High-LET-Radiation-Induced Persistent DNA Damage Response Signaling and Gastrointestinal Cancer Development

 ,
,  , and
, and
Abstract
1. Introduction
2. Role of DNA Repair Machinery in Cellular Response to IR
3. DDR Alterations in GI Cancer
4. DDR Alterations in Heavy-Ion-Radiation-Induced GI-Carcinogenesis
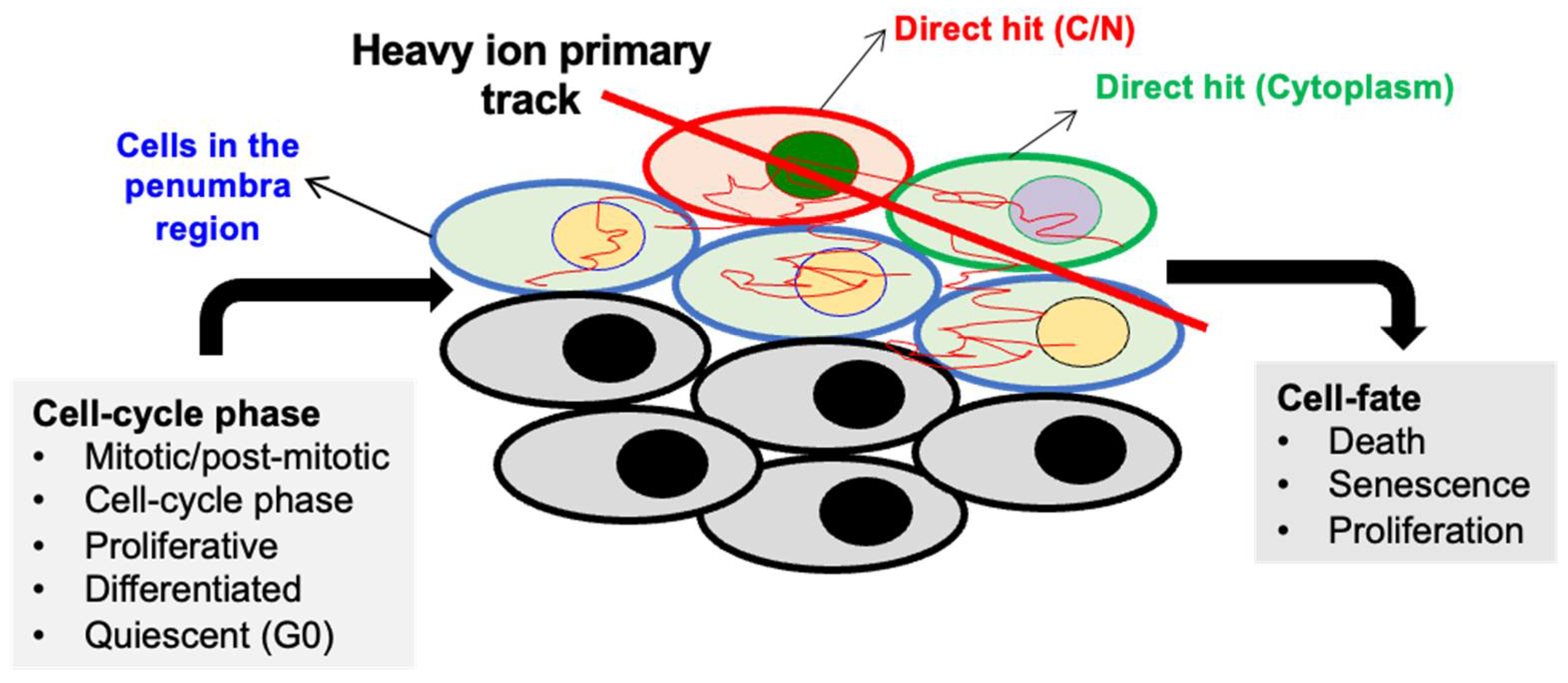
4.1. In Vitro Studies
4.2. Animal Model Studies
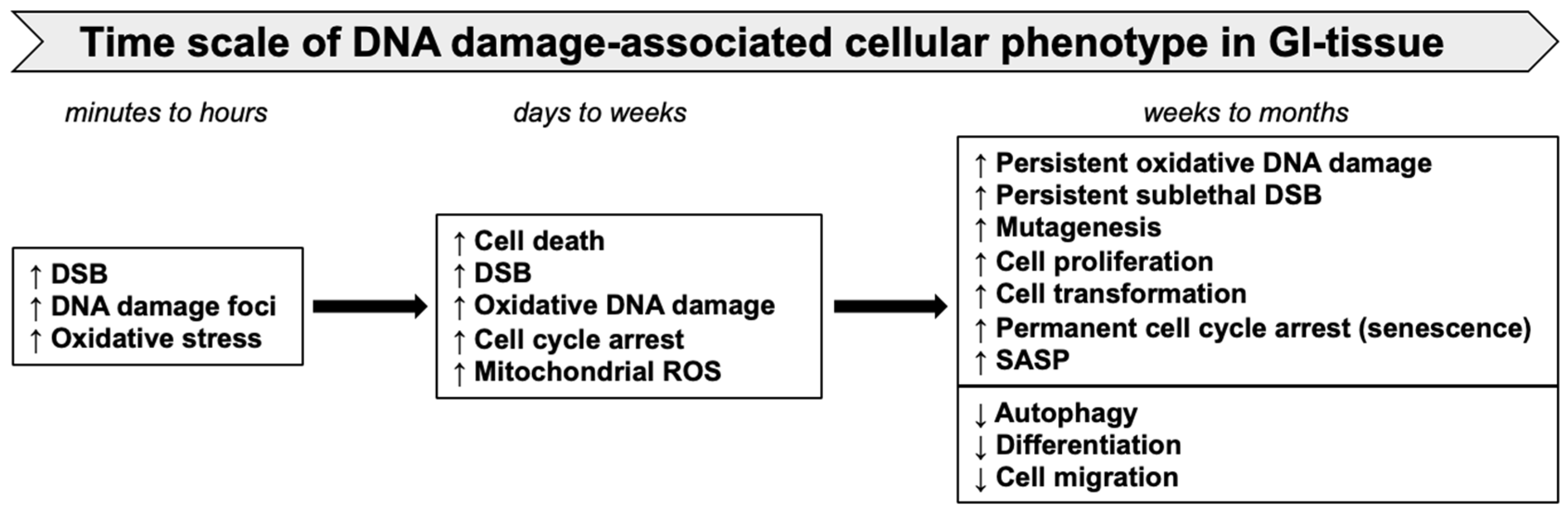
5. Persistent DDR, Cellular Senescence, and Accumulation of SASP Cells
6. SASP Signaling in GI Cancer Progression
7. Space-Radiation-Induced GI-Cancer Risk Reduction through DDR Modification
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 6th ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Joiner, M.C.; van der Kogel, A.J. Basic Clinical Radiobiology; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Tinganelli, W.; Durante, M. Carbon Ion Radiobiology. Cancers 2020, 12, 3022. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T. Particle Track Structure and Biological Implications. In Handbook of Bioastronautics; Springer: Berlin/Heidelberg, Germany, 2021; pp. 287–312. [Google Scholar]
- Allen, C.; Borak, T.B.; Tsujii, H.; Nickoloff, J.A. Heavy charged particle radiobiology: Using enhanced biological effectiveness and improved beam focusing to advance cancer therapy. Mutat. Res. 2011, 711, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Townsend, L.W. Implications of the space radiation environment for human exploration in deep space. Radiat. Prot. Dosim. 2005, 115, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Norbury, J.W.; Schimmerling, W.; Slaba, T.C.; Azzam, E.I.; Badavi, F.F.; Baiocco, G.; Benton, E.; Bindi, V.; Blakely, E.A.; Blattnig, S.R.; et al. Galactic cosmic ray simulation at the NASA Space Radiation Laboratory. Life Sci. Space Res. 2016, 8, 38–51. [Google Scholar] [CrossRef]
- Peracchi, S.; James, B.; Pagani, F.; Pan, V.; Vohradsky, J.; Bolst, D.; Prokopovich, D.A.; Guatelli, S.; Petasecca, M.; Lerch, M.L.; et al. Radiation Shielding Evaluation of Spacecraft Walls Against Heavy Ions Using Microdosimetry. IEEE Trans. Nucl. Sci. 2020, 68, 897–905. [Google Scholar] [CrossRef]
- Becker, D.; Kumar, A.; Adhikary, A.; Sevilla, M.D. Gamma- and Ion-beam DNA Radiation Damage: Theory and Experiment. In DNA Damage, DNA Repair and Disease; Dizdaroglu, M., Llyod, R.S., Eds.; Royal Society of Chemistry (RSC): London, UK, 2020; Volume 2, Chapter 31; pp. 426–457. [Google Scholar]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef]
- Suman, S.; Jaruga, P.; Dizdaroglu, M.; Fornace, A.J.; Datta, K. Heavy ion space radiation triggers ongoing DNA base damage by downregulating DNA repair pathways. Life Sci. Space Res. 2020, 27, 27–32. [Google Scholar] [CrossRef]
- Singh, V.; Johansson, P.; Torchinsky, D.; Lin, Y.-L.; Öz, R.; Ebenstein, Y.; Hammarsten, O.; Westerlund, F. Quantifying DNA damage induced by ionizing radiation and hyperthermia using single DNA molecule imaging. Transl. Oncol. 2020, 13, 100822. [Google Scholar] [CrossRef]
- Asaithamby, A.; Chen, D.J. Mechanism of cluster DNA damage repair in response to high-atomic number and energy particles radiation. Mutat. Res. 2011, 711, 87–99. [Google Scholar] [CrossRef]
- Ray, S.; Cekanaviciute, E.; Lima, I.P.; Sørensen, B.S.; Costes, S.V. Comparing Photon and Charged Particle Therapy Using DNA Damage Biomarkers. Int. J. Part. Ther. 2018, 5, 15–24. [Google Scholar] [CrossRef]
- Perez, R.L.; Nicolay, N.H.; Wolf, J.C.; Frister, M.; Schmezer, P.; Weber, K.J.; Huber, P.E. DNA damage response of clinical carbon ion versus photon radiation in human glioblastoma cells. Radiother. Oncol. 2019, 133, 77–86. [Google Scholar] [CrossRef]
- Nakano, T.; Akamatsu, K.; Tsuda, M.; Tujimoto, A.; Hirayama, R.; Hiromoto, T.; Tamada, T.; Ide, H.; Shikazono, N. Formation of clustered DNA damage in vivo upon irradiation with ionizing radiation: Visualization and analysis with atomic force microscopy. Proc. Natl. Acad. Sci. USA 2022, 119, e2119132119. [Google Scholar] [CrossRef]
- Adhikary, A.; Becker, D.; Sevilla, M.D. Electron spin resonance of radicals in irradiated, D.N.A. In Applications of EPR in Radiation Research; Lund, A., Shiotani, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 299–352. [Google Scholar]
