Advances in Immunotherapy for Hepatocellular Carcinoma (HCC)


Abstract
:1. Introduction
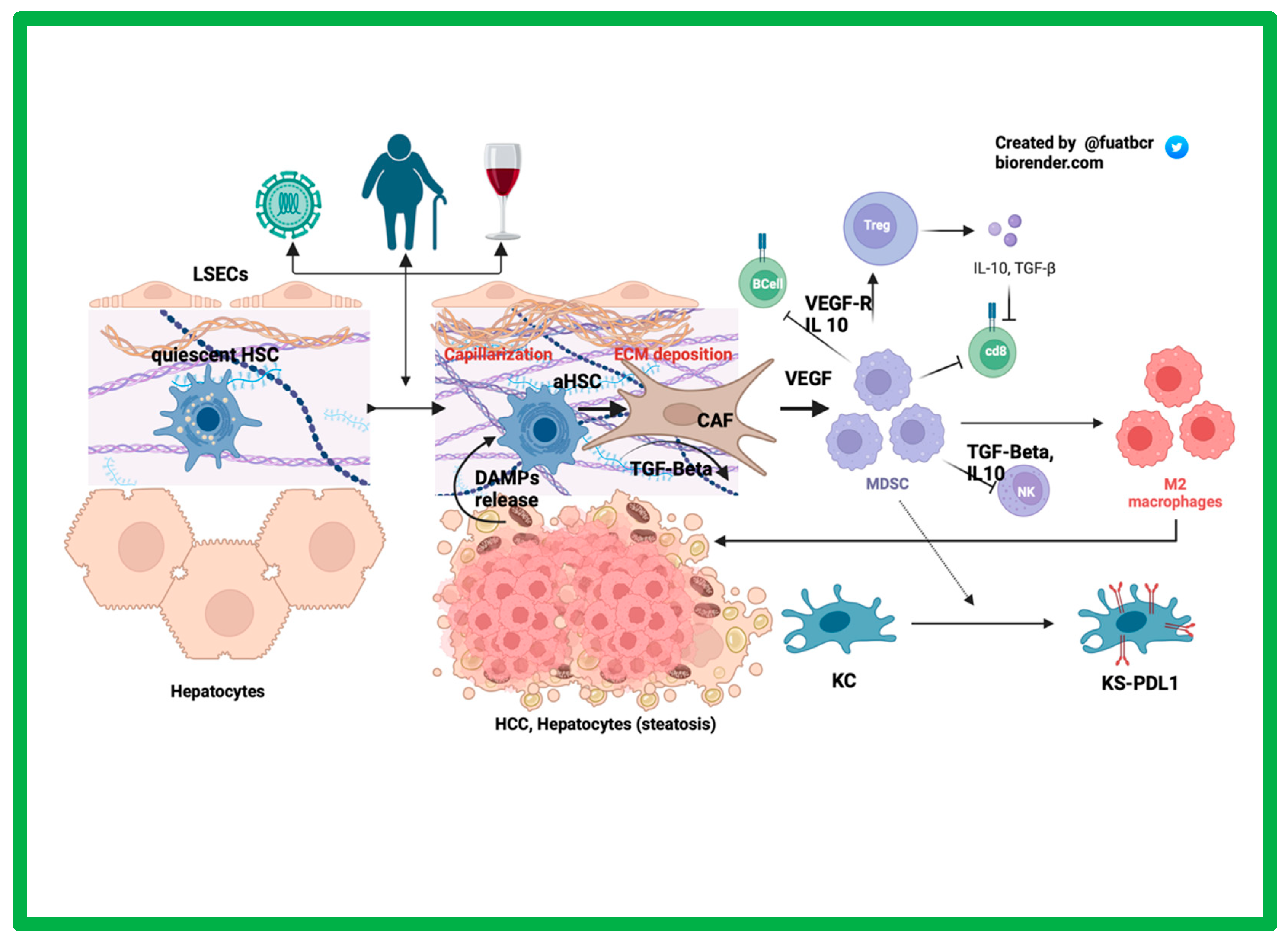
The Role of the Tumor Microenvironment in HCC
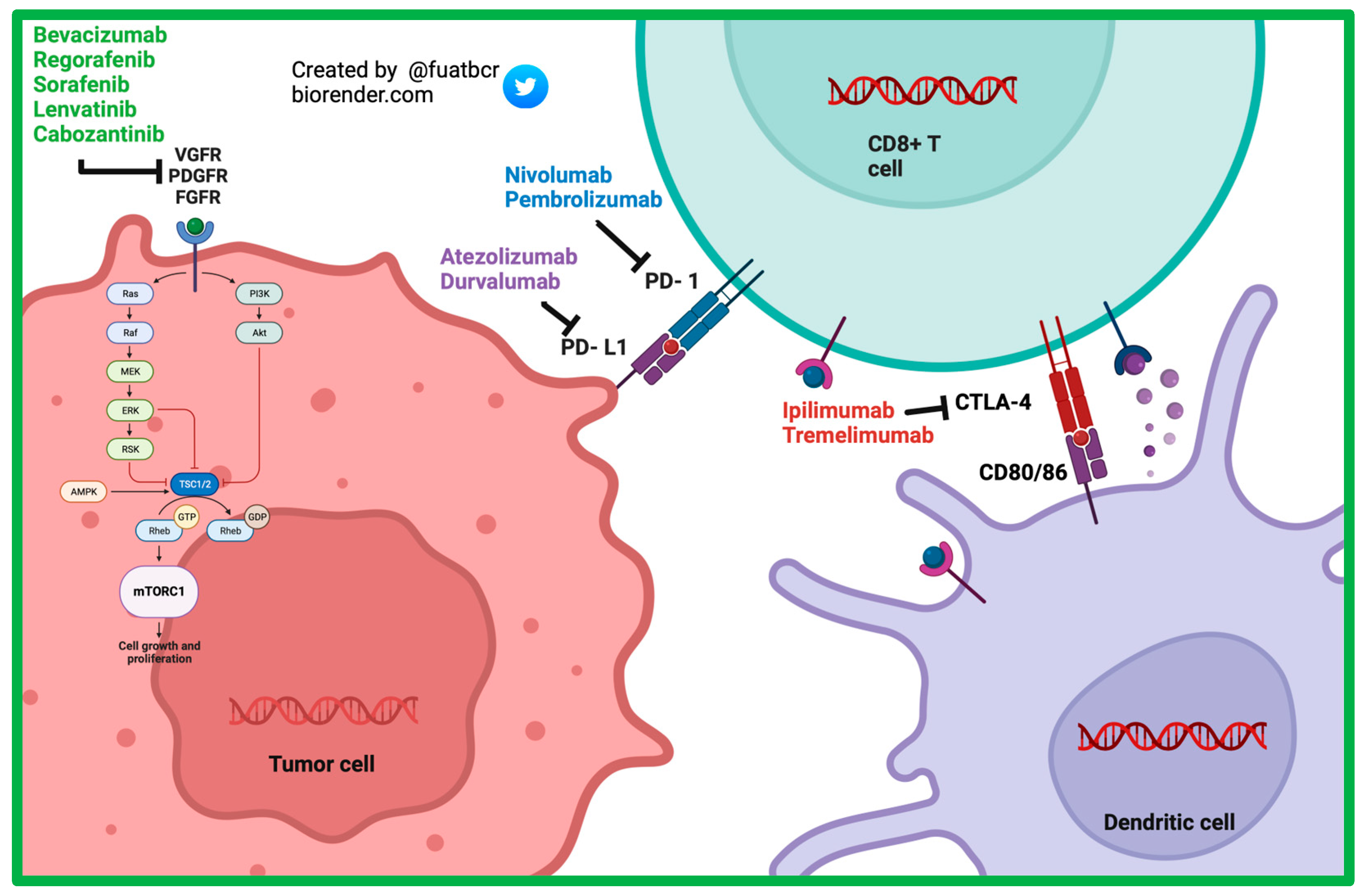
2. Current First-Line Therapies for Advanced/Metastatic HCC
2.1. TKI-Based Therapies
2.1.1. Sorafenib
2.1.2. Lenvatinib
2.2. ICI-Based Therapies
2.2.1. Atezolizumab and Bevacizumab
2.2.2. Durvalumab + Tremelimumab
3. Current Second-Line Therapies for Advanced/Metastatic HCC
3.1. ICI-Based Therapies
3.1.1. Nivolumab
3.1.2. Pembrolizumab
3.1.3. Nivolumab/Ipilimumab
4. Combination Therapy of ICI with Anti-Angiogenic Therapy and TKI
5. Chimeric Antigen Receptor (CAR)-T Cell Therapy
6. Small-Molecule Inhibitors
7. Resistance Mechanisms to ICIs in HCC and Possible Solutions
7.1. Internal Resistance
7.1.1. TMB
7.1.2. Gene Signatures and Biomarkers

{kind=link}
{kind=link}
| Internal Mechanisms | External Mechanisms |
|---|---|
|
|
|
|
|
|
7.2. External Resistance
8. Immunotherapy Combined with Locoregional Therapies
9. Potential Solutions to Overcome Resistance to Immune Checkpoint Inhibitors in HCC via Targeting Other Checkpoint Molecules
9.1. TIM-3
9.2. LAG-3
9.3. TIGIT
| TIM-3 Preclinical Studies | |
|---|---|
| Study | Findings |
| Anti-TIM-3 blockade after PD-1 failure in lung cancer mice models [110] | OS: 11.9 weeks in TIM-3 blockade after PD-1 failure versus 5.0 weeks in PD-1 blockade monotherapy (p = 0.0008) in mice |
| PD-1 and TIM-3 expression in HBV-associated HCC versus cirrhosis [127] | Greater PD-1 expression in tumor tissue compared to surrounding cirrhosis tissue (p < 0.001) Greater TIM-3 expression in tumor tissue compared to cirrhosis tissue (p < 0.001) |
| LAG-3 Preclinical Studies | |
| Mechanisms of enhanced anti-tumor immunity with dual blockade of LAG-3 and PD-1 in an ovarian murine tumor model [133] | Increase in CD8+ T cells and decrease in Treg cells after blockade CD8+ T cells were not exhausted |
| Tumor response with LAG-3 and PD-1 blockade in Sa1N fibrosarcoma and MC38-colorectal adenocarcinoma [132] | Combination: tumor resolution (% population): Sa1N fibrosarcoma: 70% MC38-colorectal adenocarcinoma: 80% Monotherapy: tumor resolution PD-1 and LAG-3 monotherapy: 0–40% |
| Outcome of PD-L1 and LAG-3 expression in HCC [137] | Patients with high LAG-3 and PD-1 had poorer overall survival compared to elevation of only LAG-3 or PD-1 |
| TIGIT Preclinical Studies | |
| Mechanisms of resistance of anti-PD-1 blockade in mice liver tumor and effects of PD-1 and TIGIT blockade in mice liver tumor [143] | Anti-PD-1 blockade led to the mice harboring many more T cells expressing PD-1, LAG-3, and TIGIT compared to the non-treatment mice After anti-PD-1 anti-TIGIT blockade, there was evidence of reduced tumor growth, increased overall survival, and more expression of CD8+ T cells |
| Effect of TIGIT and PD-1 blockade on CD8+ T cells; CD8+ T cells effect on antibody response [144] | Dual blockade enhanced proliferation of CD8+ T cells compared to single blockade (p < 0.05) Tumors with CD8+ T cell depletion did not show response to anti-TIGIT and PD-L1 blockade |
| TIGIT expression of T cells in healthy donors compared to those with chronic HBV infection [147] | TIGIT expression was highest for effector T cells in chronic HBV infection compared to healthy donors |
10. Conclusions and Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Asafo-Agyei, K.O.; Samant, H. Hepatocellular Carcinoma. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK559177/ (accessed on 3 October 2023).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.X.; Seto, W.-K.; Lai, C.-L.; Yuen, M.-F. Epidemiology of Hepatocellular Carcinoma in the Asia-Pacific Region. Gut Liver 2016, 10, 332–339. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73, 4–13. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primer 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Testino, G.; Leone, S.; Borro, P. Alcohol and hepatocellular carcinoma: A review and a point of view. World J. Gastroenterol. 2014, 20, 15943–15954. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Jain, R.K.; Duda, D.G. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma: A Review. JAMA Oncol. 2021, 7, 113–123. [Google Scholar] [CrossRef]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef]
