Abstract
Pancreatic ductal adenocarcinoma remains one of the most lethal solid tumors due to its local aggressiveness and metastatic potential, with a 5-year survival rate of only 13%. A robust connection between pancreatic cancer microenvironment and tumor progression exists, as well as resistance to current anticancer treatments. Pancreatic cancer has a complex tumor microenvironment, characterized by an intricate crosstalk between cancer cells, cancer-associated fibroblasts and immune cells. The complex composition of the tumor microenvironment is also reflected in the diversity of its acellular components, such as the extracellular matrix, cytokines, growth factors and secreted ligands involved in signaling pathways. Desmoplasia, the hallmark of the pancreatic cancer microenvironment, contributes by creating a dense and hypoxic environment that promotes further tumorigenesis, provides innate systemic resistance and suppresses anti-tumor immune invasion. We discuss the complex crosstalk among tumor microenvironment components and explore therapeutic strategies and opportunities in pancreatic cancer research. Better understanding of the tumor microenvironment and its influence on pancreatic cancer progression could lead to potential novel therapeutic options, such as integration of immunotherapy and cytokine-targeted treatments.
1. Introduction
Pancreatic cancer is the third leading cause of cancer-related death worldwide with an increasing incidence of approximately 1% annually in both sexes. PDAC is the most common histological subtype accounting for around 90% of pancreatic cancer. It is often associated with a poor prognosis due to late diagnosis and limited treatment options, with a 5-year OS rate of only 12–13% [1,2]. PDAC arises mainly from noninvasive precancerous lesions, mostly from microscopic PanIN, less frequently from IPMNs or MCN [3]. In the preneoplastic lesions, cells accumulate genetic and epigenetic alterations in oncogenes and tumor suppressor genes. Most common are alterations of the oncogene KRAS and the tumor suppressor genes CDKN2A, TP53, and SMAD4, triggering aberrant signaling cascades [4]. In addition to the extensive molecular alterations in neoplastic cells, the TME plays a crucial role in pancreatic tumorigenesis, tumor growth and metastasis as well as treatment resistance [5]. This review aims to elucidate the complex interactions within the tumor microenvironment and their therapeutic implications.
2. Characteristics of the Pancreatic Cancer Microenvironment
There is a growing interest in a better understanding of the tumor microenvironment, its role in resistance to systemic therapies and its potential as a target for novel therapeutic approaches. PDAC has a particularly prominent mesenchymal compartment within its stroma, which includes fibroblasts, ECM components, immune cells, nerves and endothelial cells. All these components might contribute to tumor progression [6] (Figure 1).
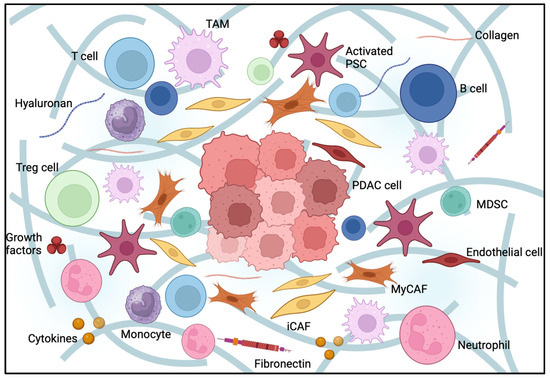
Figure 1.
Pancreatic cancer tumor microenvironment (TME). Pancreatic ductal adenocarcinoma TME composition. PDA stroma is comprised of cellular and acellular components, such as fibroblasts, myofibroblasts, pancreatic stellate cells, immune cells, blood vessels, extracellular matrix and soluble proteins, such as cytokines and growth factors. Interactions between these elements promote the desmoplastic structure, creating a physical barrier responsible for PDAC treatment resistance.
2.1. Desmoplasia
PDAC is characterized by an extensive fibrosis, termed desmoplasia, which promotes tumor progression and inhibits drug penetration and uptake [7]. In desmoplasia, the PSC transforms from a quiescent cell to that of an activated SMA expressing cell via the TGFβ and PGFGF signaling pathway, promoting ECM synthesis and deposition [7]. TGFβ has a dual nature: the tumor-intrinsic TGF-β plays a tumor suppressive function in the early stages through SMAD4-regulated genes, but the stromal-derived TGF-β promotes cancer growth and immunosuppressive mechanisms in the later stages, upon SMAD4 inactivation [8]. Desmoplasia lead to compression of microvasculature and overall hypo-vascularity in the TME, establishing a hypoxic microenvironment which contributes to pancreatic cancer aggressiveness [9]. In addition, pancreatic cancer cells can secrete antiangiogenic factors, such as angiostatin, endostatin and PEDF, supporting a hypoxic microenvironment [10,11]. Thus, desmoplasia is associated with a poor prognosis and its components might be potential therapeutic targets of pancreatic cancer.
2.2. Extracellular Matrix and Structural Proteins
The extracellular matrix consists of proteins, glycoproteins, proteoglycans, polysaccharides and soluble signaling molecules that provide a physical scaffold for its surrounding cells. The ECM can be divided into two matrices: the BM and the IM. The BM supports epithelial and endothelial cells and separates them from the IM, which makes up the main stroma and plays a major role in cell migration, cell adhesion, angiogenesis, tissue development and repair [12]. The major components of the basement membrane are the network-forming collagens, such as type IV and VIII collagen and, in the interstitial matrix, the fibrillar-forming collagens type I, II, III, V, XI, XXIV, XXVII and the beaded filament type VI collagen [13]. Cancer cells secrete MMPs, which physiologically regulate collagen production and assembly [14,15], leading to a complex of pro- and antitumor signals from BM’s degradation products [13]. These structural modifications contribute to increased stiffening of the PDAC tissue, influencing cellular behavior and regulating cell proliferation, differentiation, gene expression, migration, invasion and metastasis [16,17].
2.2.1. Hyaluronic Acid
HA is a matrix glycosaminoglycan in PDAC stroma, linked to elevated IFP. Given its large amount, HA has been identified as a promising potential target for therapeutic strategy. Hyaluronidase encapsulated by polyethylene glycol (PEGPH20) has been formulated to improve in vivo half-life and degrade HA. KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Cre) mice, a mouse model that recapitulates the human disease, treated with a single cycle of PEGPH20 in addition to the standard chemotherapy with gemcitabine experienced a substantial decrease in tumor volume, suggesting that reducing IFP enhances the tumor perfusion of chemotherapeutic agents [18]. A different study showed that PEGPH20 reduced vascular collapse and increased intratumoral delivery of gemcitabine in vivo, leading to an increase in animal survival compared with chemotherapy alone [19]. However, the administration of PEGPH20 combined with modified FOLFIRINOX (mFOLFIRINOX) resulted associated with a reduced survival in treatment-naïve patients with metastatic PDAC [20]. A phase III trial (HALO 301 study) [21] comparing gemcitabine/nab-paclitaxel with or without PEGPH20 in untreated patients with stage IV PDAC has been unsuccessful. Similarly, Atezolizumab plus PEGPH20 did not improve OS or PFS in previously treated patients with advanced pancreatic cancer [22]. Therefore, the available data on the use of this enzyme did not show a statistically significant clinical benefit in PDAC patients.
2.2.2. Fibronectin
FN, a large multidomain glycoprotein dimer assembled by cell-driven forces into a fibrillar array, is another component of TME. It serves as a scaffold for other matrix proteins and as a binding site for soluble factors within the tumor microenvironment. FN regulates proliferation and metastatic behavior of multiple cell types, largely through mechanisms involving integrin-mediated signaling [23]. Its induction during tumorigenesis can sustain proliferative signaling, promote angiogenesis, facilitate invasion and metastasis, modulate growth suppressor activity and regulate anti-tumoral immunity. Moreover, fibronectin plays a key role in the development of gemcitabine resistance through ERK1/2 activation. Thus, combining FN-blocking agents plus gemcitabine-based chemotherapy could potentially overcome chemoresistance in PDAC, providing better clinical outcomes [24].
2.3. Cells in Tumor Microenvironment
PDAC desmoplastic stroma is rich in cancer-associated fibroblasts (CAFs) and infiltrated by immune cells and their soluble functional molecules [7]. Exploring the biological and functional characteristics of TME is of great interest for identifying potential novel therapeutic options.