- Hagiwara, Y.; Oike, T.; Niimi, A.; Yamauchi, M.; Sato, H.; Limsirichaikul, S.; Held, K.D.; Nakano, T.; Shibata, A. Clustered DNA double-strand break formation and the repair pathway following heavy-ion irradiation. J. Radiat. Res. 2019, 60, 69–79. [Google Scholar] [CrossRef]
- Hada, M.; Georgakilas, A.G. Formation of clustered DNA damage after high-LET irradiation: A review. J. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef]
- Pomerantz, R.T.; Temiakov, D.; Anikin, M.; Vassylyev, D.G.; McAllister, W.T. A mechanism of nucleotide misincorporation during transcription due to template-strand misalignment. Mol. Cell 2006, 24, 245–255. [Google Scholar] [CrossRef][Green Version]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef]
- Pedroza-Garcia, J.A.; Xiang, Y.; De Veylder, L. Cell cycle checkpoint control in response to DNA damage by environmental stresses. Plant J. 2022, 109, 490–507. [Google Scholar] [CrossRef]
- Krenning, L.; van den Berg, J.; Medema, R.H. Life or death after a break: What determines the choice. Mol. Cell 2019, 76, 346–358. [Google Scholar] [CrossRef]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage-how and why we age. Elife 2021, 10, e62852. [Google Scholar] [CrossRef]
- Haque, S.; Dean, P.J. Stromal neoplasms of the rectum and anal canal. Hum. Pathol. 1992, 23, 762–767. [Google Scholar] [CrossRef]
- Dhuppar, S.; Mazumder, A. Measuring cell cycle-dependent DNA damage responses and p53 regulation on a cell-by-cell basis from image analysis. Cell Cycle 2018, 17, 1358–1371. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Hopkins, S.R.; Ju, L.; Jeggo, P.A. Distinct response of adult neural stem cells to low versus high dose ionising radiation. DNA Repair. 2019, 76, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.P.; Mather, S.J. General aspects of the cellular response to low- and high-LET radiation. Eur. J. Nucl. Med. 2001, 28, 541–561. [Google Scholar] [CrossRef] [PubMed]
- Yu, H. Typical cell signaling response to ionizing radiation: DNA damage and extranuclear damage. Chin. J. Cancer Res. 2012, 24, 83–89. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Raschellà, G.; Melino, G.; Malewicz, M. New factors in mammalian DNA repair-the chromatin connection. Oncogene 2017, 36, 4673–4681. [Google Scholar] [CrossRef]
- Jensen, R.B.; Rothenberg, E. Preserving genome integrity in human cells via DNA double-strand break repair. Mol. Biol. Cell 2020, 31, 859–865. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Negritto, M.C. Repairing double-strand DNA breaks. Nat. Educ. 2010, 3, 26. [Google Scholar]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11–RAD50–NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 2018, 87, 263. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130. [Google Scholar]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef]
- Heidenreich, E.; Novotny, R.; Kneidinger, B.; Holzmann, V.; Wintersberger, U. Non-homologous end joining as an important mutagenic process in cell cycle-arrested cells. EMBO J. 2003, 22, 2274–2283. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef]
- Melis, J.P.M.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- Preston, T.J.; Henderson, J.T.; McCallum, G.P.; Wells, P.G. Base excision repair of reactive oxygen species–initiated 7, 8-dihydro-8-oxo-2′-deoxyguanosine inhibits the cytotoxicity of platinum anticancer drugs. Mol. Cancer Ther. 2009, 8, 2015–2026. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, C.; Sun, F.; Wei, W.; Hu, B.; Wang, J. Both complexity and location of DNA damage contribute to cellular senescence induced by ionizing radiation. PLoS ONE 2016, 11, e0155725. [Google Scholar] [CrossRef] [PubMed]
- Lorković, Z.J.; Berger, F. Heterochromatin and DNA damage repair: Use different histone variants and relax. Nucleus 2017, 8, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Watts, F.Z. Repair of DNA Double-Strand Breaks in Heterochromatin. Biomolecules 2016, 6, 47. [Google Scholar] [CrossRef]
- Datta, K.; Suman, S.; Kallakury, B.V.; Fornace, A.J. Exposure to heavy ion radiation induces persistent oxidative stress in mouse intestine. PLoS ONE 2012, 7, e42224. [Google Scholar] [CrossRef]
