A Perspective on the CD47-SIRPA Axis in High-Risk Neuroblastoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Plasticity of Macrophages and Heterogeneity of Tumor-Associated Macrophages
3. Macrophage Polarization Status and Phagocytic Capacity in High-Risk Neuroblastoma
4. The CD47-SIRPA Axis in Cancer Immunotherapy
5. The CD47-SIRPA Axis in High-Risk Neuroblastoma
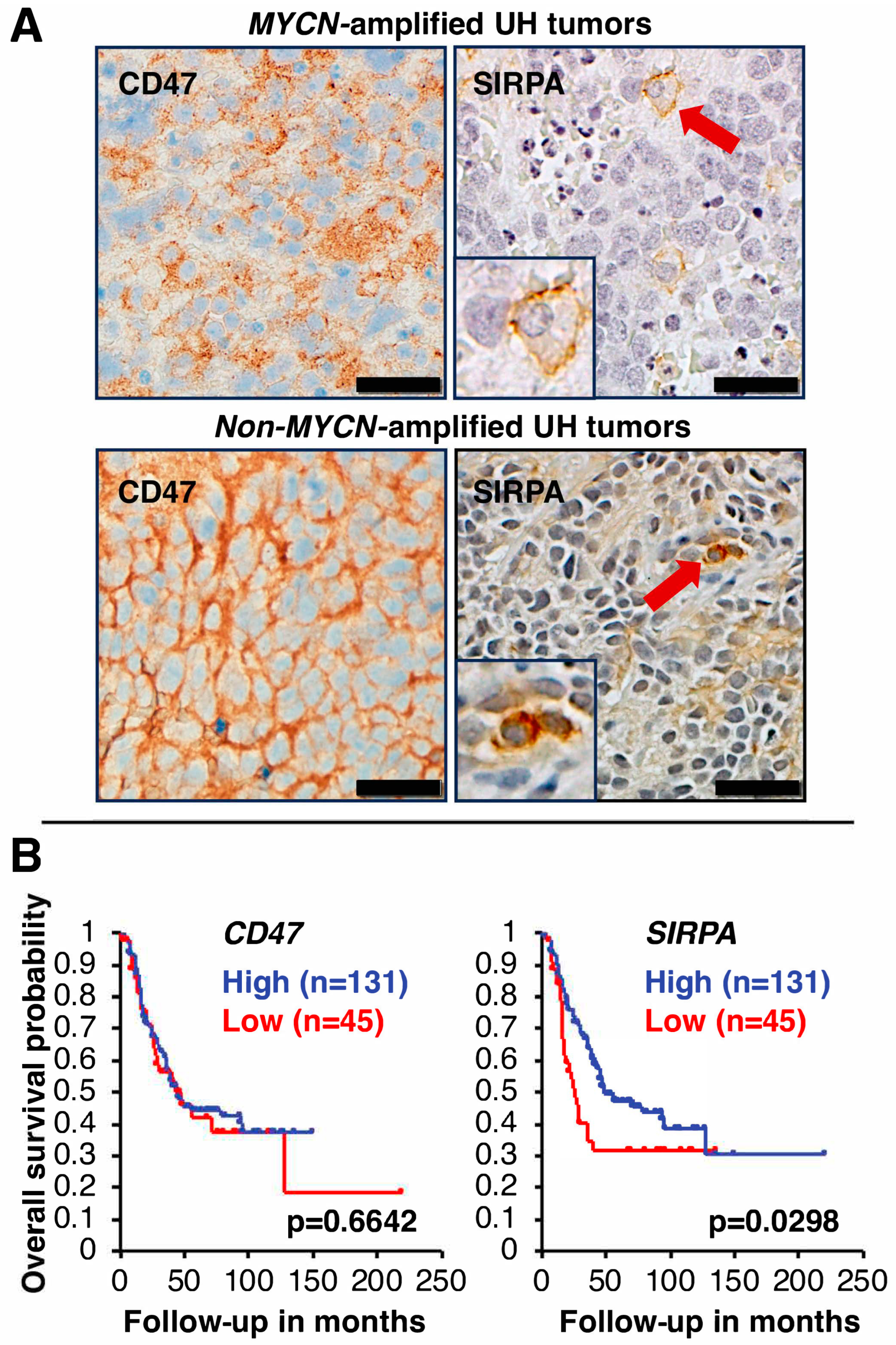
5.1. CD47 Expression in High-Risk Neuroblastoma
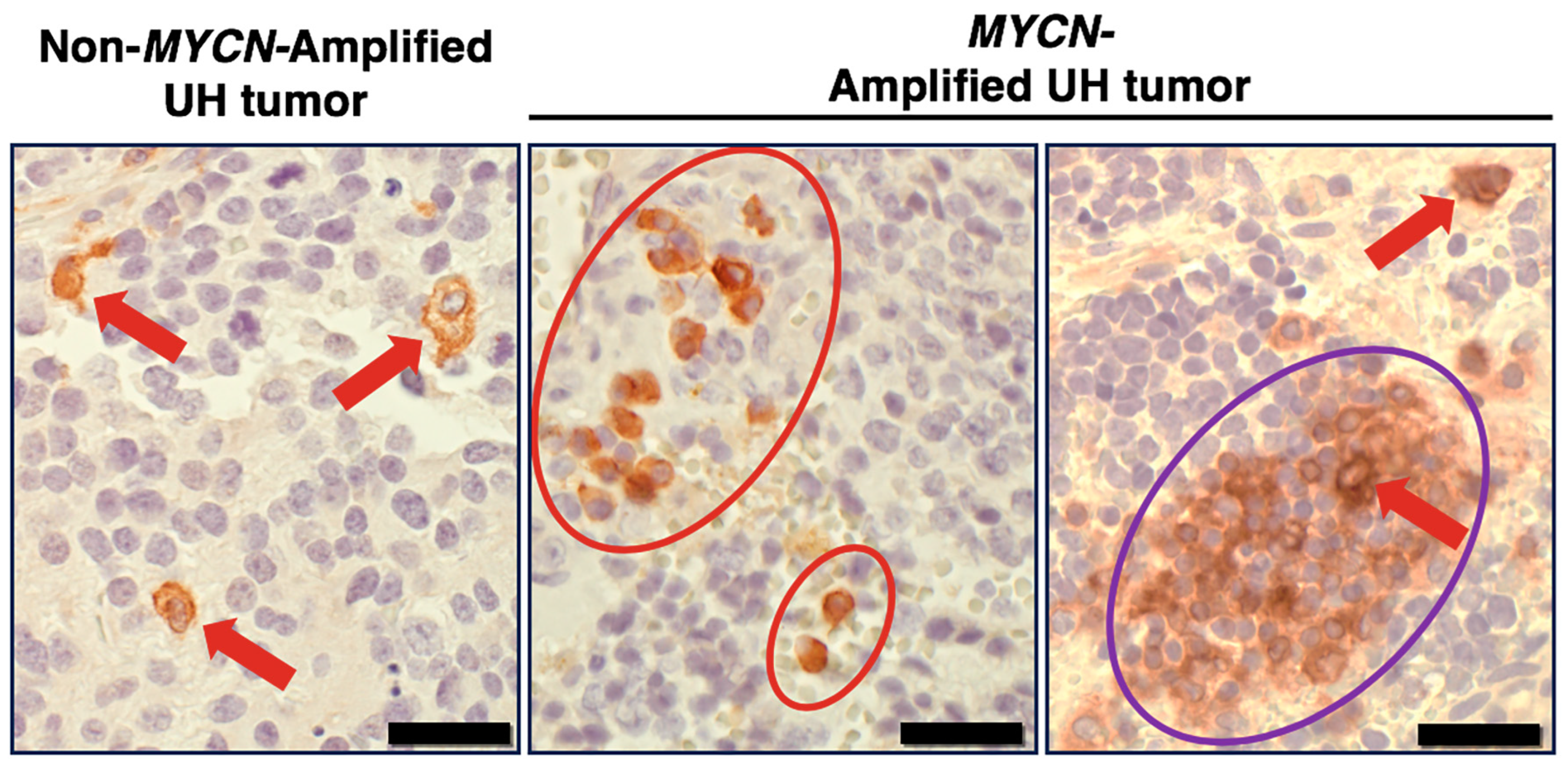
5.2. SIRPA Expression in High-Risk Neuroblastoma
5.3. Prognostic Significance of CD47 and SIRPA Expression in High-Risk Neuroblastoma
5.4. A Missing Piece of the Puzzle
5.5. A Perspective on the Biological Significance of the CD47-SIRPA Axis in Cancer Immunotherapy and Influences of the CD47/SIRPA Blockade on Macrophage Polarization
5.6. SLAMF7 as a Potential Target to Activate TAMs in High-Risk Neuroblastoma
5.7. A Proposed Combination Antibody-Based Therapy for High-Risk Neuroblastoma
6. Recent Clinical Trials with Anti-CD47 Antibody Magrolimab in Human Adult and Pediatric Cancers
7. Anti-SIRPA Antibodies as Alternatives to Target the CD47-SIRPA Axis
8. Discussion
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef]
- Finck, A.V.; Blanchard, T.; Roselle, C.P.; Golinelli, G.; June, C.H. Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 2022, 28, 678–689. [Google Scholar] [CrossRef]
- Wölfl, M.; Jungbluth, A.A.; Garrido, F.; Cabrera, T.; Meyen-Southard, S.; Spitz, R.; Ernestus, K.; Berthold, F. Expression of MHC class I, MHC class II, and cancer germline antigens in neuroblastoma. Cancer Immunol. Immunother. 2005, 54, 400–406. [Google Scholar] [CrossRef]
- Tang, X.X.; Shimada, H.; Ikegaki, N. Clinical Relevance of CD4 Cytotoxic T Cells in High-Risk Neuroblastoma. Front. Immunol. 2021, 12, 650427. [Google Scholar] [CrossRef]
- Tang, X.X.; Shimada, H.; Ikegaki, N. Macrophage-mediated anti-tumor immunity against high-risk neuroblastoma. Genes Immun. 2022, 23, 129–140. [Google Scholar] [CrossRef]