2.3.1. Cancer-Associated Fibroblasts
During tumor progression, CAFs are the main protagonists in dysregulating collagen turnover, leading to excessive collagen deposition and tumor fibrosis [25]. Due to their various origins, CAFs form a heterogeneous cell population with strong differences in morphology, cell–cell interaction and expression profile. CAFs express myofibroblast markers, such as α-SMA, vimentin, type XI collagen, fibronectin, FSP-1 and FAP [26], and promote tumor growth. Genome-wide studies have identified distinct subtypes of CAFs in PDAC: myo-fibroblastic (myCAFs) or inflammatory phenotypes (iCAFs). While the majority of fibroblasts express low levels of αSMA and high levels of FAP, a subpopulation of FAP+ cells show elevated αSMA expression. MyCAFs, characterized by high expression of α-SMA and low expression of IL6, are activated by direct contact between tumor cells and PSCs. They produce extracellular matrix components and promote stromal deposition [27]. iCAFs, with low α-SMA and high IL-6 levels and inflammatory features, are located farther away from tumor cells and activated by paracrine factors secreted from tumor cells. IL-6, IL-11, CXCL1, CXCL2 and LIF are upregulated in iCAFs, that specifically expressed HAS1 and HAS2, two enzymes responsible for the synthesis of hyaluronic acid [27]. Different pathways are enriched in iCAFs, including IFN-γ response, TNF/NF-κB, IL2/STAT 5 and IL6/JAK/STAT3 in humans. Il-1/LIF/JAK/STAT pathway could activate iCAFs, which in turn suppress the immune response and ECM deposition, accelerating the occurrence and progression of PDAC [28]. Furthermore, CAFs contribute to chemoresistance via various mechanisms. PAI-1, a cytokine produced by CAFs, activates Erk/Akt signaling and suppresses caspase-3 activation [29]. Similarly, interleukin 6 (IL6), also produced by CAFs, contributes to drug resistance mediating CXCR7 expression in tumor cells [30,31]. Especially under hypoxic conditions, CAFs produce high levels of TGFβ, conferring stem cell-like properties to tumor cells and increasing their chemoresistance [32]. Moreover, the complex ECM produced by CAFs interferes with therapeutic response by producing a protective barrier and interacting with integrins and cadherins [33,34,35,36,37]. Considering the heterogeneity of CAFs, understanding their functions might lead to develop specific strategies for each subgroup. It has been shown that chemoresistance driven by CD10+GPR77+CAFs [38] could be mitigated by the depletion of these cells with an anti-GPR77 antibody. Another approach might be reprogramming CAFs to a quiescent phenotype by inhibiting activating pathways, such as NFκb signaling, which has been shown to improve response to cisplatin in ovarian cancer xenografts [31]. Sherman et al. have demonstrated that activated CAFs also express vitamin D receptor. Calcipotriol, a vitamin D analog, can reverse CAFs back to stellate cells, normalize the TME and increase gemcitabine concentration in the tumor, resulting in improved response in PDAC models [39]. In addition, there is an invasive and migratory CAF phenotype associated with ROCK-pathway activation, that can be targeted with the ROCK inhibitor, fasudil. This has shown to reduce collagen deposition, enhance gemcitabine uptake, and improve treatment response in a transgenic PDAC model [40].
2.3.2. Pancreatic Stellate Cells
During PDAC, quiescent pancreatic stellate cells are activated and transformed into the myofibroblast-like phenotype and secrete molecules like TGFβ, IL-6, SDF-1, HGF and galectin-1, involved in pancreatic cancer progression [41]. Furthermore, PSCs can also induce desmoplasia via numerous signaling pathways, including IL-6, paracrine SHH signaling, VDR pathway and CXCL12/CXCR4 pathway [42]. The CXCL12/CXCR4 axis plays a critical role in promoting cancer cell proliferation, migration, invasion, angiogenesis and metastasis. Combined therapies with CXCR4 antagonists, such as CXCL14, have shown great promise in preclinical models, but most of the studies investigated drug administration early in the disease course, which does not always reflect clinical reality [43]. Another potential drug target is SHH signaling. Induced by KRAS via NF- kB signaling, pancreatic cancer cells express SHH to activate the target gene GLI1 and create a tumor-supportive microenvironment [44]. SHH signaling activates PSCs into activated myCAFs [45], affecting the ratio of myCAFs/iCAFs in PDAC tumors, and influences the expression of inflammatory signaling molecules from the iCAF population and the immune cell infiltration in TME. Although well-known Hh signaling antagonists, such as vismodegib (GDC-0449), have been investigated, they have not yielded clinically meaningful results [46,47]. Available data suggest that ormeloxifene, a non-hormonal, nonsteroidal oral contraceptive molecule, has a property of inhibiting the SHH pathway and, in combination with gemcitabine, could serve as a novel therapeutic strategy for PDAC [48].
2.3.3. Endothelial Cells
Solid tumors are commonly characterized by an abnormal vasculature. In response to hypoxia due to poor vasculature, tumors usually activate mechanisms to stimulate angiogenesis, supporting tumor growth and metastasis. Pancreatic cancer cells and other immunosuppressive cells in the tumor microenvironment induce angiogenesis by secreting pro-angiogenic factors, cytokines and growth factors, such as VEGF, regulated by multiple signaling pathways. STAT3 expression in pancreatic cancer cells activates VEGF expression, promoting angiogenesis [49]. Similarly, MUC1 induces hypoxia, stimulating VEGF-A and PDGF-B production and contributing to endothelial cell tube formation [50]. Additionally, activated NF-κB in pancreatic cancer cells can upregulate VEGF [51]; this pathway can be suppressed by xanthohumol, with subsequent angiogenesis inhibition [52]. Although therapeutic agents that inhibit angiogenesis or normalize the tumor vasculature are not part of the standard of care in the treatment of PDAC [53], some patients might benefit from such approaches. For example, mice with highly vascular PDAC have shown modest but significant benefit in overall survival with VEGF receptor inhibition [54,55]. Regarding lymphatic vessels, they play a fundamental role not only in transporting tumor antigens to draining lymph nodes for presentation and activation of tumor-reactive T cells by leukocytes, but also because they represent the main dissemination route in PDAC [56,57]. Lymphatic vasculature contribution to PDAC pathogenesis remains poorly understood and is likely to be complex. Chemokines are involved in lymph-angiogenesis and cell migration. For example, CCL21, secreted by lymphatic endothelial cells, attracts dendritic cells to lymph nodes via lymphatics, while tumor cells expressing CCR7 may exploit this mechanism to disseminate to lymph nodes [58,59,60]. Similarly, CXCL12 produced in lymph nodes may attract CXCR4-expressing cancer cells or leukocytes [61,62]. Thus, tumor-associated lymph-angiogenesis is a critical step for pancreatic cancer progression, especially for lymph node metastasis.
3. Neural Invasion in Pancreatic Cancer
NI consists of the presence of cancer cells along nerves and/or within the epineural, perineural and endo-neural spaces [63]. Epineural invasion is characterized by cancer cells that directly touch the epineurium without penetrating it; differently, in perineural and endo-neural invasion, cancer cells invade the perineural and the endo-neural sheet, respectively [63,64,65]. NI, particularly endo-neural invasion, correlates with increased neuropathic and inflammatory pain, which significantly impacts patient outcomes [65]. There is a reciprocal interaction between tumor and nerve cells that creates a microenvironment favorable to tumor growth. Nerve-secreted substances promote tumor invasion and growth, while tumor-secreted molecular mediators stimulate nerve axons’ growth toward tumor cells [66], supporting tumor development and influencing its microenvironment [67]. Additionally, tumor induced-neural sprouting enhances a cancer cell–nerve crosstalk, known as axonogenesis [68]. SCs, the principal glial cells of the peripheral neural system [69], regulate neural growth and pain signaling through the production of a variety of growth factors and pain-related molecules, such as the NGF, the GDNF and the BDNF [70]. Neurotrophin antibodies, including anti-NGF, anti-BDNF, anti-NT-3, and anti-NT-4/5, have shown promising results in preclinical studies [71]. A recent phase III study has demonstrated the efficacy and safety of tanezumab, a monoclonal antibody anti-NGF, in patients with cancer pain predominantly caused by bone metastasis, who were already on opioid therapy. However, the long-term efficacy of anti-NGF therapy beyond 8 weeks remains unclear; therefore, future research should focus on evaluating the extended therapeutic benefits of this treatment [72]. Lastly, understanding the molecular mechanisms behind NI could potentially lead to innovative treatments that improve outcomes and quality of life of patients.
4. Immunosuppression
The correlation between anti-tumor immunity and pancreatic cancer progression has been the subject of several studies. The immune system has a dual activity: it prevents the transformation of mutated cells into tumor cells and promotes pancreatic cancer progression by creating favorable conditions for immunosuppression and metastasis [73,74]. Tumor immunity plays an important role in cancer patient prognosis, as evidenced by the presence of CD8+ TILs involved in killing tumor cells [75,76]. Normally, APCs, such as macrophages and DCs, process tumor antigens and display them on MHC I molecules. Consequently, CD8+ T cells are activated to kill tumor cells via the granzyme, perforin, and Fas/FasL (Fas ligand) pathways. On the other side, pancreatic cancer cells promote immunosuppression by inhibiting CD8+ T cell activation and decrease HLA class I expression, escaping the tumor infiltration by cytotoxic T-cells [77]. Furthermore, pancreatic cancer cells can evade Fas-mediated immune surveillance by expressing a nonfunctional FasR (Fas receptor) to resist Fas-mediated apoptosis and by expressing functional FasL to induce apoptosis in Fas-sensitive activated T-cells [78].
5. Immunosuppressive Cells
Tumors have developed strategies to successfully evade the host immune system, utilizing various molecular and cellular mechanisms. In PDAC, MDSCs, TAMs, and Tregs contribute to establishing an immunosuppressive TME [79].
5.1. Myeloid-Derived Suppressor Cells (MDSCs)
MDSCs, a heterogeneous population of immature myeloid cells in spleen and tumor, play a critical role in immunosuppression. In the GEMM and orthotopic mouse model of PDAC, myeloid cells, including MDSCs and TAMs, are the most abundant immune cells [80]. MDSCs are characterized by the expression of CD11b and CD33 and peripheral blood MDSCs level may be a predictive biomarker of chemotherapy failure in pancreatic cancer patients [81]. Pancreatic cancer induces the proliferation and mobility of MDSCs from the bone marrow to the tumor microenvironment [82] via cytokines, especially GM–CSF, which promotes the differentiation of myeloid progenitor cells into MDSCs [83]. MDSCs exert their immunosuppressive function through different pathways. For instance, they release ROS, under the influence of cytokines, such as TGFβ and IL-10, inducing oxidative stress in T cells and inhibiting the CD3ζ chain translation [84]. PMN-MDSCs (polymorphonuclear MDSCs) take up, process and present antigens on MHC I proteins. These antigen-MHC I complexes present antigens to CD8+ T cells, inducing immune tolerance and facilitating immune evasion [85]. Furthermore, MDSCs express enzymes. like Arg1 and iNOS, which deplete L-arginine, an essential amino acid involved in T cells’ proliferation and activation [86]. The STAT3 signaling pathway, activated by IL-10, not only induces Arg1 expression but also upregulates PD-L1 expression on MDSCs [87], further suppressing T cell activation.