- Sridharan, D.M.; Asaithamby, A.; Bailey, S.M.; Costes, S.V.; Doetsch, P.W.; Dynan, W.S.; Kronenberg, A.; Rithidech, K.N.; Saha, J.; Snijders, A.M.; et al. Understanding cancer development processes after HZE-particle exposure: Roles of ROS, DNA damage repair and inflammation. Radiat. Res. 2015, 183, 1–26. [Google Scholar] [CrossRef]
- Okayasu, R.; Okada, M.; Okabe, A.; Noguchi, M.; Takakura, K.; Takahashi, S. Repair of DNA damage induced by accelerated heavy ions in mammalian cells proficient and deficient in the non-homologous end-joining pathway. Radiat. Res. 2006, 165, 59–67. [Google Scholar] [CrossRef]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The major DNA repair pathway after both proton and carbon-ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat. Res. 2015, 183, 345–356. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Zhang, P.; Wang, Y. The Ku-dependent non-homologous end-joining but not other repair pathway is inhibited by high linear energy transfer ionizing radiation. DNA Repair. 2008, 7, 725–733. [Google Scholar] [CrossRef]
- Zhao, L.; Bao, C.; Shang, Y.; He, X.; Ma, C.; Lei, X.; Mi, D.; Sun, Y. The Determinant of DNA Repair Pathway Choices in Ionising Radiation-Induced DNA Double-Strand Breaks. BioMed Res. Int. 2020, 2020, 4834965. [Google Scholar] [CrossRef]
- Gupta, A.; Hunt, C.R.; Chakraborty, S.; Pandita, R.K.; Yordy, J.; Ramnarain, D.B.; Horikoshi, N.; Pandita, T.K. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat. Res. 2014, 181, 1–8. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef]
- Bonis, V.; Rossell, C.; Gehart, H. The Intestinal Epithelium—Fluid Fate and Rigid Structure from Crypt Bottom to Villus Tip. Front. Cell Dev. Biol. 2021, 9, 661931. [Google Scholar] [CrossRef]
- Swift, M.L.; Beishline, K.; Flashner, S.; Azizkhan-Clifford, J. DSB repair pathway choice is regulated by recruitment of 53BP1 through cell cycle-dependent regulation of Sp1. Cell Rep. 2021, 34, 108840. [Google Scholar] [CrossRef]
- Swift, M.L.; Azizkhan-Clifford, J. DNA damage-induced sumoylation of Sp1 induces its interaction with RNF4 and degradation in S phase to remove 53BP1 from DSBs and permit HR. DNA Repair. 2022, 111, 103289. [Google Scholar] [CrossRef]
- Chao, H.X.; Poovey, C.E.; Privette, A.A.; Grant, G.D.; Cook, J.G.; Purvis, J.E. Orchestration of DNA Damage Checkpoint Dynamics across the Human Cell Cycle. Cell Syst. 2017, 5, 445–459.e5. [Google Scholar] [CrossRef]
- Chakraborty, U.; Alani, E. Understanding how mismatch repair proteins participate in the repair/anti-recombination decision. FEMS Yeast Res. 2016, 16, fow071. [Google Scholar] [CrossRef][Green Version]
- Zhao, L.; Mi, D.; Hu, B.; Sun, Y. A generalized target theory and its applications. Sci. Rep. 2015, 5, 14568. [Google Scholar] [CrossRef]
- Saha, J.; Wilson, P.; Thieberger, P.; Lowenstein, D.; Wang, M.; Cucinotta, F.A. Biological characterization of low-energy ions with high-energy deposition on human cells. Radiat. Res. 2014, 182, 282–291. [Google Scholar] [CrossRef]
- Yahyapour, R.; Salajegheh, A.; Safari, A.; Amini, P.; Rezaey-An, A.; Amraee, A.; Najafi, M. Radiation-induced Non-targeted Effect and Carcinogenesis; Implications in Clinical Radiotherapy. J. Biomed. Phys. Eng. 2018, 8, 435–446. [Google Scholar] [CrossRef]
- Kanagaraj, K.; Rajan, V.; Pandey, B.N.; Thayalan, K.; Venkatachalam, P. Primary and secondary bystander effect and genomic instability in cells exposed to high and low linear energy transfer radiations. Int. J. Radiat. Biol. 2019, 95, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res. 1992, 52, 6394–6396. [Google Scholar] [PubMed]
- Shuryak, I.; Sachs, R.K.; Brenner, D.J. Biophysical models of radiation bystander effects: 1. Spatial effects in three-dimensional tissues. Radiat. Res. 2007, 168, 741–749. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feringa, F.M.; Raaijmakers, J.A.; Hadders, M.A.; Vaarting, C.; Macurek, L.; Heitink, L.; Krenning, L.; Medema, R.H. Persistent repair intermediates induce senescence. Nat. Commun. 2018, 9, 3923. [Google Scholar] [CrossRef]
- Jaiswal, H.; Lindqvist, A. Bystander communication and cell cycle decisions after DNA damage. Front. Genet. 2015, 6, 63. [Google Scholar] [CrossRef]