- Chamoto, K.; Yaguchi, T.; Tajima, M.; Honjo, T. Insights from a 30-year journey: function, regulation and therapeutic modulation of PD1. Nat. Rev. Immunol. 2023, 23, 682–695. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Bied, M.; Ho, W.W.; Ginhoux, F.; Bleriot, C. Roles of macrophages in tumor development: a spatiotemporal perspective. Cell Mol. Immunol. 2023, 20, 983–992. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Tumor-associated macrophages. Curr. Biol. 2020, 30, R246–R248. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, M.; Jia, S. Macrophage: Key player in the pathogenesis of autoimmune diseases. Front. Immunol. 2023, 14, 1080310. [Google Scholar] [CrossRef]
- Kuninaka, Y.; Ishida, Y.; Ishigami, A.; Nosaka, M.; Matsuki, J.; Yasuda, H.; Kofuna, A.; Kimura, A.; Furukawa, F.; Kondo, T. Macrophage polarity and wound age determination. Sci. Rep. 2022, 12, 20327. [Google Scholar] [CrossRef]
- Hourani, T.; Holden, J.A.; Li, W.; Lenzo, J.C.; Hadjigol, S.; O’Brien-Simpson, N.M. Tumor Associated Macrophages: Origin, Recruitment, Phenotypic Diversity, and Targeting. Front. Oncol. 2021, 11, 788365. [Google Scholar] [CrossRef]
- Jahchan, N.S.; Mujal, A.M.; Pollack, J.L.; Binnewies, M.; Sriram, V.; Reyno, L.; Krummel, M.F. Tuning the Tumor Myeloid Microenvironment to Fight Cancer. Front. Immunol. 2019, 10, 1611. [Google Scholar] [CrossRef]
- Su, Z.; Fang, H.; Hong, H.; Shi, L.; Zhang, W.; Zhang, W.; Zhang, Y.; Dong, Z.; Lancashire, L.J.; Bessarabova, M.; et al. An investigation of biomarkers derived from legacy microarray data for their utility in the RNA-seq era. Genome Biol. 2014, 15, 523. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 133. [Google Scholar] [CrossRef]
- Yang, L.; Forker, L.; Irlam, J.J.; Pillay, N.; Choudhury, A.; West, C.M.L. Validation of a hypoxia related gene signature in multiple soft tissue sarcoma cohorts. Oncotarget. 2018, 9, 3946–3955. [Google Scholar] [CrossRef]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef]
- Mao, Y.; Finnemann, S.C. Regulation of phagocytosis by Rho GTPases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Sathyapalan, T.; Diboun, I.; Atkin, S.L.; Butler, A.E. Identification of macrophage activation-related biomarkers in obese type 2 diabetes that may be indicative of enhanced respiratory risk in COVID-19. Sci. Rep. 2021, 11, 6428. [Google Scholar] [CrossRef]
- Amici, S.A.; Young, N.A.; Narvaez-Miranda, J.; Jablonski, K.A.; Arcos, J.; Rosas, L.; Papenfuss, T.L.; Torrelles, J.B.; Jarjour, W.N.; Guerau-de-Arellano, M. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front. Immunol. 2018, 9, 1593. [Google Scholar] [CrossRef]