- Feola, S.; Chiaro, J.; Martins, B.; Cerullo, V. Uncovering the Tumor Antigen Landscape: What to Know about the Discovery Process. Cancers 2020, 12, 1660. [Google Scholar] [CrossRef]
- Lawal, G.; Xiao, Y.; Rahnemai-Azar, A.A.; Tsilimigras, D.I.; Kuang, M.; Bakopoulos, A.; Pawlik, T.M. The Immunology of Hepatocellular Carcinoma. Vaccines 2021, 9, 1184. [Google Scholar] [CrossRef]
- Lee, W.-C. Cell-mediated immunotherapy for hepatocellular carcinoma. J. Cancer Metastasis Treat. 2017, 3, 244. [Google Scholar] [CrossRef]
- Zhong, C.; Li, Y.; Yang, J.; Jin, S.; Chen, G.; Li, D.; Fan, X.; Lin, H. Immunotherapy for Hepatocellular Carcinoma: Current Limits and Prospects. Front. Oncol. 2021, 11, 589680. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, E.; Vaquero, J.; Férnandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF-β Pathway: A Pharmacological Target in Hepatocellular Carcinoma? Cancers 2021, 13, 3248. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Araki, K.; Kuwano, H.; Shirabe, K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6841–6850. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Zhou, S.; Zhao, Z.; Zhong, H.; Ren, Z.; Li, Y.; Wang, H.; Qiu, Y. The role of myeloid-derived suppressor cells in liver cancer. Discov. Oncol. 2023, 14, 77. [Google Scholar] [CrossRef]
- Cao, P.; Sun, Z.; Zhang, F.; Zhang, J.; Zheng, X.; Yu, B.; Zhao, Y.; Wang, W.; Wang, W. TGF-β Enhances Immunosuppression of Myeloid-Derived Suppressor Cells to Induce Transplant Immune Tolerance Through Affecting Arg-1 Expression. Front. Immunol. 2022, 13, 919674. [Google Scholar] [CrossRef]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased Regulatory T Cells Correlate with CD8 T-Cell Impairment and Poor Survival in Hepatocellular Carcinoma Patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef]
- Unitt, E.; Rushbrook, S.M.; Marshall, A.; Davies, S.; Gibbs, P.; Morris, L.S.; Coleman, N.; Alexander, G.J.M. Compromised lymphocytes infiltrate hepatocellular carcinoma: The role of T-regulatory cells. Hepatology 2005, 41, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; De Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Ruf, B.; Heinrich, B.; Greten, T.F. Immunobiology and immunotherapy of HCC: Spotlight on innate and innate-like immune cells. Cell. Mol. Immunol. 2021, 18, 112–127. [Google Scholar] [CrossRef]
- Ben Mousa, A. Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi J. Gastroenterol. 2008, 14, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Raoul, J.-L.; Sherman, M.; Mazzaferro, V.; Bolondi, L.; Craxi, A.; Galle, P.R.; Santoro, A.; Beaugrand, M.; Sangiovanni, A.; et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: Subanalyses of a phase III trial. J. Hepatol. 2012, 57, 821–829. [Google Scholar] [CrossRef]
- Su, G.L.; Altayar, O.; O’Shea, R.; Shah, R.; Estfan, B.; Wenzell, C.; Sultan, S.; Falck-Ytter, Y. AGA Clinical Practice Guideline on Systemic Therapy for Hepatocellular Carcinoma. Gastroenterology 2022, 162, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kudo, M.; Assenat, E.; Cattan, S.; Kang, Y.-K.; Lim, H.Y.; Poon, R.T.P.; Blanc, J.-F.; Vogel, A.; Chen, C.-L.; et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA 2014, 312, 57–67. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Lin, D.-Y.; Park, J.-W.; Kudo, M.; Qin, S.; Chung, H.-C.; Song, X.; Xu, J.; Poggi, G.; et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: Results of a randomized phase III trial. J. Clin. Oncol. 2013, 31, 4067–4075. [Google Scholar] [CrossRef]
- Johnson, P.J.; Qin, S.; Park, J.-W.; Poon, R.T.P.; Raoul, J.-L.; Philip, P.A.; Hsu, C.-H.; Hu, T.-H.; Heo, J.; Xu, J.; et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: Results from the randomized phase III BRISK-FL study. J. Clin. Oncol. 2013, 31, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Cainap, C.; Qin, S.; Huang, W.-T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.-K.; Chen, P.-J.; Toh, H.-C.; et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota Caremoli, E.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.J.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. SEARCH: A phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Brackenier, C.; Kinget, L.; Cappuyns, S.; Verslype, C.; Beuselinck, B.; Dekervel, J. Unraveling the Synergy between Atezolizumab and Bevacizumab for the Treatment of Hepatocellular Carcinoma. Cancers 2023, 15, 348. [Google Scholar] [CrossRef]