- Kumar, S.; Suman, S.; Fornace, A.J.; Datta, K. Space radiation triggers persistent stress response, increases senescent signaling, and decreases cell migration in mouse intestine. Proc. Natl. Acad. Sci. USA 2018, 115, E9832–E9841. [Google Scholar] [CrossRef]
- Kumar, S.; Suman, S.; Fornace, A.J.; Datta, K. Intestinal stem cells acquire premature senescence and senescence associated secretory phenotype concurrent with persistent DNA damage after heavy ion radiation in mice. Aging 2019, 11, 4145–4158. [Google Scholar] [CrossRef]
- Datta, K.; Suman, S.; Kallakury, B.V.; Fornace, A.J. Heavy ion radiation exposure triggered higher intestinal tumor frequency and greater β-catenin activation than γ radiation in APC(Min/+) mice. PLoS ONE 2013, 8, e59295. [Google Scholar] [CrossRef]
- Datta, K.; Suman, S.; Kumar, S.; Fornace, A.J. Colorectal Carcinogenesis, Radiation Quality, and the Ubiquitin-Proteasome Pathway. J. Cancer 2016, 7, 174–183. [Google Scholar] [CrossRef]
- Datta, K.; Suman, S.; Moon, B.-H.; Fornace, A.J. Space radiation-induced decline in gut autophagy and expansion of both mitotic and senescent population denotes an aging phenotype with enhanced cancer risk. Cancer Res. 2019, 79, 3736. [Google Scholar] [CrossRef]
- Suman, S.; Kumar, S.; Fornace, A.J.; Datta, K. Space radiation exposure persistently increased leptin and IGF1 in serum and activated leptin-IGF1 signaling axis in mouse intestine. Sci. Rep. 2016, 6, 31853. [Google Scholar] [CrossRef]
- Suman, S.; Kumar, S.; Fornace, A.J.; Datta, K. The effect of carbon irradiation is associated with greater oxidative stress in mouse intestine and colon relative to γ-rays. Free Radic. Res. 2018, 52, 556–567. [Google Scholar] [CrossRef]
- Song, Y.; Huang, J.; Liang, D.; Hu, Y.; Mao, B.; Li, Q.; Sun, H.; Yang, Y.; Zhang, J.; Zhang, H.; et al. DNA Damage Repair Gene Mutations Are Indicative of a Favorable Prognosis in Colorectal Cancer Treated with Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 549777. [Google Scholar] [CrossRef]
- Arai, H.; Elliott, A.; Wang, J.; Battaglin, F.; Soni, S.; Zhang, W.; Sohal, D.; Goldberg, R.M.; Hall, M.J.; Scott, A.J.; et al. The landscape of DNA damage response (DDR) pathway in colorectal cancer (CRC). J. Clin. Oncol. 2020, 38, 4064. [Google Scholar] [CrossRef]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann. Transl. Med. 2020, 8, 1685. [Google Scholar] [CrossRef]
- Chang, P.-Y.; Chang, S.-C.; Wang, M.-C.; Chen, J.-S.; Tsai, W.-S.; You, J.-F.; Chen, C.-C.; Liu, H.-L.; Chiang, J.-M. Pathogenic Germline Mutations of DNA Repair Pathway Components in Early-Onset Sporadic Colorectal Polyp and Cancer Patients. Cancers 2020, 12, 3560. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar]
- Catalano, F.; Borea, R.; Puglisi, S.; Boutros, A.; Gandini, A.; Cremante, M.; Martelli, V.; Sciallero, S.; Puccini, A. Targeting the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Colorectal Cancer. Cancers 2022, 14, 1388. [Google Scholar] [CrossRef]
- Liebl, M.C.; Hofmann, T.G. The Role of p53 Signaling in Colorectal Cancer. Cancers 2021, 13, 2125. [Google Scholar] [CrossRef]
- Lee, K.; Tosti, E.; Edelmann, W. Mouse models of DNA mismatch repair in cancer research. DNA Repair. 2016, 38, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, N.; Tierling, S.; Es, H.A.; Varkiani, M.; Mojarad, E.N.; Aghdaei, H.A.; Walter, J.; Totonchi, M. DNA methylation biomarkers in colorectal cancer: Clinical applications for precision medicine. Int. J. Cancer 2022, 151, 2068–2081. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Creighton, C.J.; Schultz, N.; Shinbrot, E.; Chang, K.; Gunaratne, P.H.; Muzny, D.; Sander, C.; Hamilton, S.R.; Gibbs, R.A.; et al. MLH1-silenced and non-silenced subgroups of hypermutated colorectal carcinomas have distinct mutational landscapes. J. Pathol. 2013, 229, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Molnár, B.; Galamb, O.; Péterfia, B.; Wichmann, B.; Csabai, I.; Bodor, A.; Kalmár, A.; Szigeti, K.A.; Barták, B.K.; Nagy, Z.B.; et al. Gene promoter and exon DNA methylation changes in colon cancer development—mRNA expression and tumor mutation alterations. BMC Cancer 2018, 18, 695. [Google Scholar] [CrossRef]
- Steine, E.J.; Ehrich, M.; Bell, G.W.; Raj, A.; Reddy, S.; van Oudenaarden, A.; Jaenisch, R.; Linhart, H.G. Genes methylated by DNA methyltransferase 3b are similar in mouse intestine and human colon cancer. J. Clin. Investig. 2011, 121, 1748–1752. [Google Scholar] [CrossRef]