5.2. Tumor-Associated Macrophages (TAMs)
Macrophages are phagocytic cells involved in the innate immune system. The PanIN regulates macrophage function via IL-13, polarizing them from M1 to M2 phenotype, which shows immunosuppressive function and promotes tumor growth [88]. In the pancreatic cancer microenvironment, various factors, such as CSF-1, IL-4, IL-13, TGFβ and IL-10, promote myeloid progenitor cell differentiation into monocytes and macrophages [89,90], which in turn secrete immunosuppressive cytokines, chemokines and enzymes, such as TGFβ, IL-10, CCL17, and CCL22 [91]. TAMs express high levels of Arg1, interfering with the metabolism of effector T cells [92], support regulatory Treg recruitment and inhibit CD8+ T cells [93]. Additionally, NLRP3 signaling in macrophages promotes the differentiation of CD4+ T cells into tumor-promoting Th2 cells, Th17 cells and Tregs, while suppressing Th1 cell polarization [94]. TAMs also contribute to the development of desmoplasia. In fact, M2 macrophages promote pancreatic fibrosis [95] and, overall, macrophages drive fibrosis, immunosuppression and metastasis through the PI3Kγ pathway [96]. Lastly, TAMs can stimulate PSC proliferation and ECM secretion via TGFβ1 and PDGF respectively [97], involved in multiple steps of pancreatic cancer progression (Figure 2).
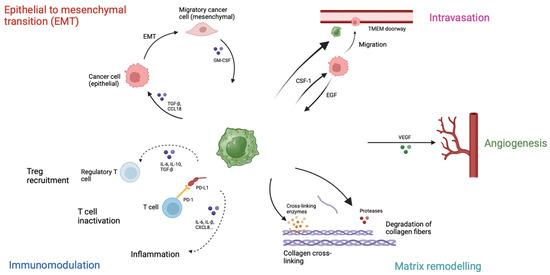
Figure 2.
Role of Tumor-associated macrophages (TAMs). TAMs inactivate cytotoxic T cells through PD-L1 expression, produce cytokines to recruit regulatory T cells and create an inflammatory environment. These macrophages are involved in extracellular matrix remodeling by producing proteases, such as metalloproteinases, that degrade collagen fibers, and cross-linking enzymes, which contribute to the stiffness of the extracellular matrix. Furthermore, TAMs secrete VEGF and promote angiogenesis, which is involved in tumor progression and metastasis. Tumor-associated macrophages migrate with cancer cells to blood vessels and create TMEM (tumor microenvironment of metastasis) doorways, permitting cancer cells to disseminate in the circulation. Lastly, TAMs play a role in epithelia to mesenchymal transition by producing TGF-β and CCL18, allowing cancer cells to migrate.
5.3. Tumor-Associated Neutrophils (TANs)
Only limited information is available on the roles of TANs. In cancer patients, peripheral blood neutrophils level and circulating neutrophil-to-lymphocyte ratio represent biomarkers of poor clinical outcome [98]. Neutrophils are attracted to the TME in response to TNF-α and IL-12 secreted by cancer cells. Via TGF-β signaling, TANs are polarized into the N1 subtype, which inhibits cancer growth by activating CD8+ T cells through releasing TNF-α, CCL-3, CXCL-9, and CXCL-10 [99]. In contrast, Treg-derived IL-35 induces the polarization of neutrophils into the N2 subtype, which has a pro-carcinogenic role due to various secreted molecules [100]. N2 neutrophil-derived ROS, RNS and NE contribute to carcinogenesis and promote cancer progression [101]. In addition, neutrophils produce HGF and MMPs, which facilitate angiogenesis, tumor invasion and metastasis [102,103].
5.4. Regulatory T Cells (Tregs)
Tregs, also known as suppressor T cells, are a subtype of T cells that play an important role in maintaining tolerance to self-antigens and preventing autoimmune disease by downregulating or suppressing effector T cells [104]. CD4+ CD25+ Tregs, recruited into the TME by pancreatic cancer cells, perform a significant role in immunosuppression during pancreatic cancer progression. However, the precise mechanism of their immunomodulatory functions in PDAC remains unclear. A high level of Tregs is associated with poor prognosis and low survival rate [105]. Intratumor Tregs express elevated levels of CTLA-4 and PD-1 compared to Tregs in pancreatic lymph nodes (Pan LNs) and peripheral inguinal lymph nodes (iLNs) [106]. The impact of Treg depletion has been investigated in GEMMs, revealing a pro-carcinogenic effect mediated by Tregs interaction with tumor-associated CD11+ DCs [106]. Prolonged binding between intratumor Tregs and CD11+ DCs selectively impacts CD8+ T cell activation, leading to reduced levels of IFN-γ, CD44, and Granzyme B, as well as decreased tumor size and increased overall survival [103]. Conversely, Treg depletion has been found to induce myeloid cell recruitment into the TME via a CCR1-dependent pathway, resulting in suppressed anti-tumor immunity [107]. Furthermore, Tregs demonstrated plasticity towards Th17 cells (Foxp3+RORγ+), promoting cancer growth and evasion through the production of IL-17, IL-23 and TGF-β [107]. However, an alternative study suggest that Treg depletion induces increased expression of chemo-attractants to myeloid cells (CCL3, CCL6, CCL8) and immune-suppression genes in fibroblasts (PD-L1, Arg1), consequently reprogramming myofibroblasts to an inflammatory phenotype and enhancing their pro-carcinogenic function [108]. Overall, Tregs exhibit complex crosstalk with CAFs and immune compartments within the TME, resulting in both pro- and anti-tumor effects in desmoplastic cancers such as PDAC.
6. Secreted Signaling Molecules
In TME, different subpopulations of cells secrete various signaling molecules, typically TGF-β or pro-inflammatory cytokines, altering the tumor microenvironment (Table 1).

Table 1.
Pro-inflammatory cytokines. Cytokines, low-molecular-weight proteins that regulate many biological processes, play a dual role in cancer: they modulate the tumor microenvironment and directly affect cancer cells. CAF cancer associated fibroblast; DC dendritic cell; EMT epithelial to mesenchymal transition; PSCs pancreatic stellate cells; TAM tumor-associated macrophage; NEU neutrophil. VEGF vascular endothelial growth factor; TGFβ transforming growth factor beta; IL interleukin; TNF tumor necrosis factor; PDAC pancreatic ductal adenocarcinoma.
6.1. Interleukin 8 (IL-8)
Interleukin 8 (IL-8), a strong pro-inflammatory cytokine, could serve as a good marker in predicting the prognosis of patients [109] and mainly acts as an angiogenic factor, associated with an increased production of VEGF, VEGFR (VEGF receptor) and NRP-2, which are crucial for angiogenesis in growing pancreatic tumor [109]. It is also involved in the MAPK pathway by enhancing the phosphorylation of ERK, implicated in cell growth, survival and tumorigenesis [110]. The TRAIL can selectively induce apoptosis, engaging its receptors TRAIL-R1 and TRAIL-R2. This leads to increased expression of uPA and IL-8, involved in invasion, metastasis and tumor progression, especially in cells overexpressing TRAF2 or Bcl-xL. However, TRIAL-receptors are also implicated in non-apoptotic pathways and it was demonstrated that non-apoptotic signaling in PDAC cells is mediated by TRAIL-R1 and enhanced by overexpression of TRAF2 or Bcl-xL [111]. Based on these data, inhibition of Bcl-xL and TRAF2 could represent a targeting strategy, improving the TRAIL-based therapy in the treatment of PDAC [112].
6.2. Interleukin 6 (IL-6)
Interleukin 6 (IL-6) is another interleukin involved in inflammation, cell proliferation and differentiation, and its gene polymorphisms are associated with pancreatic cancer. IL-6 activates JAK, a tyrosine kinase, which in turn activates MAPKs, PI3Ks and STATs signaling pathways [113]. IL-6 cooperates with oncogenic K-Ras to activate a detoxification program for ROS via the MAPK/ERK signaling cascade [114]. This detoxification process is particularly important because it enables cancer cells to survive in stressful environments and resist apoptosis. IL-6 activates the gp130 receptor, which subsequently leads to Jak2 activation. Activation of the oncogenic JAK2/STAT3 pathway enhances REG3A expression, accelerates cell cycle progression by increasing CyclinD1 expression and upregulates the anti-apoptosis Bcl-2 family [115]. Importantly, REG3A activation further enhances the JAK2/STAT3 pathway, forming a positive feedback loop that amplifies the oncogenic effects of the IL-6/JAK2/STAT3 pathway, linked to inflammation-related tumorigenesis [116]. Additionally, Jak2 phosphorylates and activates STAT3. Studies have shown that the inhibition of IL-6/STAT3 signaling pathway reduces EMT and inflammation in pancreatic cancer cells [114]. For example, the STAT3 inhibitor NSC74859 opposes PSC-induced migration and EMT-related markers (Snail and cadherin-2) expression in pancreatic cancer cells [115]. In addition, the IL-6/STAT3 signaling pathway can methylate the SOCS3 via DNMT1, leading to pancreatic cancer progression and metastasis. Therefore, targeting STAT3 or DNMT1 might represent a potential strategy in the treatment of pancreatic cancer [117]. For example, embelin, an anti-inflammatory and anti-tumor drug, reduces cell proliferation and induces apoptosis by inhibiting STAT3 pathway and activating p53 signaling [118]. Lastly, combination therapy with IL-6 blockade could modulate immunological features of PDAC and enhance the effectiveness of anti-PD-L1 (anti-programmed death-1-ligand 1) checkpoint inhibitors [119].
