood, P.; Krausz, T.; et al. BAP1 cancer syndrome: Malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J. Transl. Med. 2012, 10, 179. [Google Scholar] [CrossRef]
- Walpole, S.; Pritchard, A.L.; Cebulla, C.M.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; de la Fouchardiere, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef]
- Hirsch, T.Z.; Negulescu, A.; Gupta, B.; Caruso, S.; Noblet, B.; Couchy, G.; Bayard, Q.; Meunier, L.; Morcrette, G.; Scoazec, J.Y.; et al. BAP1 mutations define a homogeneous subgroup of hepatocellular carcinoma with fibrolamellar-like features and activated PKA. J. Hepatol. 2020, 72, 924–936. [Google Scholar] [CrossRef]
- Brugarolas, J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer J. 2013, 19, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Kapur, P.; Pena-Llopis, S.; Christie, A.; Zhrebker, L.; Pavia-Jimenez, A.; Rathmell, W.K.; Xie, X.J.; Brugarolas, J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol. 2013, 14, 159–167. [Google Scholar] [CrossRef]
- Lalloo, F.; Kulkarni, A.; Chau, C.; Nielsen, M.; Sheaff, M.; Steele, J.; van Doorn, R.; Wadt, K.; Hamill, M.; Torr, B.; et al. Clinical practice guidelines for the diagnosis and surveillance of BAP1 tumour predisposition syndrome. Eur. J. Hum. Genet. 2023, 31, 1261–1269. [Google Scholar] [CrossRef]
- Argani, P. MiT family translocation renal cell carcinoma. Semin. Diagn. Pathol. 2015, 32, 103–113. [Google Scholar] [CrossRef]
- Calio, A.; Segala, D.; Munari, E.; Brunelli, M.; Martignoni, G. MiT Family Translocation Renal Cell Carcinoma: From the Early Descriptions to the Current Knowledge. Cancers 2019, 11, 1110. [Google Scholar] [CrossRef]
- Ellis, C.L.; Eble, J.N.; Subhawong, A.P.; Martignoni, G.; Zhong, M.; Ladanyi, M.; Epstein, J.I.; Netto, G.J.; Argani, P. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: Impact of fusion subtype, age, and stage. Mod. Pathol. 2014, 27, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.N.; Clark, J.I.; Flanigan, R.C.; Picken, M.M. Xp11.2 translocation renal cell carcinoma with very aggressive course in five adults. Am. J. Clin. Pathol. 2007, 128, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Gan, W.; Zhang, G.; Li, X.; Guo, H. Clinical characteristics of XP11.2 translocation/TFE3 gene fusion renal cell carcinoma: A systematic review and meta-analysis of observational studies. BMC Urol. 2016, 16, 40. [Google Scholar] [CrossRef] [PubMed]
- Rais-Bahrami, S.; Drabick, J.J.; De Marzo, A.M.; Hicks, J.; Ho, C.; Caroe, A.E.; Argani, P. Xp11 translocation renal cell carcinoma: Delayed but massive and lethal metastases of a chemotherapy-associated secondary malignancy. Urology 2007, 70, 178.e3–178.e6. [Google Scholar] [CrossRef]
- Smith, P.S.; Whitworth, J.; West, H.; Cook, J.; Gardiner, C.; Lim, D.H.K.; Morrison, P.J.; Hislop, R.G.; Murray, E.; BioResource, N.R.D.; et al. Characterization of renal cell carcinoma-associated constitutional chromosome abnormalities by genome sequencing. Genes. Chromosomes Cancer 2020, 59, 333–347. [Google Scholar] [CrossRef]
- Andreou, A.; Yngvadottir, B.; Bassaganyas, L.; Clark, G.; Martin, E.; Whitworth, J.; Cornish, A.J.; Genomics England Research, C.; Houlston, R.S.; Rich, P.; et al. Elongin C (ELOC/TCEB1)-associated von Hippel-Lindau disease. Hum. Mol. Genet. 2022, 31, 2728–2737. [Google Scholar] [CrossRef]