- Grimm, C.; Chavez, L.; Vilardell, M.; Farrall, A.L.; Tierling, S.; Böhm, J.W.; Grote, P.; Lienhard, M.; Dietrich, J.; Timmermann, B.; et al. DNA-methylome analysis of mouse intestinal adenoma identifies a tumour-specific signature that is partly conserved in human colon cancer. PLoS Genet. 2013, 9, e1003250. [Google Scholar] [CrossRef]
- van der Heijden, M.; Vermeulen, L. Stem cells in homeostasis and cancer of the gut. Mol. Cancer 2019, 18, 66. [Google Scholar] [CrossRef]
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.; Campbell, B.J.; Pritchard, D.M. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef]
- Kamiya, K.; Ozasa, K.; Akiba, S.; Niwa, O.; Kodama, K.; Takamura, N.; Zaharieva, E.K.; Kimura, Y.; Wakeford, R. Long-term effects of radiation exposure on health. The Lancet 2015, 386, 469–478. [Google Scholar] [CrossRef]
- Gilbert, E.S. Ionising radiation and cancer risks: What have we learned from epidemiology. Int. J. Radiat. Biol. 2009, 85, 467–482. [Google Scholar] [CrossRef]
- Boice, J.D. The Million Person Study relevance to space exploration and Mars. Int. J. Radiat. Biol. 2022, 98, 551–559. [Google Scholar] [CrossRef]
- Brenner, A.V.; Preston, D.L.; Sakata, R.; Cologne, J.; Sugiyama, H.; Utada, M.; Cahoon, E.K.; Grant, E.; Mabuchi, K.; Ozasa, K. Comparison of All Solid Cancer Mortality and Incidence Dose-Response in the Life Span Study of Atomic Bomb Survivors, 1958–2009. Radiat. Res. 2022, 197, 491–508. [Google Scholar] [CrossRef]
- Cucinotta, F.A.; Alp, M.; Rowedder, B.; Kim, M.H. Safe days in space with acceptable uncertainty from space radiation exposure. Life Sci. Space Res. 2015, 5, 31–38. [Google Scholar] [CrossRef]
- Cucinotta, F.A.; To, K.; Cacao, E. Predictions of space radiation fatality risk for exploration missions. Life Sci. Space Res. 2017, 13, 1–11. [Google Scholar] [CrossRef]
- Cucinotta, F.A.; Cacao, E. Non-Targeted Effects Models Predict Significantly Higher Mars Mission Cancer Risk than Targeted Effects Models. Sci. Rep. 2017, 7, 1832. [Google Scholar] [CrossRef]
- Suman, S.; Moon, B.H.; Thakor, H.; Fornace, A.J.; Datta, K. Wip1 abrogation decreases intestinal tumor frequency in APC(Min/+) mice irrespective of radiation quality. Radiat. Res. 2014, 182, 345–349. [Google Scholar] [CrossRef]
- Suman, S.; Kumar, S.; Fornace, A.J.; Datta, K. Decreased RXRα is Associated with Increased β-Catenin/TCF4 in (56)Fe-Induced Intestinal Tumors. Front. Oncol. 2015, 5, 218. [Google Scholar] [CrossRef]
- Suman, S.; Kumar, S.; Moon, B.-H.; Strawn, S.J.; Thakor, H.; Fan, Z.; Shay, J.W.; Fornace, A.J.; Datta, K. Relative Biological Effectiveness of Energetic Heavy Ions for Intestinal Tumorigenesis Shows Male Preponderance and Radiation Type and Energy Dependence in APC(1638N/+) Mice. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 131–138. [Google Scholar] [CrossRef]
- Suman, S.; Kumar, S.; Moon, B.H.; Fornace, A.J.; Datta, K. Low and high dose rate heavy ion radiation-induced intestinal and colonic tumorigenesis in APC1638N/+ mice. Life Sci. Space Res. 2017, 13, 45–50. [Google Scholar] [CrossRef]
- Suman, S.; Kumar, S.; Moon, B.H.; Angdisen, J.; Kallakury, B.V.; Datta, K.; Fornace, A.J., Jr. Effects of dietary aspirin on high-LET radiation-induced prostaglandin E2 levels and gastrointestinal tumorigenesis in Apc1638N/+ mice. Life Sci. Space Res. 2021, 31, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Kumar, S.; Kallakury, B.V.S.; Moon, B.-H.; Angdisen, J.; Datta, K.; Fornace, A.J. Predominant contribution of the dose received from constituent heavy-ions in the induction of gastrointestinal tumorigenesis after simulated space radiation exposure. Radiat. Environ. Biophys. 2022, 61, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Roig, A.I.; Hight, S.K.; Shay, J.W. Two- and three-dimensional models for risk assessment of radiation-enhanced colorectal tumorigenesis. Radiat. Res. 2009, 171, 33–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eskiocak, U.; Kim, S.B.; Roig, A.I.; Kitten, E.; Batten, K.; Cornelius, C.; Zou, Y.S.; Wright, W.E.; Shay, J.W. CDDO-Me protects against space radiation-induced transformation of human colon epithelial cells. Radiat. Res. 2010, 174, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Eskiocak, U.; Kim, S.B.; Roig, A.I.; Kitten, E.; Batten, K.; Cornelius, C.; Zou, Y.S.; Wright, W.E.; Shay, J.W. Radiation promotes colorectal cancer initiation and progression by inducing senescence-associated inflammatory responses. Oncogene 2016, 35, 3365–3375. [Google Scholar]