- Adams, S.; van der Laan, L.J.; Vernon-Wilson, E.; Renardel de Lavalette, C.; Döpp, E.A.; Dijkstra, C.D.; Simmons, D.L.; van den Berg, T.K. Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. J. Immunol. 1998, 161, 1853–1859. [Google Scholar] [CrossRef]
- Barclay, A.N. Signal regulatory protein alpha (SIRPalpha)/CD47 interaction and function. Curr. Opin. Immunol. 2009, 21, 47–52. [Google Scholar] [CrossRef]
- Han, X.; Sterling, H.; Chen, Y.; Saginario, C.; Brown, E.J.; Frazier, W.A.; Lindberg, F.P.; Vignery, A. CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J. Biol. Chem. 2000, 275, 37984–37992. [Google Scholar] [CrossRef]
- Seiffert, M.; Cant, C.; Chen, Z.; Rappold, I.; Brugger, W.; Kanz, L.; Brown, E.J.; Ullrich, A.; Bühring, H.J. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood 1999, 94, 3633–3643. [Google Scholar] [CrossRef]
- Veillette, A.; Thibaudeau, E.; Latour, S. High expression of inhibitory receptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 in macrophages. J. Biol. Chem. 1998, 273, 22719–22728. [Google Scholar] [CrossRef]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Chen, Z.; Sures, I.; Wang, H.; Schilling, J.; Ullrich, A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997, 386, 181–186. [Google Scholar] [CrossRef]
- Fujioka, Y.; Matozaki, T.; Noguchi, T.; Iwamatsu, A.; Yamao, T.; Takahashi, N.; Tsuda, M.; Takada, T.; Kasuga, M. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol. Cell Biol. 1996, 16, 6887–6899. [Google Scholar] [CrossRef]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar] [CrossRef]
- Chao, M.P.; Tang, C.; Pachynski, R.K.; Chin, R.; Majeti, R.; Weissman, I.L. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood 2011, 118, 4890–4901. [Google Scholar] [CrossRef]
- Kim, D.; Wang, J.; Willingham, S.B.; Martin, R.; Wernig, G.; Weissman, I.L. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia 2012, 26, 2538–2545. [Google Scholar] [CrossRef]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Ozkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Guo, N.; Schnorr, P.J.; Maute, R.L.; Volkmer, J.-P.; Weissman, I.L. Direct SIRPa Blockade Augments Macrophage Responses to Therapeutic Anticancer Antibodies. Blood 2014, 124, 2729. [Google Scholar] [CrossRef]
- Theruvath, J.; Menard, M.; Smith, B.A.H.; Linde, M.H.; Coles, G.L.; Dalton, G.N.; Wu, W.; Kiru, L.; Delaidelli, A.; Sotillo, E.; et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med. 2022, 28, 333–344. [Google Scholar] [CrossRef]