- Wang, J.J.; Huang, R.R.; Cone, B.D.; Kang, S.L.; Setoodeh, R.; Sisk, A.E.; Sajed, D.P.; Shuch, B.M.; Sowalsky, A.G.; Ye, H. ELOC-Mutated Renal Cell Carcinoma is a Rare Indolent Tumor with Distinctive Genomic Characteristics. Mod. Pathol. 2025, 38, 100777. [Google Scholar] [CrossRef]
- DiNatale, R.G.; Gorelick, A.N.; Makarov, V.; Blum, K.A.; Silagy, A.W.; Freeman, B.; Chowell, D.; Marcon, J.; Mano, R.; Sanchez, A.; et al. Putative Drivers of Aggressiveness in TCEB1-mutant Renal Cell Carcinoma: An Emerging Entity with Variable Clinical Course. Eur. Urol. Focus. 2021, 7, 381–389. [Google Scholar] [CrossRef]
- Batavia, A.A.; Rutishauser, D.; Sobottka, B.; Schraml, P.; Beerenwinkel, N.; Moch, H. Biallelic ELOC-Inactivated Renal Cell Carcinoma: Molecular Features Supporting Classification as a Distinct Entity. Mod. Pathol. 2023, 36, 100194. [Google Scholar] [CrossRef]
- Bahrami, A.; Truong, L.D.; Ro, J.Y. Undifferentiated tumor: True identity by immunohistochemistry. Arch. Pathol. Lab. Med. 2008, 132, 326–348. [Google Scholar] [CrossRef]
- Centeno, B.A.; Bloom, G.; Chen, D.T.; Chen, Z.; Gruidl, M.; Nasir, A.; Yeatman, T.Y. Hybrid model integrating immunohistochemistry and expression profiling for the classification of carcinomas of unknown primary site. J. Mol. Diagn. 2010, 12, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Dietel, M.; Johrens, K.; Laffert, M.V.; Hummel, M.; Blaker, H.; Pfitzner, B.M.; Lehmann, A.; Denkert, C.; Darb-Esfahani, S.; Lenze, D.; et al. A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: A review focussing on clinical relevance. Cancer Gene Ther. 2015, 22, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Kozarewa, I.; Armisen, J.; Gardner, A.F.; Slatko, B.E.; Hendrickson, C.L. Overview of Target Enrichment Strategies. Curr. Protoc. Mol. Biol. 2015, 112, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Freedman, A.N.; Klabunde, C.N.; Wiant, K.; Enewold, L.; Gray, S.W.; Filipski, K.K.; Keating, N.L.; Leonard, D.G.B.; Lively, T.; McNeel, T.S.; et al. Use of Next-Generation Sequencing Tests to Guide Cancer Treatment: Results From a Nationally Representative Survey of Oncologists in the United States. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef]
- Wagle, N.; Berger, M.F.; Davis, M.J.; Blumenstiel, B.; Defelice, M.; Pochanard, P.; Ducar, M.; Van Hummelen, P.; Macconaill, L.E.; Hahn, W.C.; et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012, 2, 82–93. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Bokkers, K.; Vlaming, M.; Engelhardt, E.G.; Zweemer, R.P.; van Oort, I.M.; Kiemeney, L.; Bleiker, E.M.A.; Ausems, M. The Feasibility of Implementing Mainstream Germline Genetic Testing in Routine Cancer Care-A Systematic Review. Cancers 2022, 14, 1059. [Google Scholar] [CrossRef]
- Ladd, M.K.; Peshkin, B.N.; Isaacs, C.; Hooker, G.; Willey, S.; Valdimarsdottir, H.; DeMarco, T.; O’Neill, S.; Binion, S.; Schwartz, M.D.; et al. Predictors of genetic testing uptake in newly diagnosed breast cancer patients. J. Surg. Oncol. 2020, 122, 134–143. [Google Scholar] [CrossRef]
- Patel, D.; Blouch, E.L.; Rodgers-Fouche, L.H.; Emmet, M.M.; Shannon, K.M. Finding a Balance: Reconciling the Needs of the Institution, Patient, and Genetic Counselor for Optimal Resource Utilization. J. Genet. Couns. 2018, 27, 1318–1327. [Google Scholar] [CrossRef]