- Buonanno, M.; de Toledo, S.M.; Azzam, E.I. Increased frequency of spontaneous neoplastic transformation in progeny of bystander cells from cultures exposed to densely ionizing radiation. PLoS ONE 2011, 6, e21540. [Google Scholar] [CrossRef]
- Turker, M.S.; Grygoryev, D.; Lasarev, M.; Ohlrich, A.; Rwatambuga, F.A.; Johnson, S.; Dan, C.; Eckelmann, B.; Hryciw, G.; Mao, J.H.; et al. Simulated space radiation-induced mutants in the mouse kidney display widespread genomic change. PLoS ONE 2017, 12, e0180412. [Google Scholar] [CrossRef]
- Kronenberg, A.; Gauny, S.; Kwoh, E.; Connolly, L.; Dan, C.; Lasarev, M.; Turker, M.S. Comparative analysis of cell killing and autosomal mutation in mouse kidney epithelium exposed to 1 GeV/nucleon iron ions in vitro or in situ. Radiat. Res. 2009, 172, 550–557. [Google Scholar] [CrossRef]
- Aghabozorgi, A.S.; Bahreyni, A.; Soleimani, A.; Bahrami, A.; Khazaei, M.; Ferns, G.A.; Avan, A.; Hassanian, S.M. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie 2019, 157, 64–71. [Google Scholar] [CrossRef]
- Kumar, S.; Suman, S.; Kallakury, B.V.; Moon, B.-H.; Fornace, A.J.; Datta, K. Inverse effect of 28Si and 56Fe radiation on intestinal tumorigenesis vs. carcinogenesis in APC1638N/+ mice. Cancer Res. 2019, 79, 3728. [Google Scholar] [CrossRef]
- Shuryak, I.; Fornace, A.J., Jr.; Datta, K.; Suman, S.; Kumar, S.; Sachs, R.K.; Brenner, D.J. Scaling Human Cancer Risks from Low LET to High LET when Dose-Effect Relationships are Complex. Radiat. Res. 2017, 187, 476–482. [Google Scholar] [CrossRef]
- Smits, R.; van Oordt, W.V.D.H.; Luz, A.; Zurcher, C.; Jagmohan-Changur, S.; Breukel, C.; Khan, P.M.; Fodde, R. Apc1638N: A mouse model for familial adenomatous polyposis-associated desmoid tumors and cutaneous cysts. Gastroenterology 1998, 114, 275–283. [Google Scholar] [CrossRef]
- Smits, R.; Kielman, M.F.; Breukel, C.; Zurcher, C.; Neufeld, K.; Jagmohan-Changur, S.; Hofland, N.; van Dijk, J.; White, R.; Edelmann, W.; et al. Apc1638T: A mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev. 1999, 13, 1309–1321. [Google Scholar] [CrossRef]
- Kuraguchi, M.; Yang, K.; Wong, E.; Avdievich, E.; Fan, K.; Kolodner, R.D.; Lipkin, M.; Brown, A.M.; Kucherlapati, R.; Edelmann, W. The distinct spectra of tumor-associated Apc mutations in mismatch repair-deficient Apc1638N mice define the roles of MSH3 and MSH6 in DNA repair and intestinal tumorigenesis. Cancer Res. 2001, 61, 7934–7942. [Google Scholar]
- Knudson, A.G. Cancer genetics. Am. J. Med. Genet. 2002, 111, 96–102. [Google Scholar] [CrossRef]
- Lacina, L.; Plzak, J.; Kodet, O.; Szabo, P.; Chovanec, M.; Dvorankova, B.; Smetana, K., Jr. Cancer Microenvironment: What Can We Learn from the Stem Cell Niche. Int. J. Mol. Sci. 2015, 16, 24094–24110. [Google Scholar] [CrossRef]
- Vicente-Dueñas, C.; Hauer, J.; Cobaleda, C.; Borkhardt, A.; Sánchez-García, I. Epigenetic Priming in Cancer Initiation. Trends Cancer 2018, 4, 408–417. [Google Scholar] [CrossRef]
- Matsuya, Y.; Sasaki, K.; Yoshii, Y.; Okuyama, G. Integrated modelling of cell responses after irradiation for DNA-targeted effects and non-targeted effects. Sci. Rep. 2018, 8, 4849. [Google Scholar] [CrossRef]
- Suman, S.; Rodriguez, O.C.; Winters, T.A.; Fornace, A.J.; Albanese, C.; Datta, K. Therapeutic and space radiation exposure of mouse brain causes impaired DNA repair response and premature senescence by chronic oxidant production. Aging 2013, 5, 607–622. [Google Scholar] [CrossRef]
- Leach, J.K.; Van Tuyle, G.; Lin, P.S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar]
- Yoshida, T.; Goto, S.; Kawakatsu, M.; Urata, Y.; Li, T.S. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free Radic. Res. 2012, 46, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Leduc, A.; Pottier, I.; Prévost, V.; Sichel, F.; Lefaix, J.L. Dramatic increase in oxidative stress in carbon-irradiated normal human skin fibroblasts. PLoS ONE 2013, 8, e85158. [Google Scholar] [CrossRef] [PubMed]
