A Review of the Clinical Features and Management of Systemic Congenital Mastocytosis through the Presentation of An Unusual Prenatal-Onset Case

 , and
, and
Abstract
:1. Introduction
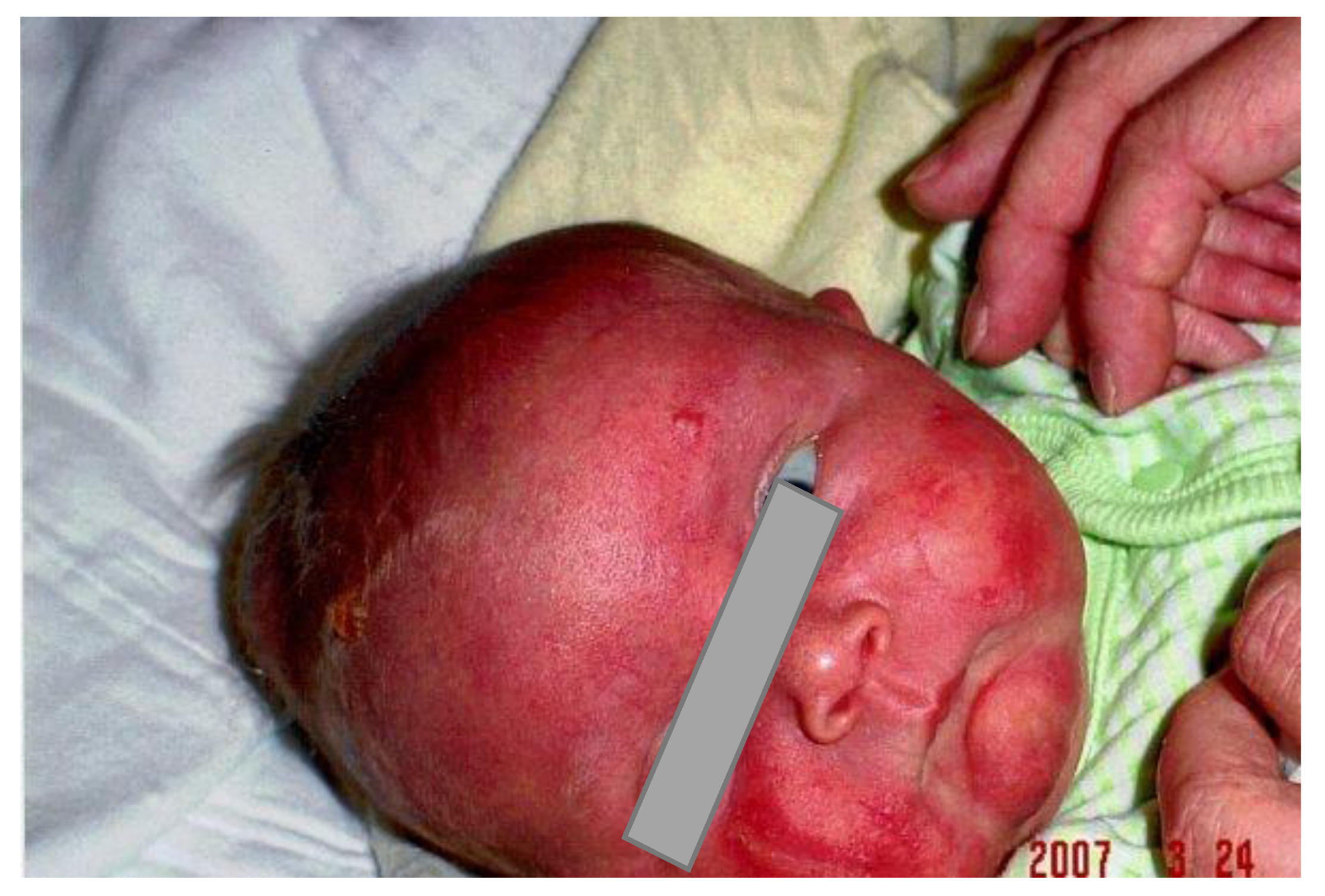
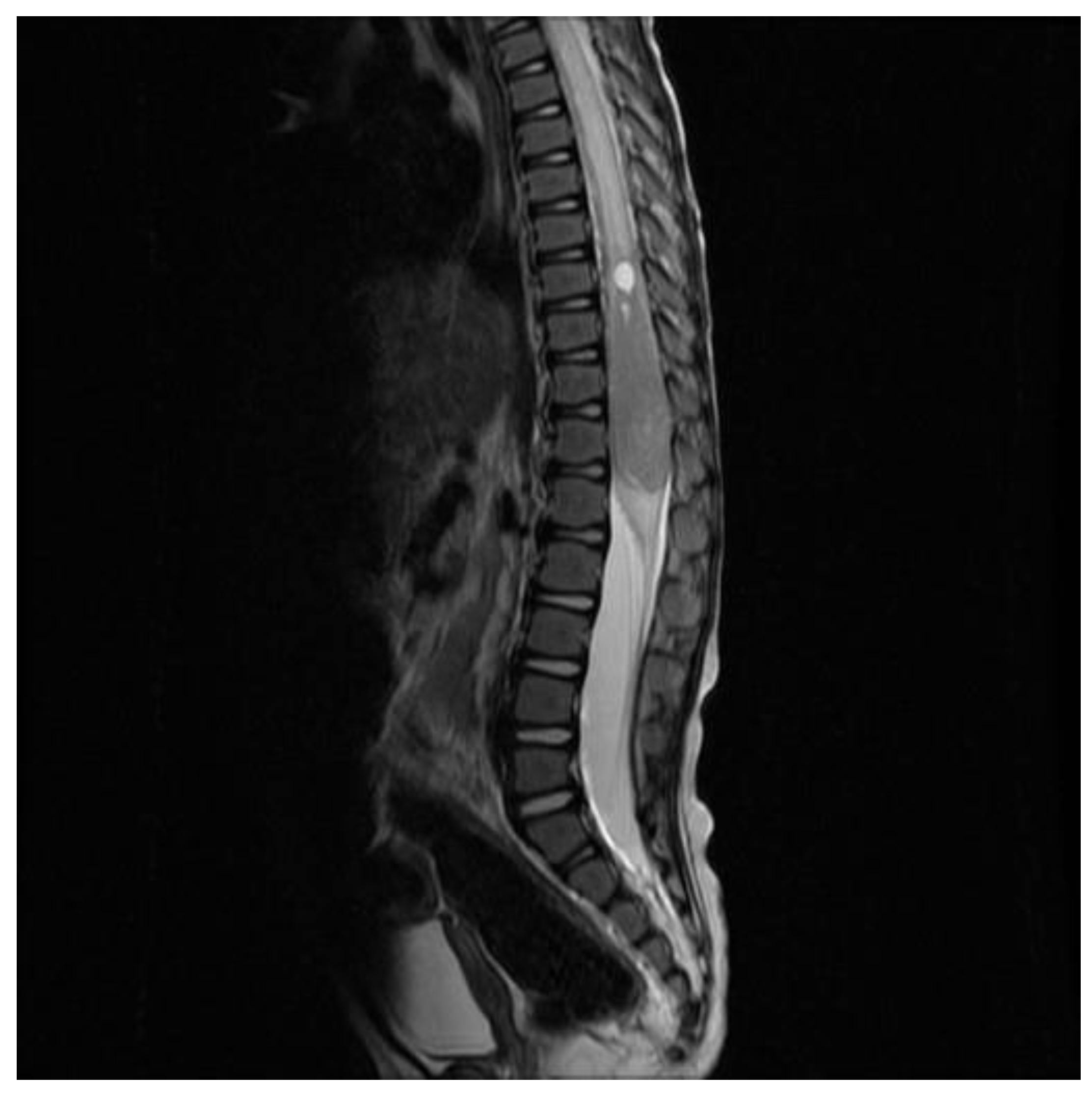