- Shuch, B.; Vourganti, S.; Ricketts, C.J.; Middleton, L.; Peterson, J.; Merino, M.J.; Metwalli, A.R.; Srinivasan, R.; Linehan, W.M. Defining early-onset kidney cancer: Implications for germline and somatic mutation testing and clinical management. J. Clin. Oncol. 2014, 32, 431–437. [Google Scholar] [CrossRef]
- Alaghehbandan, R.; Perez Montiel, D.; Luis, A.S.; Hes, O. Molecular Genetics of Renal Cell Tumors: A Practical Diagnostic Approach. Cancers 2019, 12, 85. [Google Scholar] [CrossRef]
- Tung, N.; Ricker, C.; Messersmith, H.; Balmana, J.; Domchek, S.; Stoffel, E.M.; Almhanna, K.; Arun, B.; Chavarri-Guerra, Y.; Cohen, S.A.; et al. Selection of Germline Genetic Testing Panels in Patients With Cancer: ASCO Guideline. J. Clin. Oncol. 2024, 42, 2599–2615. [Google Scholar] [CrossRef] [PubMed]
- Yngvadottir, B.; Macarthur, D.G.; Jin, H.; Tyler-Smith, C. The promise and reality of personal genomics. Genome Biol. 2009, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.; Huether, R.; White, K.; Hoskinson, D.; Beaubier, N.; Dong, H.; Adjei, A.A.; Mansfield, A.S. Tumor Mutational Burden From Tumor-Only Sequencing Compared With Germline Subtraction From Paired Tumor and Normal Specimens. JAMA Netw. Open 2020, 3, e200202. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, A.; Sholl, L.; Reardon, B.; Taylor-Weiner, A.; Amin-Mansour, A.; Miao, D.; Liu, D.; Oliver, N.; MacConaill, L.; Ducar, M.; et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016, 8, 79. [Google Scholar] [CrossRef]
- Vocke, C.D.; Ricketts, C.J.; Metwalli, A.R.; Pinto, P.A.; Gautam, R.; Raffeld, M.; Merino, M.J.; Ball, M.W.; Linehan, W.M. Differential VHL Mutation Patterns in Bilateral Clear Cell RCC Distinguishes Between Independent Primary Tumors and Contralateral Metastatic Disease. Urology 2022, 165, 170–177. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Jiang, P.; Lo, Y.M.D. The Long and Short of Circulating Cell-Free DNA and the Ins and Outs of Molecular Diagnostics. Trends Genet. 2016, 32, 360–371. [Google Scholar] [CrossRef]
- de Martino, M.; Klatte, T.; Haitel, A.; Marberger, M. Serum cell-free DNA in renal cell carcinoma: A diagnostic and prognostic marker. Cancer 2012, 118, 82–90. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Uemura, M.; Fujita, M.; Maejima, K.; Koh, Y.; Matsushita, M.; Nakano, K.; Hayashi, Y.; Wang, C.; Ishizuya, Y.; et al. Clinical significance of the mutational landscape and fragmentation of circulating tumor DNA in renal cell carcinoma. Cancer Sci. 2019, 110, 617–628. [Google Scholar] [CrossRef]
- Ball, M.W.; Gorin, M.A.; Guner, G.; Pierorazio, P.M.; Netto, G.; Paller, C.J.; Hammers, H.J.; Diaz, L.A.; Allaf, M.E. Circulating Tumor DNA as a Marker of Therapeutic Response in Patients With Renal Cell Carcinoma: A Pilot Study. Clin. Genitourin. Cancer 2016, 14, e515–e520. [Google Scholar] [CrossRef] [PubMed]
- Zengin, Z.B.; Weipert, C.; Salgia, N.J.; Dizman, N.; Hsu, J.; Meza, L.; Chehrazi-Raffle, A.; Muddasani, R.; Salgia, S.; Malhotra, J.; et al. Complementary Role of Circulating Tumor DNA Assessment and Tissue Genomic Profiling in Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2021, 27, 4807–4813. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.W.; Gill, D.M.; Maughan, B.; Agarwal, A.; Arjyal, L.; Gupta, S.; Streeter, J.; Bailey, E.; Pal, S.K.; Agarwal, N. Correlation of genomic alterations assessed by next-generation sequencing (NGS) of tumor tissue DNA and circulating tumor DNA (ctDNA) in metastatic renal cell carcinoma (mRCC): Potential clinical implications. Oncotarget 2017, 8, 33614–33620. [Google Scholar] [CrossRef]