6.3. Interleukin 1β (IL-1β)
IL-1β, another pro-inflammatory cytokine, involved in tumor growth and metastasis, activates NF-κB, upregulates COX-2 and leads to chemoresistance in pancreatic adenocarcinoma [120,121]. Its expression inhibits components of the integrin signaling pathway, such as vinculin and α5-integrin, via a JNK-dependent mechanism, promoting cells’ migratory potential [122]. Specifically, the IL-1β -511CT/-31TC genotype significantly correlates with an elevated risk for pancreatic cancer, particularly in advanced and undifferentiated tumors [123]. IL-1β also recruits proangiogenic macrophages, which explains why using an IL-1β antagonist can reduce tumor inflammation, angiogenesis and growth [124]. Additionally, IL-1R (interleukin 1 receptor) levels are elevated in pancreatic cancer, making this receptor a potential therapeutic target. Sandberg et al. demonstrated that the administration of IL-1β with IL-1RA counteracted IL-1β effects in mouse models, as evidence of IL-1RA inhibitory activity [125,126]. Z-360, a CCK2/gastrin receptor antagonist, in combination with gemcitabine, extended the survival period in a lethal pancreatic cancer xenograft mouse model [127]. Furthermore, a phase Ib/IIa clinical trial reported pain improvement in advanced pancreatic cancer patients treated with Z-360 plus gemcitabine. These findings underscore the potential therapeutic efficacy of targeting IL-1β and associated pathways in PDAC treatment.
6.4. Tumor Necrosis Factor-Alpha (TNF-α)
TNF-α, a pro-inflammatory cytokine, belongs to the TNF family, which also includes TNF-β, CD40L (CD40 ligand), TRAIL and FasL. In pancreatic cells, TNF-α activates NF-κB [128,129], inhibiting apoptosis, and increases PDGF expression, which strongly stimulates fibrogenesis during early pancreatic inflammation [130]. At high doses, TNF-α exhibits a cytotoxic effect on tumor cells, mediated by TNF-R1. However, in the pancreatic cancer microenvironment, TNF-α binds to TNF-R2, leading to upregulation of the EGFR and subsequent cancer cell proliferation [131]. TNF-R1 is prevalent in tumor and stromal cells, whereas TNF-R2 is generally present on leukocyte infiltrates. Once released, TNF-α binds to TNF-R1 and targets cells responsible for the production of inflammatory cytokines such as IL-6, IL-8, and IL-10 [132]. Several studies have explored targeting TNF-α for pancreatic cancer treatment. In a phase II study, thalidomide reduced TNF levels and improved weight after eight weeks of treatment compared to placebo [133]. Another phase II trial combined thalidomide with capecitabine in advanced or metastatic pancreatic cancer patients, inhibiting TNF mRNA stability and TNF production. This combination reported a median progression-free survival (PFS) of 2.7 months and a median overall survival (OS) of 6.1 months [134]. However, a recent phase I/II study evaluated the efficacy and tolerability of gemcitabine and etanercept, a TNF-α blocker, in 38 patients with advanced pancreatic cancer, but there was no significant improvement in survival [135]. Overall, although preclinical and early clinical trials have not yet successfully targeted TNF-α in cancer treatment, it remains a promising target for future research.
6.5. Interleukin 10 (IL-10)
IL-10 is a homo-dimeric polypeptide chain, expressed by various cells, influencing the expression of cytokines, cell surface molecules and soluble mediators, to activate and sustain immune and inflammatory responses. Generally considered an immunosuppressive cytokine, IL-10 inhibits the production of various cytokines, like IL-1α, IL-1β, IL-6, IL-12, IL-18, TNF, LIF and PAF, by activated monocytes/macrophages [136,137,138]. Additionally, IL-10 is linked with M2-polarized TAMs and their infiltration intensity correlates with pancreatic cancer prognosis. In pancreatic cancer cells, M2-polarized TAMs promote EMT through TLR4/IL-10 signaling, which in turn enhances proliferation, migration and the proteolytic activity of MMP2 and MMP9 [139]. On the other hand, evidence indicates that IL-10 also possesses immunostimulatory and anti-tumor properties [140], including the inhibition of angiogenesis and matrix metalloproteinase 2, thereby inhibiting metastasis [141]. Clinical studies have shown IL-10 upregulation in pancreatic cancer and its correlation with tumor stage and prognosis [142]. Thus, IL-10 might be a potential biomarker for predicting pancreatic cancer prognosis.
7. PDAC Therapeutics and Tumor Microenvironment
Over the past few decades, the effectiveness of PDAC treatments in the clinic has not yet improved significantly. However, the increasing understanding of PDAC biology is paving the way for novel therapeutic strategies that target the components of the TME.
7.1. Targeting the Immunosuppressive Mechanism
PDACs are generally considered as immunologically “cold” tumors, indicating that they do not induce active anti-tumor immune responses, except for MSI-high subtype [143,144]. This immunological ‘coldness’ involves several factors, including an immunosuppressive microenvironment characterized by the expression of PD-L1 and IDangioO1 and a low tumor mutation burden that limits neo-antigenicity [145,146]. Additionally, a dense desmoplastic stroma acts as a physical barrier to immune cell infiltration, playing an important role in the limited responses to immune checkpoint inhibitors with the failure of immunotherapy [147,148]. The components of the immunosuppressive system are potential therapeutic targets. Tregs are one of the most promising candidates and can be selectively depleted administering low doses of cyclophosphamide [149] which, in combination with GVAX, a GM-CSF gene-transfected tumor cell vaccine, showed an enhanced immune response to PDAC [150]. As with many solid tumors, ICIs have been tested in PDAC treatment, primarily targeting CTLA-4 and PD1/PD-L1, but we only have safety data available, making further evaluation of efficacy necessary. For example, the combination of anti-CD40 monoclonal antibody with gemcitabine, with or without anti-PD-1 antibodies, has shown promising outcomes [151]. Thus, significant research is needed in order to investigate potential remarkable benefits offered by immunotherapy in PDAC, as observed in other types of cancers.
7.2. Inhibition of Monocyte/Macrophage Recruitment
There is a significant interest in targeting TAMs due to their role in tumor initiation, invasion and metastasis. Targeting the CCL2/CCR2 axis, which plays a crucial role in recruiting monocytes in various cancer types such as pancreatic cancer [152], offers a promising therapeutic approach to hinder the recruitment of monocytes from the bone marrow into the bloodstream [153]. For example, carlumab, an antibody against CCL2, has been tested in clinical trials [154,155] and using carlumab in association with conventional chemotherapy demonstrated preliminary antitumor activity in patients with advanced cancer [156]. Furthermore, PF-04136309, a CCR2 inhibitor, was shown to reduce CCR-2+/CD14+ monocytes and macrophages in the pre-metastatic liver and primary pancreatic tumor. In a phase 1b trial, PF-04136309 administered in combination with FOLFIRINOX chemotherapy proved to be safe, tolerable and recommended for patients with pancreatic cancer [157].
7.3. Inhibition of Macrophage Activation
In macrophage activation, CSF-1/CSF-1R signaling plays a crucial role. The use of CSF-1R inhibitor or CSF-1/CSF-1R neutralizing antibody (e.g., PLX6134/PLX3397) represents one of the most advanced methods for targeting macrophage activation [158,159]. Additionally, combining a CSF-1R signaling antagonist with paclitaxel has been shown to enhance the survival of mice with mammary tumors by slowing primary tumor growth and reducing pulmonary metastasis [160]. In glioblastoma mouse models, inhibition of CSF-1R resulted in significant reduction of tumor size and long-term survival [161,162]. Ries et al. demonstrated that treating animal models with a monoclonal antibody (RG7155) significantly reduced F4/80+ TAMs and increased the CD8+/CD4+ T cell ratio. They also found that administering RG7155 to patients led to a substantial depletion of CSF-1R+CD163+ macrophages in tumor tissues, correlating with clinical responses in patients with diffuse-type giant cell tumor [158]. Furthermore, tyrosine kinase receptor inhibitors regulate the immunosuppressive and tumorigenic functions of TAMs [163,164]. Erlotinib in combination with gemcitabine showed a statistically significant, but not clinically improved, survival in advanced pancreatic cancer [165]. Otherwise, in the LAPO7 trial, which evaluated the administration of chemoradiotherapy after induction chemotherapy in locally advanced pancreatic cancer, no differences in OS and PFS were shown with gemcitabine compared to gemcitabine plus erlotinib [166]. Therefore, the inhibition of CSF-1R in vivo demonstrated a decrease in pancreatic tumor-initiating cells and improved chemotherapy efficacy [80], and the interruption CSF-1/CSF-1R signaling eliminated CD206hi TAMs within pancreatic tumors, reprogramming the remaining macrophages to support antitumor immunity [167]. These findings support the role of CSF-1/CSF-1R signaling as an effective therapeutic target for reprogramming the immunosuppressive microenvironment in PDAC. More studies targeting CSF-2/CSF-2R signaling are needed to assess the potential of immunotherapy in pancreatic cancer treatment.
7.4. Cancer Vaccine
PDACs can evade the immune system through various mechanisms, each of which presents an opportunity for therapeutic development. One approach involves addressing the limited cancer antigen presentation by exploring different cancer vaccine strategies, such as peptide-based, virus-based, DNA-based and cell-based vaccines. Despite positive outcomes in phases I and II, some vaccine strategies, including Telovac, PANVAC and algenpantucel-L, failed in phase III trials [168]. Notably, the GVAX trial, which used a tumor cell vaccine transfected with the granulocyte-macrophage colony-stimulating factor (GM-CSF) gene, demonstrated efficacy in mobilizing effector immune cells into the TME. Consequently, treatment strategies that combine GVAX with other therapies are still being pursued [169]. Therefore, personalized RNA neo-antigen vaccines hold a new hope for more efficient pancreatic cancer treatment due to their ability to activate T cells [170].