- Liu, X.; Lu, Y.; Qin, S. Atezolizumab and bevacizumab for hepatocellular carcinoma: Mechanism, pharmacokinetics and future treatment strategies. Future Oncol. 2021, 17, 2243–2256. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Roy, A. Updated Efficacy and Safety Data from IMbrave150: Atezolizumab Plus Bevacizumab vs. Sorafenib for Unresectable Hepatocellular Carcinoma. J. Clin. Exp. Hepatol. 2022, 12, 1575–1576. [Google Scholar] [CrossRef]
- Zhu, A.X.; Abbas, A.R.; De Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.-H.; He, A.R.; Ryoo, B.-Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef]
- Hack, S.P.; Zhu, A.X.; Wang, Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front. Immunol. 2020, 11, 598877. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, 8. [Google Scholar] [CrossRef]
- Helwick, C. HIMALAYA Trial: First-Line Tremelimumab Plus Durvalumab Improves Overall Survival in Unresectable Hepatocellular Carcinoma. The ASCO Post, 10 February 2022. [Google Scholar]
- Elevar Therapeutics Submits New Drug Application to FDA for Combination of Rivoceranib and Camrelizumab as First-Line Treatment Option for Unresectable Hepatocellular Carcinoma. Available online: https://elevartherapeutics.com/2023/07/17/elevar-therapeutics-announces-fda-acceptance-for-filing-of-new-drug-application-for-rivoceranib-in-combination-with-camrelizumab-as-a-first-line-treatment-for-unresectable-hepatocellular-carcinoma/ (accessed on 17 July 2023).
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Matilla, A.; Santoro, A.; Melero, I.; Gracián, A.C.; Acosta-Rivera, M.; Choo, S.-P.; El-Khoueiry, A.B.; Kuromatsu, R.; El-Rayes, B.; et al. CheckMate 040 cohort 5: A phase I/II study of nivolumab in patients with advanced hepatocellular carcinoma and Child-Pugh B cirrhosis. J. Hepatol. 2021, 75, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Qin, S.; Chen, Z.; Fang, W.; Ren, Z.; Xu, R.; Ryoo, B.-Y.; Meng, Z.; Bai, Y.; Chen, X.; Liu, X.; et al. Pembrolizumab plus best supportive care versus placebo plus best supportive care as second-line therapy in patients in Asia with advanced hepatocellular carcinoma (HCC): Phase 3 KEYNOTE-394 study. J. Clin. Oncol. 2022, 40, 383. [Google Scholar] [CrossRef]
- Foerster, F.; Gairing, S.J.; Ilyas, S.I.; Galle, P.R. Emerging immunotherapy for HCC: A guide for hepatologists. Hepatology 2022, 75, 1604–1626. [Google Scholar] [CrossRef]
- Saung, M.T.; Pelosof, L.; Casak, S.; Donoghue, M.; Lemery, S.; Yuan, M.; Rodriguez, L.; Schotland, P.; Chuk, M.; Davis, G.; et al. FDA Approval Summary: Nivolumab Plus Ipilimumab for the Treatment of Patients with Hepatocellular Carcinoma Previously Treated with Sorafenib. Oncologist 2021, 26, 797–806. [Google Scholar] [CrossRef]
- Bruix, J. Regorafenib and the RESORCE trial: A new second-line option for hepatocellular carcinoma patients. Hepatic Oncol. 2016, 3, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.V.; Merle, P.; et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 39, 267. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients with Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kudo, M.; Cheng, A.-L.; Finn, R.S.; Galle, P.R.; Kaneko, S.; Meyer, T.; Qin, S.; Dutcus, C.E.; Chen, E.; et al. Lenvatinib (len) plus pembrolizumab (pembro) for the first-line treatment of patients (pts) with advanced hepatocellular carcinoma (HCC): Phase 3 LEAP-002 study. J. Clin. Oncol. 2019, 37, TPS4152. [Google Scholar] [CrossRef]
- Kelley, R.K.; Rimassa, L.; Cheng, A.-L.; Kaseb, A.; Qin, S.; Zhu, A.X.; Chan, S.L.; Melkadze, T.; Sukeepaisarnjaroen, W.; Breder, V.; et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 995–1008. [Google Scholar] [CrossRef]
- Akce, M.; El-Rayes, B.F.; Bekaii-Saab, T.S. Frontline therapy for advanced hepatocellular carcinoma: An update. Ther. Adv. Gastroenterol. 2022, 15, 175628482210861. [Google Scholar] [CrossRef]
- A Study of Tivozanib in Combination with Durvalumab in Subjects with Advanced Hepatocellular Carcinoma (DEDUCTIVE). Available online: https://clinicaltrials.gov/study/NCT03970616#more-information (accessed on 1 July 2023).