- Braun, D.A.; Ishii, Y.; Walsh, A.M.; Van Allen, E.M.; Wu, C.J.; Shukla, S.A.; Choueiri, T.K. Clinical Validation of PBRM1 Alterations as a Marker of Immune Checkpoint Inhibitor Response in Renal Cell Carcinoma. JAMA Oncol. 2019, 5, 1631–1633. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Publisher Correction: Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 1941. [Google Scholar] [CrossRef]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [PubMed]
- Green, E.A.; Li, R.; Albiges, L.; Choueiri, T.K.; Freedman, M.; Pal, S.; Dyrskjot, L.; Kamat, A.M. Clinical Utility of Cell-free and Circulating Tumor DNA in Kidney and Bladder Cancer: A Critical Review of Current Literature. Eur. Urol. Oncol. 2021, 4, 893–903. [Google Scholar] [CrossRef]
- Gui, C.P.; Wei, J.H.; Zhang, C.; Tang, Y.M.; Shu, G.N.; Wu, R.P.; Luo, J.H. Single-cell and spatial transcriptomics reveal 5-methylcytosine RNA methylation regulators immunologically reprograms tumor microenvironment characterizations, immunotherapy response and precision treatment of clear cell renal cell carcinoma. Transl. Oncol. 2023, 35, 101726. [Google Scholar] [CrossRef]
- Li, H.; Jiang, H.; Huang, Z.; Chen, Z.; Chen, N. Prognostic Value of an m(5)C RNA Methylation Regulator-Related Signature for Clear Cell Renal Cell Carcinoma. Cancer Manag. Res. 2021, 13, 6673–6687. [Google Scholar] [CrossRef]
- Hu, M.; Xie, J.; Hou, H.; Liu, M.; Wang, J. Prognostic Value of DNA Methylation-Driven Genes in Clear Cell Renal Cell Carcinoma: A Study Based on Methylation and Transcriptome Analyses. Dis. Markers 2020, 2020, 8817652. [Google Scholar] [CrossRef]
- Popova, T.; Hebert, L.; Jacquemin, V.; Gad, S.; Caux-Moncoutier, V.; Dubois-d’Enghien, C.; Richaudeau, B.; Renaudin, X.; Sellers, J.; Nicolas, A.; et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am. J. Hum. Genet. 2013, 92, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Flaig, T.W.; Spiess, P.E.; Abern, M.; Agarwal, N.; Bangs, R.; Buyyounouski, M.K.; Chan, K.; Chang, S.S.; Chang, P.; Friedlander, T. NCCN Guidelines® Insights: Bladder Cancer, Version 3.2024: Featured Updates to the NCCN Guidelines. J. Natl. Compr. Cancer Netw. 2024, 22, 216–225. [Google Scholar]
- Directors, A.B.o. Clinical utility of genetic and genomic services: A position statement of the American College of Medical Genetics and Genomics. Genet. Med. 2015, 17, 505–507. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Altered Gene | WHO/ISUP Tumor Classification | Screening Recommendations | Management | Type of Operation | |
|---|---|---|---|---|---|
| VHL | VHL | Clear cell RCC | Contrast-enhanced cross-sectional imaging * every 2 years beginning at age 15 y | AS until largest tumor reaches 3 cm | Partial nephrectomy (enucleation) or ablative therapy + |
| HPRC | MET | Papillary RCC (formerly type I papillary RCC), Biphasic Squamoid Alveolar Papillary RCC (BSA PRCC) | Contrast-enhanced cross-sectional imaging * every 1–2 years beginning at age 30 y | AS until the largest tumor reaches 3 cm | Partial nephrectomy (enucleation) or ablative therapy + |
| HLRCC | FH | FH-deficient RCC (papillary); less common: chromophobe RCC | Contrast-enhanced cross-sectional imaging * annually beginning at age 8–10 y | Surgical management for any radiologically visible solid lesions & | Open partial nephrectomy with wide margin, consider radical for large lesions, and retroperitoneal lymph node dissection for large and/or complex lesions |