8. Conclusions
The tumor microenvironment is characterized by high heterogeneity, with cancerous cells, immune cells and CAFs, as well as acellular components. We explored the relationship between cellular populations and acellular factors and the important role it has in tumor growth, metastasis and drug resistance. Pancreatic cancer cells produce signaling molecules, recruiting or activating stromal and immunosuppressive cells, such as Tregs, MDSCs, TAMs, and PSCs, within the microenvironment. Over the past two decades, attention has focused on understanding the complex stromal microenvironment of PDAC and its potential impact on cancer treatment. However, further research is needed to understand the precise mechanisms underlying TME–tumor interactions and improve the efficacy of novel therapeutic strategies. Moreover, future directions will be more focused on multiple approaches, targeting different aspects of tumors, to improve patient outcomes and advance a personalized medicine approach to PDAC.
Author Contributions
Conceptualization, I.G. and F.P.; methodology, I.G. and F.P.; resources, I.G. and F.P.; writing—original draft preparation, I.G. and F.P.; writing—review and editing, I.G. and F.P.; supervision, I.G.; funding acquisition, I.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by European Organization for Research and Treatment of Cancer (EORTC RP-2146).
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results. The authors declare no conflicts of interest.
Abbreviations
APC, antigen-presenting cell; Arg, arginase; BDNF, brain-derived neurotrophic factor; BM, basement membrane; CAF, cancer-associated fibroblast; CCK, cholecystokinin; CXCL12, CXC chemokine ligand 12; CXCR4, CXC chemokine receptor type 4; DC, dendritic cell; DNMT, DNA Methyltransferase; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; EMT, epithelial-mesenchymal transition; FAP, fibroblast activating protein; FAS, first apoptosis signal; FN, fibronectin; FSP-1, fibroblast-specific protein 1; GDNF, glial cell line-derived neurotrophic factor; GEMM, genetically engineered mouse model; GM-CSF, granulocyte macrophage colony-stimulating factor; HA, Hyaluronic acid; HAS, hyaluronic acid synthase; HGF, hepatocyte growth factor; HLA, human leukocyte antigen; ICI, immune checkpoint inhibitor; IFN-γ, interferon γ; IFP, interstitial fluid pressure; IM, interstitial matrix; iNOS, inducible nitric oxide synthase; IPMN, intraductal papillary mucinous neoplasm; JAK, Janus kinase; LIF, leukemia inhibitory factor; MCN, mucinous cystic neoplasm; MDSC, myeloid-derived suppressor cell; MHC I, major histocompatibility complex I; MMP, metalloproteinase; MUC1, Mucin 1; NE, neutrophil elastase; NF-κB, nuclear factor kappa-B; NGF, nerve growth factor; NI, neural invasion; NRP-2, neuropilin-2; OS, overall survival; PEGPH20, pegylated recombinant human hyaluronidase 20; PSC, pancreatic stellate cell; PanIN, pancreatic intraepithelial neoplasia; PDAC, pancreatic ductal adenocarcinoma; PEDF, pigment epithelium-derived factors; PGFGF, platelet-derived growth factor; RNS, reactive nitrogen species; ROS, reactive oxygen species; SC, Schwann cell; SDF-1, stromal cell-derived factor-1; SHH, sonic hedgehog; SMA, smooth muscle actin; SOCS3, suppressor of cytokine signaling 3; STAT, signal transducer and activator of transcription; TAM, tumor-associated macrophage; TAN, intratumoral neutrophil; TGFβ, transforming growth factor-beta; Th cell, T helper cell; TIL, tumor-infiltrating lymphocyte; TLR, Toll-like receptor; TME, tumor microenvironment; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand; Treg, regulatory T cell; uPA, urokinase-type plasminogen activator; VDR, vitamin D receptor; VEGF, vascular endothelial growth factor.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Garajová, I.; Peroni, M.; Gelsomino, F.; Leonardi, F. A Simple Overview of Pancreatic Cancer Treatment for Clinical Oncologists. Curr. Oncol. 2023, 30, 9587–9601. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e1. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Dunne, R.F.; Hezel, A.F. Genetics and Biology of Pancreatic Ductal Adenocarcinoma. Hematol. Clin. North Am. 2015, 29, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Werba, G.; Lyssiotis, C.A.; Simeone, D.M. The Biological Underpinnings of Therapeutic Resistance in Pancreatic Cancer. Genes Dev. 2021, 35, 940–962. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef]
- Iglesias, M.; Frontelo, P.; Gamallo, C.; Quintanilla, M. Blockade of Smad4 in Transformed Keratinocytes Containing a Ras Oncogene Leads to Hyperactivation of the Ras-Dependent Erk Signalling Pathway Associated with Progression to Undifferentiated Carcinomas. Oncogene 2000, 19, 4134–4145. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Kurtoglu, M.; Kleeff, J. The Role of Hypoxia in Pancreatic Cancer: A Potential Therapeutic Target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 301–316. [Google Scholar] [CrossRef]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-Stellate Cell Interactions Perpetuate the Hypoxia-Fibrosis Cycle in Pancreatic Ductal Adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef]
- Samkharadze, T.; Erkan, M.; Reiser-Erkan, C.; Demir, I.E.; Kong, B.; O Ceyhan, G.; Michalski, C.W.; Esposito, I.; Friess, H.; Kleeff, J. Pigment Epithelium-Derived Factor Associates With Neuropathy and Fibrosis in Pancreatic Cancer. Am. J. Gastroenterol. 2011, 106, 968–980. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer Associated Fibroblasts in the Reactive Stroma and Its Relation to Cancer Biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the Extracellular Matrix in Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.D.; Joyce, J.A. Proteolytic Networks in Cancer. Trends Cell Biol. 2011, 21, 228–237. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. Embo Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic Analyses of ECM during Pancreatic Ductal Adenocarcinoma Progression Reveal Different Contributions by Tumor and Stromal Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Ko, A.H.; Kim, K.-P.; Siveke, J.T.; Lopez, C.D.; Lacy, J.; O’reilly, E.M.; Macarulla, T.; A Manji, G.; Lee, J.; Ajani, J.; et al. Atezolizumab Plus PEGPH20 Versus Chemotherapy in Advanced Pancreatic Ductal Adenocarcinoma and Gastric Cancer: MORPHEUS Phase Ib/II Umbrella Randomized Study Platform. Oncology 2023, 28, 553-e472. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, G.; Saint, A.; Ruff, M.; Rekad, Z.; Ciais, D.; Van Obberghen-Schilling, E. Shaping Up the Tumor Microenvironment With Cellular Fibronectin. Front. Oncol. 2020, 10, 641. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of Fibronectin by Human Pancreatic Stellate Cells Promotes Chemoresistance to Gemcitabine in Pancreatic Cancer Cells. BMC Cancer 2019, 19, 596. [Google Scholar] [CrossRef] [PubMed]
- Pankova, D.; Chen, Y.; Terajima, M.; Schliekelman, M.J.; Baird, B.N.; Fahrenholtz, M.; Sun, L.; Gill, B.J.; Vadakkan, T.J.; Kim, M.P.; et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol. Cancer Res. 2016, 14, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and Mechano-Regulation of Connective Tissue Remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-Activated PAI-1 Secretion in the Cancer-Associated Fibroblasts with Paracrine Effects Promoting Esophageal Squamous Cell Carcinoma Progression and Causing Chemoresistance. Cell Death Dis. 2018, 9, 759. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 Derived from Cancer-Associated Fibroblasts Promotes Chemoresistance via CXCR7 in Esophageal Squamous Cell Carcinoma. Oncogene 2017, 37, 873–883. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Z.; Jin, P.; Yang, X.; Li, X.; Wei, X.; Wang, Y.; Long, S.; Zhang, T.; Chen, G.; et al. Metformin Suppresses Tumor Progression by Inactivating Stromal Fibroblasts in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-A.; Chen, Y.-F.; Bao, Y.; Mahara, S.; Yatim, S.M.J.M.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic Tumor Microenvironment Activates GLI2 via HIF-1α and TGF-β2 to Promote Chemoresistance in Colorectal Cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Koch, U.; Hehlgans, S.; Sandfort, V.; Stanchi, F.; Zips, D.; Baumann, M.; Shevchenko, A.; Pilarsky, C.; Haase, M.; et al. PINCH1 Regulates Akt1 Activation and Enhances Radioresistance by Inhibiting PP1α. J. Clin. Investig. 2010, 120, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Naci, D.; El Azreq, M.-A.; Chetoui, N.; Lauden, L.; Sigaux, F.; Charron, D.; Al-Daccak, R.; Aoudjit, F. α2β1 Integrin Promotes Chemoresistance against Doxorubicin in Cancer Cells through Extracellular Signal-regulated Kinase (ERK). J. Biol. Chem. 2012, 287, 17065–17076. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Khambhati, N.N.; Qi, M.X.; Patel, K.S.; Ravikumar, N.; Brandenburg, C.P.; Dawson, M.R. Alterations in Ovarian Cancer Cell Adhesion Drive Taxol Resistance by Increasing Microtubule Dynamics in a FAK-dependent Manner. Sci. Rep. 2015, 5, srep09529. [Google Scholar] [CrossRef]