- Koster, J.; Volckmann, R.; Zwijnenburg, D.; Molenaar, P.; Versteeg, R. Abstract 2490: R2: Genomics analysis and visualization platform. Cancer Res. 2019, 79, 2490. [Google Scholar] [CrossRef]
- Green, C.E.; Liu, T.; Montel, V.; Hsiao, G.; Lester, R.D.; Subramaniam, S.; Gonias, S.L.; Klemke, R.L. Chemoattractant signaling between tumor cells and macrophages regulates cancer cell migration, metastasis and neovascularization. PLoS ONE 2009, 4, e6713. [Google Scholar] [CrossRef]
- Sugimura-Nagata, A.; Koshino, A.; Inoue, S.; Matsuo-Nagano, A.; Komura, M.; Riku, M.; Ito, H.; Inoko, A.; Murakami, H.; Ebi, M.; et al. Expression and Prognostic Significance of CD47-SIRPA Macrophage Checkpoint Molecules in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 2690. [Google Scholar] [CrossRef]
- Kazama, R.; Miyoshi, H.; Takeuchi, M.; Miyawaki, K.; Nakashima, K.; Yoshida, N.; Kawamoto, K.; Yanagida, E.; Yamada, K.; Umeno, T.; et al. Combination of CD47 and signal-regulatory protein-α constituting the “don’t eat me signal” is a prognostic factor in diffuse large B-cell lymphoma. Cancer Sci. 2020, 111, 2608–2619. [Google Scholar] [CrossRef]
- Chen, Y.P.; Kim, H.J.; Wu, H.; Price-Troska, T.; Villasboas, J.C.; Jalali, S.; Feldman, A.L.; Novak, A.J.; Yang, Z.Z.; Ansell, S.M. SIRPα expression delineates subsets of intratumoral monocyte/macrophages with different functional and prognostic impact in follicular lymphoma. Blood Cancer J. 2019, 9, 84. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Mitrakas, A.; Anestopoulos, I.; Kontosis, A.; Koukourakis, I.M.; Pappa, A.; Panayiotidis, M.I.; Koukourakis, M.I. Expression of CD47 and SIRPα Macrophage Immune-Checkpoint Pathway in Non-Small-Cell Lung Cancer. Cancers 2022, 14, 1801. [Google Scholar] [CrossRef]
- Koga, N.; Hu, Q.; Sakai, A.; Takada, K.; Nakanishi, R.; Hisamatsu, Y.; Ando, K.; Kimura, Y.; Oki, E.; Oda, Y.; et al. Clinical significance of signal regulatory protein alpha (SIRPα) expression in esophageal squamous cell carcinoma. Cancer Sci. 2021, 112, 3018–3028. [Google Scholar] [CrossRef]
- Timms, J.F.; Carlberg, K.; Gu, H.; Chen, H.; Kamatkar, S.; Nadler, M.J.; Rohrschneider, L.R.; Neel, B.G. Identification of major binding proteins and substrates for the SH2-containing protein tyrosine phosphatase SHP-1 in macrophages. Mol. Cell Biol. 1998, 18, 3838–3850. [Google Scholar] [CrossRef]