| BHD | FLCN | Most common: hybrid oncocytic and chromophobe RCC; less common: clear cell, oncocytoma, papillary RCC (formerly type I papillary) | Contrast-enhanced cross-sectional imaging * every 3 years beginning at age 20 y | AS until the largest tumor reaches 3 cm | Partial nephrectomy (enucleation) or ablative therapy + |
| TSC | TSC1 or TSC2 | Angiomyolipoma RCC: TSC-associated RCC with fibromyomatosis stroma, TSC-associated oncocytic tumor, eosinophilic solid and cystic tumor | Contrast-enhanced cross-sectional imaging * every 1–3 years beginning at age 12 y | AS until the largest tumor reaches 3 cm (RCC) and 4 cm (AML); angiography to determine the presence of intra-tumoral aneurysm | Tumor enucleation for RCC, selective angioembolization or tumor enucleation for AML or ablative therapy + |
| SDH | SDHAF2, SDHA, SDHB, SDHC, SDHD | SDH-deficient RCC (diverse histology pattern) | Contrast-enhanced cross-sectional imaging * every 2 years beginning at age 12 y, concurrently with paraganglioma screening | Surgical management for any radiologically visible solid lesions & | Partial nephrectomy with wide margin, consider radical for large lesions, open surgery for cystic lesions, retroperitoneal lymph node dissection for large and/or complex lesions |
| BAP1 | BAP1 | Clear cell RCC | Contrast-enhanced cross-sectional imaging * every 2 years beginning at age 30 y | Surgical management for any radiologically visible solid lesions | Partial nephrectomy (wedge) or radical nephrectomy |
| Translocation RCC | TFE3, TFEB, MITF, chromosome 3 | Molecularly defined translocation RCC with features similar to clear cell RCC | No recommended screening protocol reported; long-term follow-up after diagnosis | Surgical management for any radiologically visible solid lesions | Partial nephrectomy (wedge) or radical nephrectomy |
| ELOC | Elongin C mutated (formerly TCEB1) | Molecularly defined RCC with features similar to clear cell RCC | No recommended screening protocol reported | No currently recommended approach | No currently recommended approach |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millan, B.; Loebach, L.; Blachman-Braun, R.; Patel, M.H.; Saini, J.; Linehan, W.M.; Ball, M.W. Molecular Genetics of Renal Cell Carcinoma: A Narrative Review Focused on Clinical Relevance. Curr. Oncol. 2025, 32, 359. https://doi.org/10.3390/curroncol32060359
Millan B, Loebach L, Blachman-Braun R, Patel MH, Saini J, Linehan WM, Ball MW. Molecular Genetics of Renal Cell Carcinoma: A Narrative Review Focused on Clinical Relevance. Current Oncology. 2025; 32(6):359. https://doi.org/10.3390/curroncol32060359
Chicago/Turabian StyleMillan, Braden, Lauren Loebach, Ruben Blachman-Braun, Milan H. Patel, Jaskirat Saini, W. Marston Linehan, and Mark W. Ball. 2025. "Molecular Genetics of Renal Cell Carcinoma: A Narrative Review Focused on Clinical Relevance" Current Oncology 32, no. 6: 359. https://doi.org/10.3390/curroncol32060359
APA StyleMillan, B., Loebach, L., Blachman-Braun, R., Patel, M. H., Saini, J., Linehan, W. M., & Ball, M. W. (2025). Molecular Genetics of Renal Cell Carcinoma: A Narrative Review Focused on Clinical Relevance. Current Oncology, 32(6), 359. https://doi.org/10.3390/curroncol32060359