- Jakubzig, B.; Baltes, F.; Henze, S.; Schlesinger, M.; Bendas, G. Mechanisms of Matrix-Induced Chemoresistance of Breast Cancer Cells—Deciphering Novel Potential Targets for a Cell Sensitization. Cancers 2018, 10, 495. [Google Scholar] [CrossRef]
- Naik, A.; Al-Yahyaee, A.; Abdullah, N.; Sam, J.-E.; Al-Zeheimi, N.; Yaish, M.W.; Adham, S.A. Neuropilin-1 Promotes the Oncogenic Tenascin-C/Integrin β3 Pathway and Modulates Chemoresistance in Breast Cancer Cells. BMC Cancer 2018, 18, 533. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Ng, S.; Barrett, M.T.; Hostetter, G.; Von Hoff, D.D.; Han, H. Inhibition of ROCK1 Kinase Modulates Both Tumor Cells and Stromal Fibroblasts in Pancreatic Cancer. PLoS ONE 2017, 12, e0183871. [Google Scholar] [CrossRef]
- Wu, Q.; Tian, Y.; Zhang, J.; Zhang, H.; Gu, F.; Lu, Y.; Zou, S.; Chen, Y.; Sun, P.; Xu, M.; et al. Functions of Pancreatic Stellate Cell-Derived Soluble Factors in the Microenvironment of Pancreatic Ductal Carcinoma. Oncotarget 2017, 8, 102721–102738. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. The Critical Roles of Activated Stellate Cells-Mediated Paracrine Signaling, Metabolism and Onco-Immunology in Pancreatic Ductal Adenocarcinoma. Mol. Cancer 2018, 17, 62. [Google Scholar] [CrossRef]
- Hara, T.; Tanegashima, K. CXCL14 antagonizes the CXCL12-CXCR4 signaling axis. Biomol. Concepts 2014, 5, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic Hedgehog Promotes Desmoplasia in Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot Clinical Trial of Hedgehog Pathway Inhibitor GDC-0449 (Vismodegib) in Combination with Gemcitabine in Patients with Metastatic Pancreatic Adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I Trial of Hedgehog Pathway Inhibitor Vismodegib (GDC-0449) in Patients with Refractory, Locally Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ebeling, M.C.; Chauhan, N.; Thompson, P.A.; Gara, R.K.; Ganju, A.; Yallapu, M.M.; Behrman, S.W.; Zhao, H.; Zafar, N.; et al. Ormeloxifene Suppresses Desmoplasia and Enhances Sensitivity of Gemcitabine in Pancreatic Cancer. Cancer Res. 2015, 75, 2292–2304. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 Activation Regulates the Expression of Vascular Endothelial Growth Factor and Human Pancreatic Cancer Angiogenesis and Metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Kitamoto, S.; Yokoyama, S.; Higashi, M.; Yamada, N.; Takao, S.; Yonezawa, S. MUC1 Enhances Hypoxia-Driven Angiogenesis through the Regulation of Multiple Proangiogenic Factors. Oncogene 2012, 32, 4614–4621. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, W.; Zhong, Y.; Huo, Y.; Fan, P.; Zhan, S.; Xiao, J.; Jin, X.; Gou, S.; Yin, T.; et al. Overexpression of G Protein-Coupled Receptor GPR87 Promotes Pancreatic Cancer Aggressiveness and Activates NF-κB Signaling Pathway. Mol. Cancer 2017, 16, 1–14. [Google Scholar] [CrossRef]
- Saito, K.; Matsuo, Y.; Imafuji, H.; Okubo, T.; Maeda, Y.; Sato, T.; Shamoto, T.; Tsuboi, K.; Morimoto, M.; Takahashi, H.; et al. Xanthohumol Inhibits Angiogenesis by Suppressing Nuclear Factor-κB Activation in Pancreatic Cancer. Cancer Sci. 2017, 109, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine Plus Bevacizumab Compared With Gemcitabine Plus Placebo in Patients With Advanced Pancreatic Cancer: Phase III Trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- Katsuta, E.; Qi, Q.; Peng, X.; Hochwald, S.N.; Yan, L.; Takabe, K. Pancreatic Adenocarcinomas with Mature Blood Vessels Have Better Overall Survival. Sci. Rep. 2019, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.M.; Steele, M.M.; Hollingsworth, M.A. The Lymphatic System and Pancreatic Cancer. Cancer Lett. 2015, 381, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-N.; Goh, K.-S.; Huang, C.-R.; Chiang, T.-C.; Lee, C.-Y.; Jeng, Y.-M.; Peng, S.-J.; Chien, H.-J.; Chung, M.-H.; Chou, Y.-H.; et al. Lymphatic Vessel Remodeling and Invasion in Pancreatic Cancer Progression. EBioMedicine 2019, 47, 98–113. [Google Scholar] [CrossRef]
- Zhao, B.; Cui, K.; Wang, C.; Wang, A.; Zhang, B.; Zhou, W.; Zhao, W.; Li, S. The Chemotactic Interaction between CCL21 and Its Receptor, CCR7, Facilitates the Progression of Pancreatic Cancer via Induction of Angiogenesis and Lymphangiogenesis. J. Hepato-Biliary-Pancreatic Sci. 2011, 18, 821–828. [Google Scholar] [CrossRef]
- Guo, J.; Lou, W.; Ji, Y.; Zhang, S. Effect of CCR7, CXCR4 and VEGF-C on the Lymph Node Metastasis of Human Pancreatic Ductal Adenocarcinoma. Oncol. Lett. 2013, 5, 1572–1578. [Google Scholar] [CrossRef]
- Sperveslage, J.; Frank, S.; Heneweer, C.; Egberts, J.; Schniewind, B.; Buchholz, M.; Bergmann, F.; Giese, N.; Munding, J.; Hahn, S.A.; et al. Lack of CCR7 Expression Is Rate Limiting for Lymphatic Spread of Pancreatic Ductal Adenocarcinoma. Int. J. Cancer 2011, 131, E371–E381. [Google Scholar] [CrossRef]
- Wehler, T.; Wolfert, F.; Schimanski, C.C.; Gockel, I.; Herr, W.; Biesterfeld, S.; Seifert, J.K.; Adwan, H.; Berger, M.R.; Junginger, T.; et al. Strong Expression of Chemokine Receptor CXCR4 by Pancreatic Cancer Correlates with Advanced Disease. Oncol. Rep. 2006, 16, 1159–1164. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cui, K.; Zhao, W.; Wang, C.; Wang, A.; Zhang, B.; Zhou, W.; Yu, J.; Sun, Z.; Li, S. The CXCR4-CXCL12 Pathway Facilitates the Progression of Pancreatic Cancer Via Induction of Angiogenesis and Lymphangiogenesis. J. Surg. Res. 2011, 171, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, G.; Pellegatta, M.; Crippa, S.; Lena, M.S.; Belfiori, G.; Doglioni, C.; Taveggia, C.; Falconi, M. Nerves and Pancreatic Cancer: New Insights into A Dangerous Relationship. Cancers 2019, 11, 893. [Google Scholar] [CrossRef] [PubMed]
- Garajová, I.; Trentini, F.; Leonardi, F.; Giovannetti, E. Unraveling the Connection: Pancreatic Cancer Cells and Schwann Cells. J. Clin. Med. 2024, 13, 1785. [Google Scholar] [CrossRef] [PubMed]
- Selvaggi, F.; Melchiorre, E.; Casari, I.; Cinalli, S.; Cinalli, M.; Aceto, G.M.; Cotellese, R.; Garajova, I.; Falasca, M. Perineural Invasion in Pancreatic Ductal Adenocarcinoma: From Molecules towards Drugs of Clinical Relevance. Cancers 2022, 14, 5793. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kang, R.; Tang, D. Cellular and Molecular Mechanisms of Perineural Invasion of Pancreatic Ductal Adenocarcinoma. Cancer Commun. 2021, 41, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ma, Z.; Cao, Q.; Zhao, H.; Guo, Y.; Liu, T.; Li, J. Perineural Invasion-Associated Biomarkers for Tumor Development. Biomed. Pharmacother. 2022, 155, 113691. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G. Neuroepithelial Interactions in Cancer. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 493–514. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, S.; Chen, M. Schwann Cells in the Tumor Microenvironment: Need More Attention. J. Oncol. 2022, 2022, 1058667. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, C.; Liu, J. Schwann Cells as a Target Cell for the Treatment of Cancer Pain. Glia 2023, 71, 2309–2322. [Google Scholar] [CrossRef]
- Hirose, M.; Kuroda, Y.; Murata, E. NGF/TrkA Signaling as a Therapeutic Target for Pain. Pain Pr. 2015, 16, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Fallon, M.; Sopata, M.; Dragon, E.; Brown, M.T.; Viktrup, L.; West, C.R.; Bao, W.; Agyemang, A. A Randomized Placebo-Controlled Trial of the Anti-Nerve Growth Factor Antibody Tanezumab in Subjects With Cancer Pain Due to Bone Metastasis. Oncology 2023, 28, e1268–e1278. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.G.; Geissmann, F. Identifying the Infiltrators. Science 2014, 344, 801–802. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune Cell Promotion of Metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ Tumor-Infiltrating Lymphocytes Together with CD4+ Tumor-Infiltrating Lymphocytes and Dendritic Cells Improve the Prognosis of Patients with Pancreatic Adenocarcinoma. Pancreas 2004, 28, e26–e31. [Google Scholar] [CrossRef] [PubMed]
- Carstens, J.L.; de Sampaio, P.C.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial Computation of Intratumoral T Cells Correlates with Survival of Patients with Pancreatic Cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef] [PubMed]