- Tower, J. Stress and stem cells. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar]
- Buonanno, M.; De Toledo, S.M.; Howell, R.W.; Azzam, E.I. Low-dose energetic protons induce adaptive and bystander effects that protect human cells against DNA damage caused by a subsequent exposure to energetic iron ions. J. Radiat. Res. 2015, 56, 502–508. [Google Scholar] [CrossRef]
- Li, Z.; Doho, G.; Zheng, X.; Jella, K.K.; Li, S.; Wang, Y.; Dynan, W.S. Co-culturing with High-Charge and Energy Particle Irradiated Cells Increases Mutagenic Joining of Enzymatically Induced DNA Double-Strand Breaks in Nonirradiated Cells. Radiat. Res. 2015, 184, 249–258. [Google Scholar] [CrossRef]
- Yang, H.; Asaad, N.; Held, K.D. Medium-mediated intercellular communication is involved in bystander responses of X-ray-irradiated normal human fibroblasts. Oncogene 2005, 24, 2096–2103. [Google Scholar] [CrossRef]
- Mao, L.; Guo, C.; Zheng, S. Elevated urinary 8-oxo-7,8-dihydro-2′-deoxyguanosine and serum uric acid are associated with progression and are prognostic factors of colorectal cancer. OncoTargets Ther. 2018, 11, 5895–5902. [Google Scholar] [CrossRef]
- Oizumi, T.; Ohno, R.; Yamabe, S.; Funayama, T.; Nakamura, A.J. Repair Kinetics of DNA Double Strand Breaks Induced by Simulated Space Radiation. Life 2020, 10, 341. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Niimi, A.; Isono, M.; Yamauchi, M.; Yasuhara, T.; Limsirichaikul, S.; Oike, T.; Sato, H.; Held, K.D.; Nakano, T.; et al. 3D-structured illumination microscopy reveals clustered DNA double-strand break formation in widespread γH2AX foci after high LET heavy-ion particle radiation. Oncotarget 2017, 8, 109370–109381. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Armitage, P. Multistage models of carcinogenesis. Environ. Health Perspect. 1985, 63, 195–201. [Google Scholar] [CrossRef]
- Alrawi, S.J.; Schiff, M.; Carroll, R.E.; Dayton, M.; Gibbs, J.F.; Kulavlat, M.; Tan, D.; Berman, K.; Stoler, D.L.; Anderson, G.R. Aberrant crypt foci. Anticancer Res. 2006, 26, 107–119. [Google Scholar]
- Suman, S.; Johnson, M.D.; Fornace, A.J.; Datta, K. Exposure to ionizing radiation causes long-term increase in serum estradiol and activation of PI3K-Akt signaling pathway in mouse mammary gland. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 500–507. [Google Scholar] [CrossRef]
- Guo, Y.; Ayers, J.L.; Carter, K.T.; Wang, T.; Maden, S.K.; Edmond, D.; Newcomb, P.P.; Li, C.; Ulrich, C.; Yu, M.; et al. Senescence-associated tissue microenvironment promotes colon cancer formation through the secretory factor GDF15. Aging Cell 2019, 18, e13013. [Google Scholar] [CrossRef]
- Werneth, C.M.; Slaba, T.C.; Huff, J.L.; Patel, Z.S.; Simonsen, L.C. Medical Countermeasure Requirements to Meet NASA’s Space Radiation Permissible Exposure Limits for a Mars Mission Scenario. Health Phys. 2022, 123, 116–127. [Google Scholar] [CrossRef]
- Suman, S.; Khaitan, D.; Pati, U.; Seth, R.K.; Chandna, S. Stress response of a p53 homologue in the radioresistant Sf9 insect cells. Int. J. Radiat. Biol. 2009, 85, 238–249. [Google Scholar] [CrossRef]
- Chandna, S. RE: Multiple factors conferring high radioresistance in insect Sf9 cells. (Mutagenesis, 24, 259-269, 2009). Mutagenesis 2010, 25, 431–432. [Google Scholar] [CrossRef][Green Version]
- Gladyshev, E.; Meselson, M. Extreme resistance of bdelloid rotifers to ionizing radiation. Proc. Natl. Acad. Sci. USA 2008, 105, 5139–5144. [Google Scholar] [CrossRef]
- van Haaften, G.; Vastenhouw, N.L.; Nollen, E.A.; Plasterk, R.H.; Tijsterman, M. Gene interactions in the DNA damage-response pathway identified by genome-wide RNA-interference analysis of synthetic lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 12992–12996. [Google Scholar] [CrossRef]
- Kim, S.; Shen, S.; Moore, D.F.; Shih, W.; Lin, Y.; Li, H.; Dolan, M.; Shao, Y.-H.; Lu-Yao, G.L. Late gastrointestinal toxicities following radiation therapy for prostate cancer. Eur. Urol. 2011, 60, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Pariset, E.; Bertucci, A.; Petay, M.; Malkani, S.; Macha, A.L.; Lima, I.G.P.; Gonzalez, V.G.; Tin, A.S.; Tang, J.; Plante, I.; et al. DNA Damage Baseline Predicts Resilience to Space Radiation and Radiotherapy. Cell Rep. 2020, 33, 108434. [Google Scholar] [CrossRef] [PubMed]