- Foucher, E.D.; Blanchard, S.; Preisser, L.; Garo, E.; Ifrah, N.; Guardiola, P.; Delneste, Y.; Jeannin, P. IL-34 induces the differentiation of human monocytes into immunosuppressive macrophages. antagonistic effects of GM-CSF and IFNγ. PLoS ONE 2013, 8, e56045. [Google Scholar] [CrossRef]
- Jones, C.V.; Ricardo, S.D. Macrophages and CSF-1: implications for development and beyond. Organogenesis 2013, 9, 249–260. [Google Scholar] [CrossRef]
- Hamilton, T.A.; Zhao, C.; Pavicic, P.G., Jr.; Datta, S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front. Immunol. 2014, 5, 554. [Google Scholar] [CrossRef]
- Boulakirba, S.; Pfeifer, A.; Mhaidly, R.; Obba, S.; Goulard, M.; Schmitt, T.; Chaintreuil, P.; Calleja, A.; Furstoss, N.; Orange, F.; et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep. 2018, 8, 256. [Google Scholar] [CrossRef]
- Trus, E.; Basta, S.; Gee, K. Who’s in charge here? Macrophage colony stimulating factor and granulocyte macrophage colony stimulating factor: Competing factors in macrophage polarization. Cytokine 2020, 127, 154939. [Google Scholar] [CrossRef]
- Desgeorges, T.; Caratti, G.; Mounier, R.; Tuckermann, J.; Chazaud, B. Glucocorticoids Shape Macrophage Phenotype for Tissue Repair. Front. Immunol. 2019, 10, 1591. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab Plus Pomalidomide and Dexamethasone for Relapsed/Refractory Multiple Myeloma: Final Overall Survival Analysis From the Randomized Phase II ELOQUENT-3 Trial. J. Clin. Oncol. 2023, 41, 568–578. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Sait, S.; Modak, S. Anti-GD2 immunotherapy for neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 889–904. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; Naranjo, A.; Diccianni, M.B.; Gan, J.; Hank, J.A.; Batova, A.; London, W.B.; Tenney, S.C.; et al. Long-Term Follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin. Cancer Res. 2021, 27, 2179–2189. [Google Scholar] [CrossRef]
- Mora, J.; Castañeda, A.; Gorostegui, M.; Santa-María, V.; Garraus, M.; Muñoz, J.P.; Varo, A.; Perez-Jaume, S.; Mañe, S. Naxitamab combined with granulocyte-macrophage colony-stimulating factor as consolidation for high-risk neuroblastoma patients in complete remission. Pediatr. Blood Cancer 2021, 68, e29121. [Google Scholar] [CrossRef]