- Regorafenib Plus Tislelizumab as First-Line Systemic Therapy for Patients with Advanced Hepatocellular Carcinoma NCT04183088. Available online: https://www.clinicaltrials.gov/study/NCT04183088 (accessed on 1 July 2023).
- Phase 3 Study of Tislelizumab Versus Sorafenib in Participants with Unresectable HCC. ClinicalTrials.Gov ID NCT03412773. Available online: https://clinicaltrials.gov/study/NCT03412773 (accessed on 1 July 2023).
- A Study of Nivolumab in Combination with Ipilimumab in Participants with Advanced Hepatocellular Carcinoma (CheckMate 9DW) ClinicalTrials.Gov ID NCT04039607. Available online: https://clinicaltrials.gov/study/NCT04039607 (accessed on 27 June 2023).
- Efficacy and Safety of IBI310 Combined with Sintilimab in Patients with Advanced Hepatocellular Carcinoma. ClinicalTrials.Gov ID NCT04401813. Available online: https://clinicaltrials.gov/study/NCT04401813 (accessed on 27 June 2023).
- MGD013 Monotherapy and Combination With Brivanib Dose Escalation and Expansion Study in Advanced Liver Cancer Patients. Available online: https://clinicaltrials.gov/study/NCT04212221 (accessed on 27 June 2023).
- Acoba, J.D.; Rho, Y.; Fukaya, E. Phase II study of cobolimab in combination with dostarlimab for the treatment of advanced hepatocellular carcinoma. J. Clin. Oncol. 2023, 41, 580. [Google Scholar] [CrossRef]
- Immunotherapy With Nivolumab in Combination with Lenvatinib for Advanced Stage Hepatocellular Carcinoma. ClinicalTrials.Gov ID NCT03841201. Available online: https://clinicaltrials.gov/study/NCT03841201 (accessed on 27 July 2023).
- Combination of Regorafenib and Nivolumab in Unresectable Hepatocellular Carcinoma (RENOBATE). ClinicalTrials.Gov ID NCT04310709. Available online: https://clinicaltrials.gov/study/NCT04310709 (accessed on 25 July 2023).
- Study of ET140203 T Cells in Adults With Advanced Hepatocellular Carcinoma (ARYA-1). ClinicalTrials.Gov ID NCT04502082. Available online: https://clinicaltrials.gov/study/NCT04502082 (accessed on 15 June 2023).
- Study Evaluating the Benefit of Adding Ipilimumab to the Combination of Atezolizumab and Bevacizumab in Patients with Hepatocellular Carcinoma Receiving First-Line Systemic Therapy (TRIPLET). ClinicalTrials.Gov ID NCT05665348. Available online: https://clinicaltrials.gov/study/NCT05665348 (accessed on 27 July 2023).
- A Phase I Study of ERY974 in Patients with Hepatocellular Carcinoma ClinicalTrials.Gov ID NCT05022927. Available online: https://clinicaltrials.gov/study/NCT05022927 (accessed on 16 July 2023).
- Personalized Neoantigen Peptide-Based Vaccine in Combination with Pembrolizumab for Treatment of Advanced Solid Tumors (PNeoVCA) ClinicalTrials.Gov ID NCT05269381. Available online: https://clinicaltrials.gov/study/NCT05269381 (accessed on 1 June 2023).
- Sangro, B.; Yau, T.; Harding, J.J.; Acosta Rivera, M.; Kazushi, N.; El-Khoueiry, A.B.; Cruz-Correa, M.; Perez-Callejo, D.; McLean, S.; Sparks, J.; et al. RELATIVITY-106: A phase 1/2 trial of nivolumab (NIVO) + relatlimab (RELA) in combination with bevacizumab (BEV) in first-line (1L) hepatocellular carcinoma (HCC). J. Clin. Oncol. 2023, 41, TPS636. [Google Scholar] [CrossRef]
- Li, D.; Qin, J.; Zhou, T.; Li, Y.; Cheng, X.; Chen, Z.; Chen, J.; Zheng, W. Bispecific GPC3/PD-1 CAR-T cells for the treatment of HCC. Int. J. Oncol. 2023, 62, 53. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, T.; Guo, J.; Wang, J.; Jia, L.; Shi, X.; Yang, T.; Jiao, R.; Wei, X.; Feng, Z.; et al. Bispecific c-Met/PD-L1 CAR-T Cells Have Enhanced Therapeutic Effects on Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 546586. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Sun, Z.; Yuan, Q.; Hou, W.; Liang, Q.; Wang, Y.; Mo, W.; Wang, H.; Yu, M. Dual-function chimeric antigen receptor T cells targeting c-Met and PD-1 exhibit potent anti-tumor efficacy in solid tumors. Investig. New Drugs 2021, 39, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Zheng, Y.; Fang, W.; Zhao, Q.; Zhao, P.; Liu, L.; Zhai, Y.; Tong, Z.; Zhang, H.; Lin, M.; et al. RUNX-3-expressing CAR T cells targeting glypican-3 in patients with heavily pretreated advanced hepatocellular carcinoma: A phase I trial. eClinicalMedicine 2023, 63, 102175. [Google Scholar] [CrossRef] [PubMed]
- Acúrcio, R.C.; Scomparin, A.; Conniot, J.; Salvador, J.A.R.; Satchi-Fainaro, R.; Florindo, H.F.; Guedes, R.C. Structure–Function Analysis of Immune Checkpoint Receptors to Guide Emerging Anticancer Immunotherapy. J. Med. Chem. 2018, 61, 10957–10975. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Powderly, J.D.; Patel, M.R.; Brody, J.; Hamilton, E.P.; Infante, J.R.; Falchook, G.S.; Wang, H.; Adams, L.; Gong, L.; et al. Phase 1 trial of CA-170, a novel oral small molecule dual inhibitor of immune checkpoints PD-1 and VISTA, in patients (pts) with advanced solid tumor or lymphomas. J. Clin. Oncol. 2017, 35, TPS3099. [Google Scholar] [CrossRef]
- A Study of CA-170 (Oral PD-L1, PD-L2 and VISTA Checkpoint Antagonist) in Patients with Advanced Tumors and Lymphomas. 2020. Available online: https://clinicaltrials.gov/study/NCT02812875 (accessed on 30 September 2023).