- Aengenvoort, J.; Sekeres, M.; Proksch, P.; Fritz, G. Targeting Mechanisms of the DNA Damage Response (DDR) and DNA Repair by Natural Compounds to Improve cAT-Triggered Tumor Cell Death. Molecules 2022, 27, 3567. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxid. Med. Cell. Longev. 2018, 2018, 2389523. [Google Scholar] [CrossRef]
- Roy, M.; Sinha, D.; Mukherjee, S.; Biswas, J. Curcumin prevents DNA damage and enhances the repair potential in a chronically arsenic-exposed human population in West Bengal, India. J. Cancer Prev. 2011, 20, 123–131. [Google Scholar] [CrossRef]
- Denissova, N.G.; Nasello, C.M.; Yeung, P.L.; Tischfield, J.A.; Brenneman, M.A. Resveratrol protects mouse embryonic stem cells from ionizing radiation by accelerating recovery from DNA strand breakage. Carcinogenesis 2012, 33, 149–155. [Google Scholar] [CrossRef]
- Caputo, F.; Vegliante, R.; Ghibelli, L. Redox modulation of the DNA damage response. Biochem. Pharmacol. 2012, 84, 1292–1306. [Google Scholar] [CrossRef]
- Kennedy, A.R.; Guan, J.; Ware, J.H. Countermeasures against space radiation induced oxidative stress in mice. Radiat. Environ. Biophys. 2007, 46, 201–203. [Google Scholar] [CrossRef]
- Elbialy, A. The role of antioxidants in restoring MAPK 14 and a DNA damage marker level following autophagy suppression. Open Biol. 2020, 10, 200253. [Google Scholar] [CrossRef]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 52, 662–668. [Google Scholar] [CrossRef]
- Li, H.; Busquets, O.; Verma, Y.; Syed, K.M.; Kutnowski, N.; Pangilinan, G.R.; Gilbert, L.A.; Bateup, H.S.; Rio, D.C.; Hockemeyer, D.; et al. Highly efficient generation of isogenic pluripotent stem cell models using prime editing. Elife 2022, 11, e79208. [Google Scholar] [CrossRef]
- Doman, J.L.; Sousa, A.A.; Randolph, P.B.; Chen, P.J.; Liu, D.R. Designing and executing prime editing experiments in mammalian cells. Nat. Protoc. 2022, 17, 2431–2468. [Google Scholar] [CrossRef]
- Suman, S.; Fornace, A.J. Countermeasure development against space radiation-induced gastrointestinal carcinogenesis: Current and future perspectives. Life Sci. Space Res. 2022, 35, 53–59. [Google Scholar] [CrossRef]
- Fornace, A.; Suman, S. Metformin prevents heavy-ion radiation-induced gastrointestinal (GI) tumorigenesis in Apc1638N/+ mice via regulation of IGF1-mTOR signaling. In Proceedings of the 44th COSPAR Scientific Assembly, Athens, Greece, 16–24 July 2022; Volume 44, p. 2663. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IR-Types | Physical Characteristics | |||
|---|---|---|---|---|
| Energy | Mass | LET (keV/μm) | Charge | |
| Photon (X-or γ-rays) | Yes | No | Low | X-ray (Negative); γ-rays (Neutral) |
| Proton (H+, i.e., nucleus of H atom) | Yes | Yes (Equivalent to proton) | Low to intermediate | Positive |
| Alpha particle (He2+) | Yes | Yes (Equivalent to helium nucleus) | Intermediate to high | Positive |
| Heavy-ion (Z > 2) | Yes | Yes (Equivalent to nucleus of an atom) | Intermediate to high | Positive |
| Neutron | Yes | Yes (Equivalent to neutron) | Intermediate to high | Neutral |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, K.; Kumar, S.; Datta, K.; Fornace, A.J., Jr.; Suman, S. High-LET-Radiation-Induced Persistent DNA Damage Response Signaling and Gastrointestinal Cancer Development. Curr. Oncol. 2023, 30, 5497-5514. https://doi.org/10.3390/curroncol30060416
Kumar K, Kumar S, Datta K, Fornace AJ Jr., Suman S. High-LET-Radiation-Induced Persistent DNA Damage Response Signaling and Gastrointestinal Cancer Development. Current Oncology. 2023; 30(6):5497-5514. https://doi.org/10.3390/curroncol30060416
Chicago/Turabian StyleKumar, Kamendra, Santosh Kumar, Kamal Datta, Albert J. Fornace, Jr., and Shubhankar Suman. 2023. "High-LET-Radiation-Induced Persistent DNA Damage Response Signaling and Gastrointestinal Cancer Development" Current Oncology 30, no. 6: 5497-5514. https://doi.org/10.3390/curroncol30060416
APA StyleKumar, K., Kumar, S., Datta, K., Fornace, A. J., Jr., & Suman, S. (2023). High-LET-Radiation-Induced Persistent DNA Damage Response Signaling and Gastrointestinal Cancer Development. Current Oncology, 30(6), 5497-5514. https://doi.org/10.3390/curroncol30060416