- Furman, W.L.; Federico, S.M.; McCarville, M.B.; Shulkin, B.L.; Davidoff, A.M.; Krasin, M.J.; Sahr, N.; Sykes, A.; Wu, J.; Brennan, R.C.; et al. A Phase II Trial of Hu14.18K322A in Combination with Induction Chemotherapy in Children with Newly Diagnosed High-Risk Neuroblastoma. Clin. Cancer Res. 2019, 25, 6320–6328. [Google Scholar] [CrossRef]
- Qu, T.; Li, B.; Wang, Y. Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed. Biomark. Res. 2022, 10, 20. [Google Scholar] [CrossRef]
- Wu, Z.H.; Li, N.; Mei, X.F.; Chen, J.; Wang, X.Z.; Guo, T.T.; Chen, G.; Nie, L.; Chen, Y.; Jiang, M.Z.; et al. Preclinical characterization of the novel anti-SIRPα antibody BR105 that targets the myeloid immune checkpoint. J. Immunother. Cancer 2022, 10. [Google Scholar] [CrossRef]
- Chan, H.; Trout, C.V.; Mikolon, D.; Adams, P.; Guzman, R.; Mavrommatis, K.; Abbasian, M.; Hadjivassiliou, H.; Dearth, L.; Fox, B.A.; et al. Discovery and Preclinical Activity of BMS-986351, an Antibody to SIRPα That Enhances Macrophage-mediated Tumor Phagocytosis When Combined with Opsonizing Antibodies. Cancer Res. Commun. 2024, 4, 505–515. [Google Scholar] [CrossRef]
- Voets, E.; Paradé, M.; Lutje Hulsik, D.; Spijkers, S.; Janssen, W.; Rens, J.; Reinieren-Beeren, I.; van den Tillaart, G.; van Duijnhoven, S.; Driessen, L.; et al. Functional characterization of the selective pan-allele anti-SIRPα antibody ADU-1805 that blocks the SIRPα-CD47 innate immune checkpoint. J. Immunother. Cancer 2019, 7, 340. [Google Scholar] [CrossRef]
- Gutierrez, M.; Jamal, R.; Yamamoto, N.; Doi, T.; Elgadi, M.; Ferrada, J.L.; Wojciekowski, S.M.; Patel, M.R. 697P Open-label, phase I, dose escalation/expansion trial of the anti-SIRPα monoclonal antibody BI 770371 in patients with advanced solid tumours, alone or in combination with the anti-PD-1 monoclonal antibody ezabenlimab. Ann. Oncol. 2023, 34, S485. [Google Scholar] [CrossRef]
- Wang, X.; Mathieu, M.; Brezski, R.J. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018, 9, 63–73. [Google Scholar] [CrossRef]
- Stewart, R.; Hammond, S.A.; Oberst, M.; Wilkinson, R.W. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J. ImmunoTher. Cancer 2014, 2, 29. [Google Scholar] [CrossRef]