- Ryschich, E.; Nötzel, T.; Hinz, U.; Autschbach, F.; Ferguson, J.; Simon, I.; Schmidt, J. Control of T-Cell-Mediated Immune Response by HLA Class I in Human Pancreatic Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 498–504. [Google Scholar] [CrossRef]
- Bernstorff, W.V.; Spanjaard, R.A.; Chan, A.K.; Lockhart, D.C.; Sadanaga, N.; Wood, I.; Peiper, M.; Goedegebuure, P.S.; Eberlein, T.J. Pancreatic Cancer Cells Can Evade Immune Surveillance via Nonfunctional Fas (APO-1/CD95) Receptors and Aberrant Expression of Functional Fas Ligand. Surgery 1999, 125, 0073–0084. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Markowitz, J.; Brooks, T.R.; Duggan, M.C.; Paul, B.K.; Pan, X.; Wei, L.; Abrams, Z.; Luedke, E.; Lesinski, G.B.; Mundy-Bosse, B.; et al. Patients with Pancreatic Adenocarcinoma Exhibit Elevated Levels of Myeloid-Derived Suppressor Cells upon Progression of Disease. Cancer Immunol. Immunother. 2014, 64, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Porembka, M.R.; Mitchem, J.B.; Belt, B.A.; Hsieh, C.-S.; Lee, H.-M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic Adenocarcinoma Induces Bone Marrow Mobilization of Myeloid-Derived Suppressor Cells Which Promote Primary Tumor Growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Torroella-Kouri, M.; Rodríguez, D.; Caso, R. Alterations in Macrophages and Monocytes from Tumor-Bearing Mice: Evidence of Local and Systemic Immune Impairment. Immunol. Res. 2013, 57, 86–98. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gabrilovich, D.I. Regulation of Suppressive Function of Myeloid-Derived Suppressor Cells by CD4+ T Cells. Semin. Cancer Biol. 2012, 22, 282–288. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 Regulates Arginase-I in Myeloid-Derived Suppressor Cells from Cancer Patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T Cells Sustain Myeloid-Derived Suppressor Cell-Mediated Immune Suppression. Oncotarget 2015, 7, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Bastea, L.; Fleming, A.; Döppler, H.; Edenfield, B.H.; Dawson, D.W.; Zhang, L.; Bardeesy, N.; Storz, P. The Presence of Interleukin-13 at Pancreatic ADM/PanIN Lesions Alters Macrophage Populations and Mediates Pancreatic Tumorigenesis. Cell Rep. 2017, 19, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef]
- Hao, N.-B.; Lü, M.-H.; Fan, Y.-H.; Cao, Y.-L.; Zhang, Z.-R.; Yang, S.-M. Macrophages in Tumor Microenvironments and the Progression of Tumors. J. Immunol. Res. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and Cancer: From Mechanisms to Therapeutic Implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 Signaling Drives Macrophage-Induced Adaptive Immune Suppression in Pancreatic Carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively Activated Macrophages Promote Pancreatic Fibrosis in Chronic Pancreatitis. Nat. Commun. 2015, 6, 948098. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kγ Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, Y.; Li, Z.; Huang, C.; Yang, Y.; Lang, M.; Cao, J.; Jiang, W.; Xu, Y.; Dong, J.; et al. Hypoxia Inducible Factor 1 (HIF-1) Recruits Macrophage to Activate Pancreatic Stellate Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2016, 17, 799. [Google Scholar] [CrossRef]
- Reid, M.D.; Basturk, O.; Thirabanjasak, D.; Hruban, R.H.; Klimstra, D.S.; Bagci, P.; Altinel, D.; Adsay, V. Tumor-Infiltrating Neutrophils in Pancreatic Neoplasia. Mod. Pathol. 2011, 24, 1612–1619. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Zou, J.-M.; Qin, J.; Li, Y.-C.; Wang, Y.; Li, D.; Shu, Y.; Luo, C.; Wang, S.-S.; Chi, G.; Guo, F.; et al. IL-35 Induces N2 Phenotype of Neutrophils to Promote Tumor Growth. Oncotarget 2017, 8, 33501–33514. [Google Scholar] [CrossRef]
- Felix, K.; Gaida, M.M. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression. Int. J. Biol. Sci. 2016, 12, 302–313. [Google Scholar] [CrossRef]
- Wislez, M.; Rabbe, N.; Marchal, J.; Milleron, B.; Crestani, B.; Mayaud, C.; Cadranel, J. Hepatocyte Growth Factor Production by Neutrophils Infiltrating Bronchioloalveolar Subtype Pul-monary Adenocarcinoma: Role in Tumor Progression and Death. Cancer Res. 2003, 63, 1405–1412. [Google Scholar]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human Neutrophils Uniquely Release TIMP-Free MMP-9 to Provide a Potent Catalytic Stimulator of Angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An Increased Abundance of Tumor-Infiltrating Regulatory T Cells Is Correlated with the Progression and Prognosis of Pancreatic Ductal Adenocarcinoma. PLoS ONE 2014, 9, e91551. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, S.; Hugenschmidt, H.; Hagness, M.; Line, P.D.; Labori, K.J.; Wiedswang, G.; Taskén, K.; Aandahl, E.M. Regulatory T Cells that Co-Express RORγt and FOXP3 Are Pro-Inflammatory and Immunosuppressive and Expand in Human Pancreatic Cancer. OncoImmunology 2015, 5, e1102828. [Google Scholar] [CrossRef]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.-J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-Cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL-8/CXCR1 Axis is Associated with Cancer Stem Cell-Like Properties and Correlates with Clinical Prognosis in Human Pancreatic Cancer Cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.; Feurino, L.W.; Wang, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q. Interleukin-8 Increases Vascular Endothelial Growth Factor and Neuropilin Expression and Stimulates ERK Activation in Human Pancreatic Cancer. Cancer Sci. 2008, 99, 733–737. [Google Scholar] [CrossRef]
- Zhou, D.-H.; Yang, L.-N.; Röder, C.; Kalthoff, H.; Trauzold, A. TRAIL-Induced Expression of uPA and IL-8 Strongly Enhanced by Overexpression of TRAF2 and Bcl-xL in Pancreatic Ductal Adenocarcinoma Cells. Hepatobiliary Pancreat. Dis. Int. 2013, 12, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Dall’Ora, M.; Rovesti, G.; Bonetti, L.R.; Casari, G.; Banchelli, F.; Fabbiani, L.; Veronesi, E.; Petrachi, T.; Magistri, P.; Di Benedetto, F.; et al. TRAIL Receptors Are Expressed in Both Malignant and Stromal Cells in Pancreatic Ductal Adenocarcinoma. Am. J. Cancer Res. 2021, 11, 4500–4514. [Google Scholar] [PubMed]
- Yu, Y.; Wang, W.; Zhai, S.; Dang, S.; Sun, M. IL6 Gene Polymorphisms and Susceptibility to Colorectal Cancer: A Meta-Analysis and Review. Mol. Biol. Rep. 2012, 39, 8457–8463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; di Magliano, M.P. Interleukin-6 Is Required for Pancreatic Cancer Progression by Promoting MAPK Signaling Activation and Oxidative Stress Resistance. Cancer Res. 2013, 73, 6359–6374. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Masamune, A.; Yoshida, N.; Takikawa, T.; Shimosegawa, T. IL-6/STAT3 Plays a Regulatory Role in the Interaction Between Pancreatic Stellate Cells and Cancer Cells. Dig. Dis. Sci. 2016, 61, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, J.; Wang, H.; Yin, G.; Liu, Y.; Lei, X.; Xiang, M. REG3A Accelerates Pancreatic Cancer Cell Growth under IL-6-Associated Inflammatory Condition: Involvement of a REG3A–JAK2/STAT3 Positive Feedback Loop. Cancer Lett. 2015, 362, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Huang, B.; Zhang, Q.; Fu, S.; Wang, D.; Cheng, X.; Wu, X.; Xue, Z.; Zhang, L.; Zhang, D.; et al. Embelin Inhibits Pancreatic Cancer Progression by Directly Inducing Cancer Cell Apoptosis and Indirectly Restricting IL-6 Associated Inflammatory and Immune Suppressive cells11These Authors Contributed Equally to this Work. Cancer Lett. 2014, 354, 407–416. [Google Scholar] [CrossRef]
- A Mace, T.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 Antibody Blockade Combination Therapy Reduces Tumour Progression in Murine Models of Pancreatic Cancer. Gut 2016, 67, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Vorndamm, J.; Müerköster, S.; Yu, H.; Schmidt, W.E.; Fölsch, U.R.; Schäfer, H. Autocrine Production of Interleukin 1beta Confers Constitutive Nuclear Factor kappaB Activity and Chemoresistance in Pancreatic Carcinoma Cell Lines. Cancer Res. 2002, 62, 910–916. [Google Scholar]
- Angst, E.; Reber, H.A.; Hines, O.J.; Eibl, G. Mononuclear Cell-Derived Interleukin-1 Beta Confers Chemoresistance in Pancreatic Cancer Cells by Upregulation of Cyclooxygenase-2. Surgery 2008, 144, 57–65. [Google Scholar] [CrossRef]
- Burke, S.J.; Stadler, K.; Lu, D.; Gleason, E.; Han, A.; Donohoe, D.R.; Rogers, R.C.; Hermann, G.E.; Karlstad, M.D.; Collier, J.J. IL-1β Reciprocally Regulates Chemokine and Insulin Secretion in Pancreatic β-Cells via NF-κB. Am. J. Physiol. Metab. 2015, 309, E715–E726. [Google Scholar] [CrossRef]