- Radhakrishnan, V.; Banavali, S.; Gupta, S.; Kumar, A.; Deshmukh, C.D.; Nag, S.; Beniwal, S.K.; Gopichand, M.; Naik, R.; Lakshmaiah, K.C.; et al. Excellent CBR and prolonged PFS in non-squamous NSCLC with oral CA-170, an inhibitor of VISTA and PD-L1. Ann. Oncol. 2019, 30, v494. [Google Scholar] [CrossRef]
- Koblish, H.K.; Wu, L.; Wang, L.-C.S.; Liu, P.C.C.; Wynn, R.; Rios-Doria, J.; Spitz, S.; Liu, H.; Volgina, A.; Zolotarjova, N.; et al. Characterization of INCB086550: A Potent and Novel Small-Molecule PD-L1 Inhibitor. Cancer Discov. 2022, 12, 1482–1499. [Google Scholar] [CrossRef]
- Pelizzaro, F.; Farinati, F.; Trevisani, F. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance. Biomedicines 2023, 11, 1020. [Google Scholar] [CrossRef]
- Sullivan, K.M.C.; Vilalta, M.; Ertl, L.S.; Wang, Y.; Dunlap, C.; Ebsworth, K.; Zhao, B.N.; Li, S.; Zeng, Y.; Miao, Z.; et al. CCX559 is a potent, orally-administered small molecule PD-L1 inhibitor that induces anti-tumor immunity. PLoS ONE 2023, 18, e0286724. [Google Scholar] [CrossRef]
- Tapia, G.; Lundy, J.; Richardson, G.E.; Zhao, N.; Ebsworth, K.; Yue, H.; Miao, S.; deGoma, E.; Jain, R.; Schall, T.J.; et al. Preliminary data from an ongoing phase 1 dose-escalation study of CCX559, an orally administered small molecule PD-L1 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2022, 40, 2593. [Google Scholar] [CrossRef]
- Wang, C.-L.; Gao, M.-Z.; Gao, D.-M.; Guo, Y.-H.; Gao, Z.; Gao, X.-J.; Wang, J.-Q.; Qiao, M.-Q. Tubeimoside-1: A review of its antitumor effects, pharmacokinetics, toxicity, and targeting preparations. Front. Pharmacol. 2022, 13, 941270. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yin, M.; Dong, J.; Mao, G.; Min, W.; Kuang, Z.; Yang, P.; Liu, L.; Zhang, N.; Deng, H. Tubeimoside-1 induces TFEB-dependent lysosomal degradation of PD-L1 and promotes antitumor immunity by targeting mTOR. Acta Pharm. Sin. B 2021, 11, 3134–3149. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yang, L.; Liu, J.; Wang, J.; Fan, L.; Duangmano, S.; Liu, H.; Liu, M.; Wang, J.; Zhong, X.; et al. The role of mitochondria in the resistance of melanoma to PD-1 inhibitors. J. Transl. Med. 2023, 21, 345. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, A.; Zhang, X.; Li, Y.; Li, B.; Jin, S.; Dong, Q.; Niu, X.; Zhang, L.; Zhou, X.; et al. Identification of a novel small-molecule inhibitor targeting TIM-3 for cancer immunotherapy. Biochem. Pharmacol. 2023, 212, 115583. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.M.; Purvis, I.J.; Bomstad, C.N.; Labak, C.M.; Velpula, K.K.; Tsung, A.J.; Regan, J.N.; Venkataraman, S.; Vibhakar, R.; Asuthkar, S. Therapeutic targeting of immune checkpoints with small molecule inhibitors. Am. J. Transl. Res. 2019, 11, 529–541. [Google Scholar]
- Zhou, W.; Fang, D.; He, Y.; Wei, J. Correlation analysis of tumor mutation burden of hepatocellular carcinoma based on data mining. J. Gastrointest. Oncol. 2021, 12, 1117–1131. [Google Scholar] [CrossRef]
- Wang, P.; Chen, Y.; Wang, C. Beyond Tumor Mutation Burden: Tumor Neoantigen Burden as a Biomarker for Immunotherapy and Other Types of Therapy. Front. Oncol. 2021, 11, 672677. [Google Scholar] [CrossRef]
- Liu, S.; Tang, Q.; Huang, J.; Zhan, M.; Zhao, W.; Yang, X.; Li, Y.; Qiu, L.; Zhang, F.; Lu, L.; et al. Prognostic analysis of tumor mutation burden and immune infiltration in hepatocellular carcinoma based on TCGA data. Aging 2021, 13, 11257–11280. [Google Scholar] [CrossRef]
- Gabbia, D.; De Martin, S. Tumor Mutational Burden for Predicting Prognosis and Therapy Outcome of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 3441. [Google Scholar] [CrossRef]
- Xu, C.; Xu, Z.; Zhang, Y.; Evert, M.; Calvisi, D.F.; Chen, X. β-Catenin signaling in hepatocellular carcinoma. J. Clin. Investig. 2022, 132, e154515. [Google Scholar] [CrossRef] [PubMed]
- Krutsenko, Y.; Singhi, A.D.; Monga, S.P. β-Catenin Activation in Hepatocellular Cancer: Implications in Biology and Therapy. Cancers 2021, 13, 1830. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Lachenmayer, A.; Alsinet, C.; Savic, R.; Cabellos, L.; Toffanin, S.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Newell, P.; Tsai, H.-W.; et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 2012, 18, 4997–5007. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Liu, B.-R.; Du, Y.-J.; Chen, J.; Cheng, Y.-Q.; Xu, W.; Wang, X.-H. Wnt/β-catenin signaling pathway may regulate the expression of angiogenic growth factors in hepatocellular carcinoma. Oncol. Lett. 2014, 7, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhou, B.; Cao, M.; Shao, Q.; Pan, Y.; Zhao, T. CTEN Inhibits Tumor Angiogenesis and Growth by Targeting VEGFA Through Down-Regulation of β-Catenin in Breast Cancer. Technol. Cancer Res. Treat. 2021, 20, 153303382110455. [Google Scholar] [CrossRef]
- Yan, L.; Chen, Y.; Zhou, J.; Zhao, H.; Zhang, H.; Wang, G. Diagnostic value of circulating cell-free DNA levels for hepatocellular carcinoma. Int. J. Infect. Dis. 2018, 67, 92–97. [Google Scholar] [CrossRef]
- Xie, Q.; Zhang, P.; Wang, Y.; Mei, W.; Zeng, C. Overcoming resistance to immune checkpoint inhibitors in hepatocellular carcinoma: Challenges and opportunities. Front. Oncol. 2022, 12, 958720. [Google Scholar] [CrossRef]
- De Lorenzo, S.; Tovoli, F.; Trevisani, F. Mechanisms of Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma. Cancers 2022, 14, 4616. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, Q.; Liu, J.; Zhang, W. CTNNB1 Alternation Is a Potential Biomarker for Immunotherapy Prognosis in Patients With Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 759565. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Zhou, X.; Ni, Y.; Liang, X.; Lin, Y.; An, B.; He, X.; Zhao, X. Mechanisms of tumor resistance to immune checkpoint blockade and combination strategies to overcome resistance. Front. Immunol. 2022, 13, 915094. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Qi, F.; Jiao, R.; Zheng, L.; Zhang, Y.; Hou, D.; Liu, Y.; Kang, Z. Prognostic and clinicopathological value of high expression of TIM -3 in different cancer types: A meta-analysis. Precis. Med. Sci. 2020, 9, 31–42. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, Y.; Chen, Z. Tim-3 expression and its role in hepatocellular carcinoma. J. Hematol. Oncol. 2018, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Ganjalikhani Hakemi, M.; Jafarinia, M.; Azizi, M.; Rezaeepoor, M.; Isayev, O.; Bazhin, A.V. The Role of TIM-3 in Hepatocellular Carcinoma: A Promising Target for Immunotherapy? Front. Oncol. 2020, 10, 601661. [Google Scholar] [CrossRef]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: Challenges and opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Viveiros, P.; Riaz, A.; Lewandowski, R.J.; Mahalingam, D. Current State of Liver-Directed Therapies and Combinatory Approaches with Systemic Therapy in Hepatocellular Carcinoma (HCC). Cancers 2019, 11, 1085. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.P.; Khakoo, S.I. Immunotherapy for hepatocellular carcinoma: Current and future. World J. Gastroenterol. 2019, 25, 2977–2989. [Google Scholar] [CrossRef] [PubMed]