- Galvez-Cancino, F.; Simpson, A.P.; Costoya, C.; Matos, I.; Qian, D.; Peggs, K.S.; Litchfield, K.; Quezada, S.A. Fcγ receptors and immunomodulatory antibodies in cancer. Nat. Rev. Cancer 2024, 24, 51–71. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Brooke, G.; Holbrook, J.D.; Brown, M.H.; Barclay, A.N. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J. Immunol. 2004, 173, 2562–2570. [Google Scholar] [CrossRef]
- Ho, C.C.; Guo, N.; Sockolosky, J.T.; Ring, A.M.; Weiskopf, K.; Özkan, E.; Mori, Y.; Weissman, I.L.; Garcia, K.C. “Velcro” engineering of high affinity CD47 ectodomain as signal regulatory protein α (SIRPα) antagonists that enhance antibody-dependent cellular phagocytosis. J. Biol. Chem. 2015, 290, 12650–12663. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Wissler, H.L.; Ehlerding, E.B.; Lyu, Z.; Zhao, Y.; Zhang, S.; Eshraghi, A.; Buuh, Z.Y.; McGuth, J.C.; Guan, Y.; Engle, J.W.; et al. Site-Specific Immuno-PET Tracer to Image PD-L1. Mol. Pharm. 2019, 16, 2028–2036. [Google Scholar] [CrossRef]
- Quintero-Hernández, V.; Juárez-González, V.R.; Ortíz-León, M.; Sánchez, R.; Possani, L.D.; Becerril, B. The change of the scFv into the Fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol. Immunol. 2007, 44, 1307–1315. [Google Scholar] [CrossRef]
- Bahri, M.; Kailayangiri, S.; Vermeulen, S.; Galopin, N.; Rossig, C.; Paris, F.; Fougeray, S.; Birklé, S. SIRPα-specific monoclonal antibody enables antibody-dependent phagocytosis of neuroblastoma cells. Cancer Immunol. Immunother. 2022, 71, 71–83. [Google Scholar] [CrossRef]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef]
- Asare, P.F.; Roscioli, E.; Hurtado, P.R.; Tran, H.B.; Mah, C.Y.; Hodge, S. LC3-Associated Phagocytosis (LAP): A Potentially Influential Mediator of Efferocytosis-Related Tumor Progression and Aggressiveness. Front. Oncol. 2020, 10, 1298. [Google Scholar] [CrossRef]
- Werfel, T.A.; Cook, R.S. Efferocytosis in the tumor microenvironment. Semin. Immunopathol. 2018, 40, 545–554. [Google Scholar] [CrossRef]
- Chen, J.; Zhong, M.C.; Guo, H.; Davidson, D.; Mishel, S.; Lu, Y.; Rhee, I.; Pérez-Quintero, L.A.; Zhang, S.; Cruz-Munoz, M.E.; et al. SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature 2017, 544, 493–497. [Google Scholar] [CrossRef]
- Delord, J.-P.; Kotecki, N.; Marabelle, A.; Vinceneux, A.; Korakis, I.; Jungels, C.; Champiat, S.; Huhn, R.D.; Poirier, N.; Costantini, D.; et al. A Phase 1 Study Evaluating BI 765063, a First in Class Selective Myeloid Sirpa Inhibitor, As Stand-Alone and in Combination with BI 754091, a Programmed Death-1 (PD-1) Inhibitor, in Patients with Advanced Solid Tumours. Blood 2019, 134, 1040. [Google Scholar] [CrossRef]