- Hamacher, R.; Diersch, S.; Scheibel, M.; Eckel, F.; Mayr, M.; Rad, R.; Bajbouj, M.; Schmid, R.M.; Saur, D.; Schneider, G. Interleukin 1 Beta Gene Promoter SNPs Are Associated with Risk of Pancreatic Cancer. Cytokine 2009, 46, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Avraamides, C.J.; Foubert, P.; Shaked, Y.; Kang, S.W.; Kerbel, R.S.; Varner, J.A. Combined Blockade of Integrin-α4β1 Plus Cytokines SDF-1α or IL-1β Potently Inhibits Tumor Inflammation and Growth. Cancer Res. 2011, 71, 6965–6975. [Google Scholar] [CrossRef]
- Ebrahimi, B.; Tucker, S.L.; Li, D.; Abbruzzese, J.L.; Kurzrock, R. Cytokines in Pancreatic Carcinoma. Cancer 2004, 101, 2727–2736. [Google Scholar] [CrossRef]
- Sandberg, J.; Eizirik, D.L.; Sandler, S. IL-1 Receptor Antagonist Inhibits Recurrence of Disease after Syngeneic Pancreatic Islet Transplantation to Spontaneously Diabetic Non-Obese Diabetic (NOD) Mice. Clin. Exp. Immunol. 1997, 108, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, D.; Emori, Y.; Eta, R.; Iino, Y.; Hamano, H.; Yoshinaga, K.; Tanaka, T.; Takei, M.; Watson, S.A. Effect of Z-360, a Novel Orally Active CCK-2/Gastrin Receptor Antagonist on Tumor Growth in Human Pancreatic Adenocarcinoma Cell Lines In Vivo and Mode of Action Determinations In Vitro. Cancer Chemother. Pharmacol. 2007, 61, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.-M. Tumor necrosis Factor. Cancer Lett. 2012, 328, 222–225. [Google Scholar] [CrossRef]
- Farrow, B.; Evers, B. Inflammation and the Development of Pancreatic Cancer. Surg. Oncol. 2002, 10, 153–169. [Google Scholar] [CrossRef]
- Friess, H.; Guo, X.; Nan, B.; Kleeff, Ö.; Büchler, M.W. Growth Factors and Cytokines in Pancreatic Carcinogenesis. Ann. N. Y. Acad. Sci. 1999, 880, 110–121. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour Necrosis Factor and Cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Adelman, J.S.; Martin, L.B. Vertebrate Sickness Behaviors: Adaptive and Integrated Neuroendocrine Immune Responses. Integr. Comp. Biol. 2009, 49, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.N.; Trebble, T.M.; Ellis, R.D.; Duncan, H.D.; Johns, T.; Goggin, P.M. Thalidomide in the Treatment of Cancer Cachexia: A Randomised Placebo Controlled Trial. Gut 2005, 54, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-B.; Wang, M.; Niu, Z.-X.; Tang, X.-Y.; Liu, Q.-Y. Phase II Trial of Capecitabine Combined with Thalidomide in Second-Line Treatment of Advanced Pancreatic Cancer. Pancreatology 2012, 12, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Fernandez, S.A.; Criswell, T.B.; Chidiac, T.A.; Guttridge, D.; Villalona-Calero, M.; Bekaii-Saab, T.S. Disrupting Cytokine Signaling in Pancreatic Cancer. Pancreas 2013, 42, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the Interleukin-10 Receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- von Bernstorff, W.; Voss, M.; Freichel, S.; Schmid, A.; Vogel, I.; Johnk, C.; Kalthoff, H. Systemic and Local Immunosuppression in Pancreatic Cancer Patients. Clin. Cancer Res. Off. J. Am. Asso-Ciation Cancer Res. 2001, 7, 925s–932s. [Google Scholar]
- Bellone, G.; Turletti, A.; Artusio, E.; Mareschi, K.; Carbone, A.; Tibaudi, D.; Robecchi, A.; Emanuelli, G.; Rodeck, U. Tumor-Associated Transforming Growth Factor-β and Interleukin-10 Contribute to a Systemic Th2 Immune Phenotype in Pancreatic Carcinoma Patients. Am. J. Pathol. 1999, 155, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-Polarized Tumor-Associated Macrophages Promoted Epithelial–Mesenchymal Transition in Pancreatic Cancer Cells, Partially through TLR4/IL-10 Signaling Pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef]
- Berman, R.M.; Suzuki, T.; Tahara, H.; Robbins, P.D.; Narula, S.K.; Lotze, M.T. Systemic Administration of Cellular IL-10 Induces an Effective, Specific, and Long-Lived Immune Response against Established Tumors in Mice. J. Immunol. 1996, 157, 231–238. [Google Scholar] [CrossRef]
- Richter, G.; Krüger-Krasagakes, S.; Hein, G.; Hüls, C.; Schmitt, E.; Diamantstein, T.; Blankenstein, T. Interleukin 10 Transfected into Chinese Hamster Ovary Cells Prevents Tumor Growth and Macrophage Infiltration. Cancer Res. 1993, 53, 4134–4137. [Google Scholar] [PubMed]
- Smirne, C.; Camandona, M.; Alabiso, O.; Bellone, G.; Emanuelli, G. High serum levels of Transforming Growth Factor-beta1, Interleukin-10 and Vascular Endothelial Growth Factor in pancreatic adenocarcinoma patients. Minerva Gastroenterol. Dietol. 1999, 45, 21–27. [Google Scholar] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Ho, W.J.; Zhu, Q.; Vithayathil, T.; De Jesus-Acosta, A.; Azad, N.S.; Laheru, D.A.; Fertig, E.J.; Anders, R.; Jaffee, E.M.; et al. Programmed Cell Death Ligand-1 (PD-L1) and CD8 Expression Profiling Identify an Immunologic Subtype of Pancreatic Ductal Adenocarcinomas with Favorable Survival. Cancer Immunol. Res. 2019, 7, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.; Lim, S.J.; Popovic, A.; Azad, N.S.; Laheru, D.A.; Zheng, L.; Jaffee, E.M.; Wang, H.; Yarchoan, M. Tumor Mutational Burden, Toxicity, and Response of Immune Checkpoint Inhibitors Targeting PD(L)1, CTLA-4, and Combination: A Meta-regression Analysis. Clin. Cancer Res. 2020, 26, 4842–4851. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Jaffee, E.M. Regulatory T-cell Modulation Using Cyclophosphamide in Vaccine Approaches: A Current Perspective. Cancer Res. 2012, 72, 3439–3444. [Google Scholar] [CrossRef]
- Laheru, D.; Lutz, E.; Burke, J.; Biedrzycki, B.; Solt, S.; Onners, B.; Tartakovsky, I.; Nemunaitis, J.; Le, D.; Sugar, E.; et al. Allogeneic Granulocyte Macrophage Colony-Stimulating Factor–Secreting Tumor Immunotherapy Alone or in Sequence with Cyclophosphamide for Metastatic Pancreatic Cancer: A Pilot Study of Safety, Feasibility, and Immune Activation. Clin. Cancer Res. 2008, 14, 1455–1463. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic Delivery of Neutralizing Antibody Targeting CCL2 for Glioma Therapy. J. Neuro-Oncol. 2011, 104, 83–92. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Papadopoulos, K.; Fong, P.C.; Patnaik, A.; Messiou, C.; Olmos, D.; Wang, G.; Tromp, B.J.; Puchalski, T.A.; Balkwill, F.; et al. A First-in-Human, First-in-Class, Phase I Study of Carlumab (CNTO 888), a Human Monoclonal Antibody against CC-Chemokine Ligand 2 in Patients with Solid Tumors. Cancer Chemother. Pharmacol. 2013, 71, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 Study of Carlumab (CNTO 888), a Human Monoclonal Antibody against CC-Chemokine Ligand 2 (CCL2), in Metastatic Castration-Resistant Prostate Cancer. Investig. New Drugs 2012, 31, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Brana, I.; Calles, A.; Lorusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; De Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an Anti-C-C Chemokine Ligand 2 Monoclonal Antibody, in Combination with Four Chemotherapy Regimens for the Treatment of Patients with Solid Tumors: An Open-Label, Multicenter Phase 1b Study. Target. Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; E Sanford, D.; A Belt, B.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; A Worley, L.; Yano, M.; et al. Targeting Tumour-Associated Macrophages with CCR2 Inhibition in Combination with FOLFIRINOX in Patients with Borderline Resectable and Locally Advanced Pancreatic Cancer: A Single-Centre, Open-Label, Dose-Finding, Non-Randomised, Phase 1b Trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Mouchemore, K.A.; Sampaio, N.G.; Murrey, M.W.; Stanley, E.R.; Lannutti, B.J.; Pixley, F.J. Specific Inhibition of PI3K p110δ Inhibits CSF-1-Induced Macrophage Spreading and Invasive Capacity. FEBS J. 2013, 280, 5228–5236. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The Tumor Microenvironment Underlies Acquired Resistance to CSF-1R Inhibition in Gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef]
- Dewar, A.L.; Cambareri, A.C.; Zannettino, A.C.W.; Miller, B.L.; Doherty, K.V.; Hughes, T.P.; Lyons, A.B. Macrophage Colony-Stimulating Factor Receptor c-Fms Is A Novel Target of Imatinib. Blood 2005, 105, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Demetri, G.; Sargent, W.; Raymond, E. Molecular Basis for Sunitinib Efficacy and Future Clinical Development. Nat. Rev. Drug Discov. 2007, 6, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Hammel, P.; Huguet, F.; van Laethem, J.-L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouché, O.; Shannon, J.; André, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled after 4 Months of Gemcitabine with or without Erlotinib. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed]
- Salman, B.; Zhou, D.; Jaffee, E.M.; Edil, B.H.; Zheng, L. Vaccine Therapy for Pancreatic Cancer. OncoImmunology 2013, 2, e26662. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA Neoantigen Vaccines Stimulate T Cells in Pancreatic Cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).