- Charles, J.; Vrionis, A.; Mansur, A.; Mathias, T.; Shaikh, J.; Ciner, A.; Jiang, Y.; Nezami, N. Potential Immunotherapy Targets for Liver-Directed Therapies, and the Current Scope of Immunotherapeutics for Liver-Related Malignancies. Cancers 2023, 15, 2624. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, N.; Zhao, H.; Jin, H.; Wang, G.; Niu, C.; Terunuma, H.; He, H.; Li, W. Combination of radiofrequency ablation and sequential cellular immunotherapy improves progression-free survival for patients with hepatocellular carcinoma. Int. J. Cancer 2014, 134, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Zheng, J.-S.; Nan, G.; Li, R.; Chen, C.; Hu, C.-X.; Zhang, Y.; Sun, B.; Wang, X.-L.; Cui, S.-C.; et al. Randomized trial of [131I] metuximab in treatment of hepatocellular carcinoma after percutaneous radiofrequency ablation. J. Natl. Cancer Inst. 2014, 106, dju239. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.-H.; Lim, Y.-S.; Yeon, J.E.; Song, T.-J.; Yu, S.J.; Gwak, G.-Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391.e6. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, J.H.; Lim, Y.-S.; Yeon, J.E.; Song, T.-J.; Yu, S.J.; Gwak, G.-Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Sustained efficacy of adjuvant immunotherapy with cytokine-induced killer cells for hepatocellular carcinoma: An extended 5-year follow-up. Cancer Immunol. Immunother. 2019, 68, 23–32. [Google Scholar] [CrossRef]
- Wang, X.; Liu, G.; Chen, S.; Bi, H.; Xia, F.; Feng, K.; Ma, K.; Ni, B. Combination therapy with PD-1 blockade and radiofrequency ablation for recurrent hepatocellular carcinoma: A propensity score matching analysis. Int. J. Hyperth. 2021, 38, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Waidmann, O.; Müller, T.; Siegler, G.M.; Goetze, T.O.; De Toni, E.N.; Gonzalez-Carmona, M.A.; Hausner, G.; Geissler, M.; Fischer Von Weikersthal, L.; et al. IMMULAB: A phase II trial of immunotherapy with pembrolizumab in combination with local ablation for patients with early-stage hepatocellular carcinoma (HCC). J. Clin. Oncol. 2023, 41, 555. [Google Scholar] [CrossRef]
- Ablation Plus Tislelizumab Versus Ablation Alone for Intrahepatic Recurrent Early-Stage HCC. ClinicalTrials.Gov ID NCT04663035. Available online: https://clinicaltrials.gov/study/NCT04663035 (accessed on 1 October 2023).
- Neoadjuvant Atezo, Adjuvant Atezo + Beva Combined with RF Ablation of Small HCC: A Multicenter Randomized Phase II Trial (AB-LATE02). ClinicalTrials.Gov ID NCT04727307. Available online: https://clinicaltrials.gov/study/NCT04727307 (accessed on 1 October 2023).
- Ablation Combined With PD-1 in HCC: Phase II Study. ClinicalTrials.Gov ID NCT04652440. Available online: https://clinicaltrials.gov/study/NCT04652440 (accessed on 1 October 2023).
- Wu, Y.-C. (Jasmine); Wakil, A.; Salomon, F.; Pyrsopoulos, N. Issue on combined locoregional and systemic treatment for hepatocellular carcinoma. Hepatoma Res. 2023, 9, 6. [Google Scholar] [CrossRef]
- Li, Z.; Li, N.; Li, F.; Zhou, Z.; Sang, J.; Chen, Y.; Han, Q.; Lv, Y.; Liu, Z. Immune checkpoint proteins PD-1 and TIM-3 are both highly expressed in liver tissues and correlate with their gene polymorphisms in patients with HBV-related hepatocellular carcinoma. Medicine 2016, 95, e5749. [Google Scholar] [CrossRef]
- Desai, J.; Meniawy, T.; Beagle, B.; Li, Z.; Mu, S.; Wu, J.; Denlinger, C.S.; Messersmith, W.A. Bgb-A425, an investigational anti-TIM-3 monoclonal antibody, in combination with tislelizumab, an anti-PD-1 monoclonal antibody, in patients with advanced solid tumors: A phase I/II trial in progress. J. Clin. Oncol. 2020, 38, TPS3146. [Google Scholar] [CrossRef]
- Li, F.-J.; Zhang, Y.; Jin, G.-X.; Yao, L.; Wu, D.-Q. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8+ T cell in HCC patients. Immunol. Lett. 2013, 150, 116–122. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Young, G.D.; McMiller, T.L.; Chen, S.; Salas, J.T.; Pritchard, T.S.; Xu, H.; Meeker, A.K.; Fan, J.; Cheadle, C.; et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clin. Cancer Res. 2015, 21, 3969–3976. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Huang, R.-Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yuan, F.; Qi, F.; Sun, J.; Rao, Q.; Zhao, Z.; Huang, P.; Fang, T.; Yang, B.; Xia, J. Expression and clinical significance of LAG-3, FGL1, PD-L1 and CD8+T cells in hepatocellular carcinoma using multiplex quantitative analysis. J. Transl. Med. 2020, 18, 306. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yu, H.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Suda, K.; Ren, S.; Wu, C.; Hou, L.; et al. LAG-3 Protein Expression in Non-Small Cell Lung Cancer and Its Relationship with PD-1/PD-L1 and Tumor-Infiltrating Lymphocytes. J. Thorac. Oncol. 2017, 12, 814–823. [Google Scholar] [CrossRef]
- Lee, W.J.; Lee, Y.J.; Choi, M.E.; Yun, K.A.; Won, C.H.; Lee, M.W.; Choi, J.H.; Chang, S.E. Expression of lymphocyte-activating gene 3 and T-cell immunoreceptor with immunoglobulin and ITIM domains in cutaneous melanoma and their correlation with programmed cell death 1 expression in tumor-infiltrating lymphocytes. J. Am. Acad. Dermatol. 2019, 81, 219–227. [Google Scholar] [CrossRef]
- Guo, M.; Qi, F.; Rao, Q.; Sun, J.; Du, X.; Qi, Z.; Yang, B.; Xia, J. Serum LAG-3 Predicts Outcome and Treatment Response in Hepatocellular Carcinoma Patients With Transarterial Chemoembolization. Front. Immunol. 2021, 12, 754961. [Google Scholar] [CrossRef]
- Wei, J.; Liao, Z.; Tao, Y.; Liu, S. Evaluation of the possible association of PDCD-1 and LAG3 gene polymorphisms with hepatocellular carcinoma risk. BMC Med. Genomics 2023, 16, 92. [Google Scholar] [CrossRef]
- Sangro, B. Relatlimab + Nivolumab in Patients with Advanced Hepatocellular Carcinoma Who Are Naive to Immuno- Oncology Therapy but Progressed on Tyrosine Kinase Inhibitors, a Phase 2, Randomized, Open-Label Study: RELATIVITY-073. VOLUME 32, SUPPLEMENT 3, S117, JULY 2021. Available online: https://www.annalsofoncology.org/article/S0923-7534(21)01305-3/fulltext#%20 (accessed on 1 October 2023).