- van Helden, M.J.; Zwarthoff, S.A.; Arends, R.J.; Reinieren-Beeren, I.M.J.; Paradé, M.; Driessen-Engels, L.; de Laat-Arts, K.; Damming, D.; Santegoeds-Lenssen, E.W.H.; van Kuppeveld, D.W.J.; et al. BYON4228 is a pan-allelic antagonistic SIRPα antibody that potentiates destruction of antibody-opsonized tumor cells and lacks binding to SIRPγ on T cells. J. Immunother. Cancer 2023, 11. [Google Scholar] [CrossRef]
- Mayumi, S.; Takuya, T.; Yoko, I.; Shinko, H.; Saori, I.; Yoshitaka, I.; Jun, I.; Reimi, K.; Toshiaki, O.; Teiji, W.; et al. 808 Blocking “don’t-eat-me” signal of CD47-SIRPα by anti-SIRPα antibody enhances anti-tumor efficacy of trastuzumab deruxtecan. J. ImmunoTher. Cancer 2022, 10, A844. [Google Scholar] [CrossRef]
- Xu, C.A.; Feng, A.Z.; Ramineni, C.K.; Wallace, M.R.; Culyba, E.K.; Guay, K.P.; Mehta, K.; Mabry, R.; Farrand, S.; Xu, J.; et al. L(445)P mutation on heavy chain stabilizes IgG(4) under acidic conditions. MAbs 2019, 11, 1289–1299. [Google Scholar] [CrossRef]
- Bloom, J.W.; Madanat, M.S.; Marriott, D.; Wong, T.; Chan, S.Y. Intrachain disulfide bond in the core hinge region of human IgG4. Protein Sci. 1997, 6, 407–415. [Google Scholar] [CrossRef]
- Hezareh, M.; Hessell, A.J.; Jensen, R.C.; van de Winkel, J.G.; Parren, P.W. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 2001, 75, 12161–12168. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, X.X.; Shimada, H.; Ikegaki, N. A Perspective on the CD47-SIRPA Axis in High-Risk Neuroblastoma. Curr. Oncol. 2024, 31, 3212-3226. https://doi.org/10.3390/curroncol31060243
Tang XX, Shimada H, Ikegaki N. A Perspective on the CD47-SIRPA Axis in High-Risk Neuroblastoma. Current Oncology. 2024; 31(6):3212-3226. https://doi.org/10.3390/curroncol31060243
Chicago/Turabian StyleTang, Xao X., Hiroyuki Shimada, and Naohiko Ikegaki. 2024. "A Perspective on the CD47-SIRPA Axis in High-Risk Neuroblastoma" Current Oncology 31, no. 6: 3212-3226. https://doi.org/10.3390/curroncol31060243
APA StyleTang, X. X., Shimada, H., & Ikegaki, N. (2024). A Perspective on the CD47-SIRPA Axis in High-Risk Neuroblastoma. Current Oncology, 31(6), 3212-3226. https://doi.org/10.3390/curroncol31060243