- Huo, J.-L.; Wang, Y.-T.; Fu, W.-J.; Lu, N.; Liu, Z.-S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef]
- Ren, Z.; Guo, Y.; Bai, Y.; Ying, J.; Meng, Z.; Chen, Z.; Gu, S.; Zhang, J.; Liang, J.; Hou, X.; et al. Tebotelimab, a PD-1/LAG-3 bispecific antibody, in patients with advanced hepatocellular carcinoma who had failed prior targeted therapy and/or immunotherapy: An open-label, single-arm, phase 1/2 dose-escalation and expansion study. J. Clin. Oncol. 2023, 41, 578. [Google Scholar] [CrossRef]
- Sung, E.; Ko, M.; Won, J.-Y.; Jo, Y.; Park, E.; Kim, H.; Choi, E.; Jung, U.-J.; Jeon, J.; Kim, Y.; et al. LAG-3xPD-L1 bispecific antibody potentiates antitumor responses of T cells through dendritic cell activation. Mol. Ther. 2022, 30, 2800–2816. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.K.-C.; Yuen, V.W.-H.; Cheu, J.W.-S.; Wei, L.L.; Ting, V.; Fehlings, M.; Sumatoh, H.; Nardin, A.; Newell, E.W.; Ng, I.O.-L.; et al. Hepatocellular Carcinoma Cells Up-regulate PVRL1, Stabilizing PVR and Inhibiting the Cytotoxic T-Cell Response via TIGIT to Mediate Tumor Resistance to PD1 Inhibitors in Mice. Gastroenterology 2020, 159, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Zhou, G.; Campos Carrascosa, L.; Gausvik, E.; Boor, P.P.C.; Noordam, L.; Doukas, M.; Polak, W.G.; Terkivatan, T.; Pan, Q.; et al. TIGIT and PD1 Co-blockade Restores ex vivo Functions of Human Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 443–464. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Wang, X.; Dang, Z.; Jiang, Y.; Wang, X.; Kong, Y.; Yang, Z. PD-1+ TIGIT+ CD8+ T cells are associated with pathogenesis and progression of patients with hepatitis B virus-related hepatocellular carcinoma. Cancer Immunol. Immunother. CII 2019, 68, 2041–2054. [Google Scholar] [CrossRef]
- Wei, Y.-Y.; Fan, J.; Shan, M.-X.; Yin, D.-D.; Wang, L.-L.; Ye, W.; Zhao, W. TIGIT marks exhausted T cells and serves as a target for immune restoration in patients with chronic HBV infection. Am. J. Transl. Res. 2022, 14, 942–954. [Google Scholar]
- Finn, R.S.; Ryoo, B.-Y.; Hsu, C.-H.; Li, D.; Burgoyne, A.; Cotter, C.; Badhrinarayanan, S.; Wang, Y.; Yin, A.; Rao Edubilli, T.; et al. Results from the MORPHEUS-liver study: Phase Ib/II randomized evaluation of tiragolumab (tira) in combination with atezolizumab (atezo) and bevacizumab (bev) in patients with unresectable, locally advanced or metastatic hepatocellular carcinoma (uHCC). J. Clin. Oncol. 2023, 41, 4010. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.) (2023, July). A Study Evaluating Atezolizumab and Bevaci-Zumab, with or without Tiragolumab, in Participants with Untreated Locally Advanced or Met-Astatic Hepatocellular Carcinoma (IMbrave152) (SKYSCRAPER-14). NCT05904886. Available online: https://clinicaltrials.gov/study/NCT05904886 (accessed on 9 September 2023).

| GO30140 Cohort A: Atezolizumab + Bevacizumab | ||
|---|---|---|
| Gene Alterations or Immune Signatures | Immune Cell Types | TMB |
| Gene alterations or immune signatures associated with greater response: CD274 (PD-L1 mRNA): high expression associated with longer PFS compared to those with low expression (p = 0.0011). Teff: high expression associated with longer PFS in combination compared to those with low expression (0.0035). | Higher density of CD8+ T associated with better response (p = 0.007). | Greater ORR in TMB-high group (56%) compared to TMB-low group (35%). |
| Phase III IMbrave 150 Trial: Atezolizumab + Bevacizumab vs. Sorafenib | ||
| Gene Alterations or Immune Signatures | Immune Cell Types | TMB |
| Gene alterations or signatures associated with greater response: CD274 (PD-L1 mRNA): high expression associated with longer PFS in combination group versus sorafenib (p = 0.015), as well as greater OS (0.002) Teff: high expression associated with longer PFS in combination group versus sorafenib (p = 0.047), as well as greater OS (0.0002) | Higher density of intra-tumoral CD8+ T cells showed longer PFS (0.053) and OS (0.001) Low ratio of Treg/Teff signatures had higher PFS and OS compared to sorafenib Higher density of CD8+ T cells associated with longer OS and PFS compared with sorafenib | No associations of TMB with outcome |
| GO30140 Cohort F: Atezolizumab + Bevacizumab vs. Atezolizumab | ||
| Gene Alterations or Immune Signatures | Blood Vessel Density | |
| Genes or signatures associated with greater response: Myeloid inflammation: high expression associated with greater PFS (p = 0.036 versus monotherapy Gene signatures of Teff: high expression associated with greater PFS (p = 0.034 versus monotherapy KDR (VEGF receptor 2): high expression associated with greater PFS in combination group compared to monotherapy (p = 0.011) | High vessel density in baseline tumors associated with longer PFS in combination group compared to monotherapy (p = 0.0018) | |
| Clinical Trials in HCC | Phase | Line of Therapy | Arms | Primary Outcome(s) | Median OS (Months) | ORR (%) | Year Approved |
|---|---|---|---|---|---|---|---|
| Multikinase inhibitors and monoclonal antibody against VEGFR2 | |||||||
| SHARP [26] | III | First | Sorafenib (S) Placebo (P) | OS | S: 10.7 P: 7.9 (HR = 0.69; 95% confidence interval (CI) = 0.55–0.87; p < 0.001) | S: 43 P: 32 p = 0.002 | 2007 |
| RESORCE [54] | III | Second (post-SOR) | Regorafenib (R) Placebo | OS | R: 10.6 P: 7.8 (HR = 0.63; 95% CI = 0.50–0.79; p < 0.0001) | R:11 P: 4 p = 0.0047 | 2017 |
| REFLECT [36] | III | First | Lenvatinib (L), Sorafenib | OS | L: 13.6 S: 12.3 (HR = 0.92; 95% CI = 0.79–1.06) | L: 18.8 S: 6.5 p < 0.0001 | 2018 |
| CELESTIAL [55] | III | Second (post-SOR or other) | Cabozantinib (C) Placebo | OS | C: 10.2 P: 8.0 (HR = 0.76; p < 0.005) | C:4 P < 1 p = 0.009 | 2019 |
| REACH-2 [56] | III | Second | Ramucirumab (Ra), Placebo (AFP ≥ 400 ng/mL) | OS | Ra: 8.5 P: 7.3 (HR = 0.71; p < 0.019) | R:5 P:1 p = 0·1697 | 2019 |
| Immunotherapy (monotherapy) | |||||||
| Keynote-224 [49] | II | Second | Pembrolizumab (Pem) (post-SOR) | ORR |
Pem: 12.9 months (95% CI = 9.7–15.5) |
17 (95% CI = 11–26) | 2018 |
| Checkmate 040 (cohorts 1–3 in dose expansion phase) [46] | I/II | Second | Nivolumab (N) (post-SOR) | ORR |
6 months:83% 9 months:74% | 20 (CI = 15–26) | 2017 |
| MKI with ICI | |||||||
| IMbrave150 (2020) [57] | III | First | Atezolizumab + Bevacizumab (AB), Sorafenib | AB: 19.2 S: 13.4 | A + B:30 S:11 | 2020 | |
| Dual checkpoint inhibitors | |||||||
| Checkmate 040 (cohort 4) | I/II | Second | Nivolumab + ipilimumab | ORR |
Arm A: 22.8 months (95% CI, 9.4-not reached) Arm B: 12.5 months (95% CI, 7.6–16.4) Arm C: 12.7 months (95% CI, 7.4–33.0) | ARM A: 32 (95 = CI 20–47) ARM B: 27 (95% CI = 15–41) ARM C: 29 (95% CI = 29 (17–43) | 2020 |
| HIMALAYA [44] | III | First |
Durvalumab + Tremelimumab (STRIDE), Durvalumab (D), Sorafenib | OS | STRIDE: 16.4 S: 13.8 (HR = 0.78; 96% CI = 0.65–0.92; p = 0.0035) Durvalumab did not demonstrate superiority to sorafenib (p = 0.0674) |
STRIDE:20.1 D: 17 S: 5.1 | 2022 |
| Trial Name and ID | Cancer Type | Estimated Enrollment | Targeting Mechanism | Control Arm | Phase | Start and Completion Dates | Primary Measures |
|---|---|---|---|---|---|---|---|
| RATIONALE—301 [64] NCT03412773 | HCC | December 2017 | Tislelizumab (anti-PD-1 antibody) | Sorafenib | III | December 2017 July 2023 | OS |
| Checkmate 9DW [65] NCT04039607 | HCC | September 2019 | Nivolumab + Ipilimumab | Sorafenib or lenvatinib | III | September 2019 June 2025 | OS |
| NCT03764293 [45] (CARES-310) | Locally advanced or metastatic and unresectable HCC | June 2019 | Camrelizumab (anti-PD-1 antibody) + Apatinib (VEGF inhibitor) | Sorafenib | III | June 2019 April 2023 | OS PFS |
| DEDUCTIVE [62] NCT03970616 | Advanced HCC | September 2019 | Tivozanib (selective VEGFR 1,2,3 TKI) + Durvalumab (PD-L1 inhibitor) | N/A | 1/IIb | September 2019 March 2023 | TEAEs |
| NCT04183088 [63] | Advanced HCC | December 2020 | Tislelizumab (anti-PD-1 antibody) + regorafenib (TKI) | N/A | II | December 2020 March 2025 | TRAE ORR PFS |
| NCT04401813 [66] | Advanced HCC | June 2020 | IBI308 (anti-CTLA4 antibody) + Sintilizumab (anti-PD-1 antibody) | N/A | I | June 2020 April 2023 | AE ORR |
| NCT04212221 [67] | Advanced HCC | April 2020 | MGD013 (anti PD 1 antiobdy and anti-LAG-3 antibody) + Brivanib | N/A | I/II | Completed Pending results | DLTs ORR |
| NCT03680508 [68] | Advanced HCC | December 2019 | Cobolimab (TIM-3 binding antibody) + Dostarlimab (anti PD-1 antibody) | N/A | II | December 2019 October 2025 | ORR |
| NCT03841201 [69] | Advanced HCC | June 2019 | Lenvatinib (TKI) + Nivolumab (anti-PD-1 antibody) | N/A | II | June 2019 March 2023 | ORR AE SAE |
| RENOBATE [70] NCT04310709 | Advanced HCC | June 2020 | Regorafenib + Nivolumab | Completed Pending results | Response Rate | ||
| ARYA-1 [71] NCT04502082 | Advanced HCC | April 2021 | ET140203 autologous T cell product | N/A | I/II | April 2021 June 2024 | Incidence of AE and severity rates of AE Incidence rates of DLT RP2D |
| TRIPLET [72] NCT05665348 | HCC—Hepatocellular Carcinoma | September 2021 | Atezolizumab (anti-PD-L1 antibody) + Bevacizumab (VEGF inhibitor) + Ipilimumab (anti-CTLA-4 antibody) | Atezolizumab + Bevacizumab | II/III | September 2021 April 2026 | Objective response Overall survival |
| NCT05022927 [73] | Advanced HCC | June 2021 | ERY974 + Tocilicumab + Atezolizumab + Bevacizumab | N/A | I | June 2021 September 2024 | Incidence of treatment-emergent adverse events (TEAE) |
| The PNeoVCA Study [74] NCT05269381 | Various advanced solid tumors including HCC | March 2022 | Cyclophosphamide (alkylating agent) + Neoantigen vaccine (containing sargramostim (GM-CSF)) + Pembrolizumab (anti-PD-1 antibody) | N/A | I | March 2022 February 2025 | Incidence of AE |
| RELATIVITY—106 [75] NCT05337137 | Advanced HCC | April 2022 | Relatinib + Nivolumab + Bevacizumab | Nivolumab + Bevacizumab | 1/2 | April 2022 March 2023 | Incidence of DLT PFS |
| Agent | Target | Clinical Trial | Cancer Type | Primary Objective |
|---|---|---|---|---|
| CA-170 Trial IDs: CTRI/2017/12/011026 (phase II) CTRI/2020/07/026870 (phase IIb/III) | PD-L1 | Phase II | Lymphoma | ORR: 30% |
| PD-L1 | Phase IIb/III | Non-squamous, non-small cell lung cancer | ORR: ongoing | |
| INCB086550 Trial ID: NCT04629339 | PD-L1 | Phase II | Select solid tumors | ORR: ongoing |
| CCX559 Trial ID: ACTRN12621001342808 | PD-L1 | Phase I | Solid tumors | Safety |
| Small-Molecule Inhibitors in Preclinical Studies | ||
|---|---|---|
| Molecule | Immune Checkpoint | Pathways |
| SMI402 in tumor-bearing mice [91] | TIM-3 | Inhibition of tumor growth by increasing CD8+ T cell infiltration at tumor site |
| “Compounds 8 and 9” [92] | B7-1, preventing interaction with CTLA-4 | Lack of inhibition in a cell adhesion assay |
| Tubeimoside-1 (TBM-1) [89] | PD-L1 | Lysosomal degradation of PD-L1 in cancer cells via mTOR inactivation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bicer, F.; Kure, C.; Ozluk, A.A.; El-Rayes, B.F.; Akce, M. Advances in Immunotherapy for Hepatocellular Carcinoma (HCC). Curr. Oncol. 2023, 30, 9789-9812. https://doi.org/10.3390/curroncol30110711
Bicer F, Kure C, Ozluk AA, El-Rayes BF, Akce M. Advances in Immunotherapy for Hepatocellular Carcinoma (HCC). Current Oncology. 2023; 30(11):9789-9812. https://doi.org/10.3390/curroncol30110711
Chicago/Turabian StyleBicer, Fuat, Catrina Kure, Anil A. Ozluk, Bassel F. El-Rayes, and Mehmet Akce. 2023. "Advances in Immunotherapy for Hepatocellular Carcinoma (HCC)" Current Oncology 30, no. 11: 9789-9812. https://doi.org/10.3390/curroncol30110711
APA StyleBicer, F., Kure, C., Ozluk, A. A., El-Rayes, B. F., & Akce, M. (2023). Advances in Immunotherapy for Hepatocellular Carcinoma (HCC). Current Oncology, 30(11), 9789-9812. https://doi.org/10.3390/curroncol30110711