2. Case Report
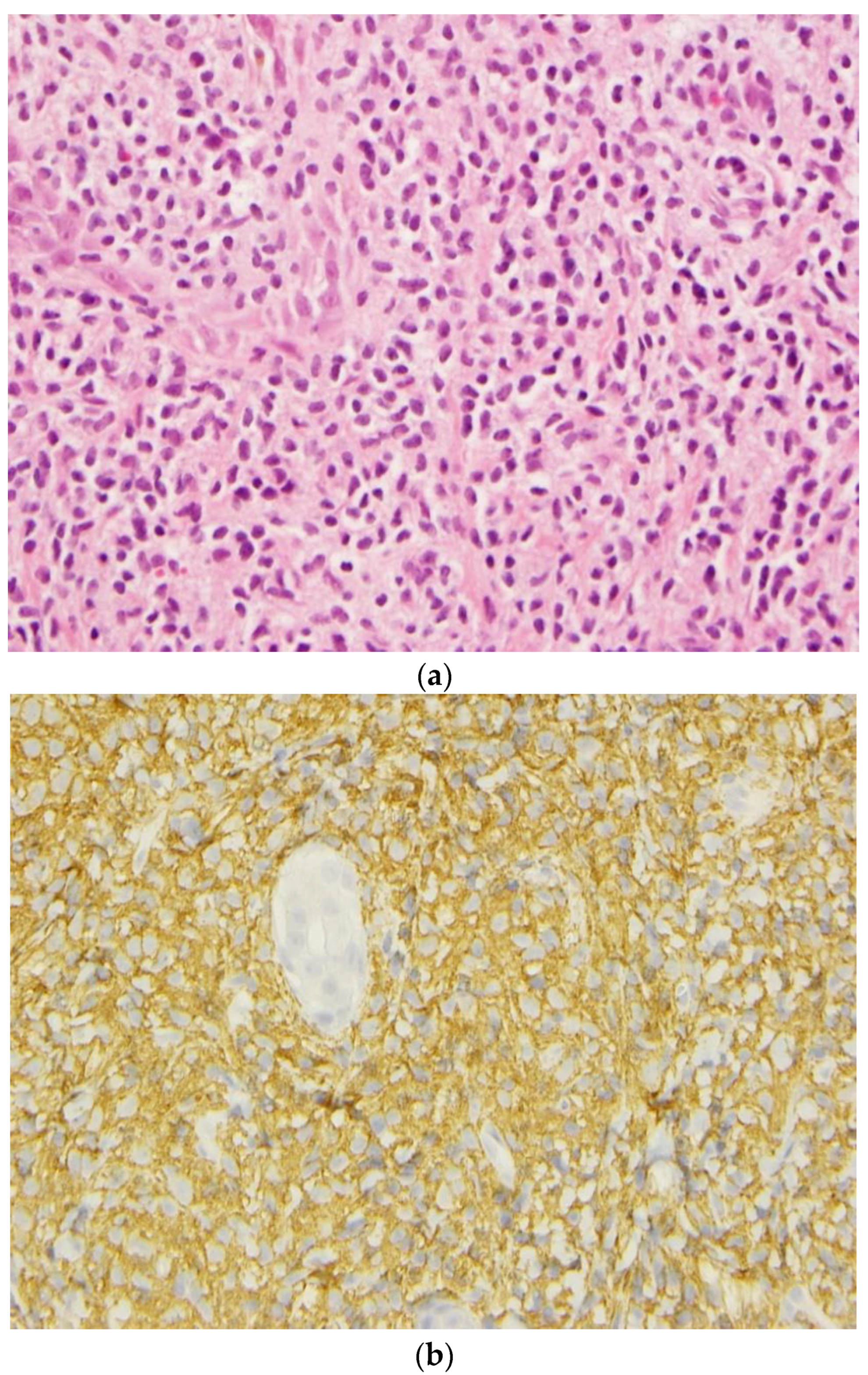
3. Pathological Evaluations
4. Genetic Evaluations
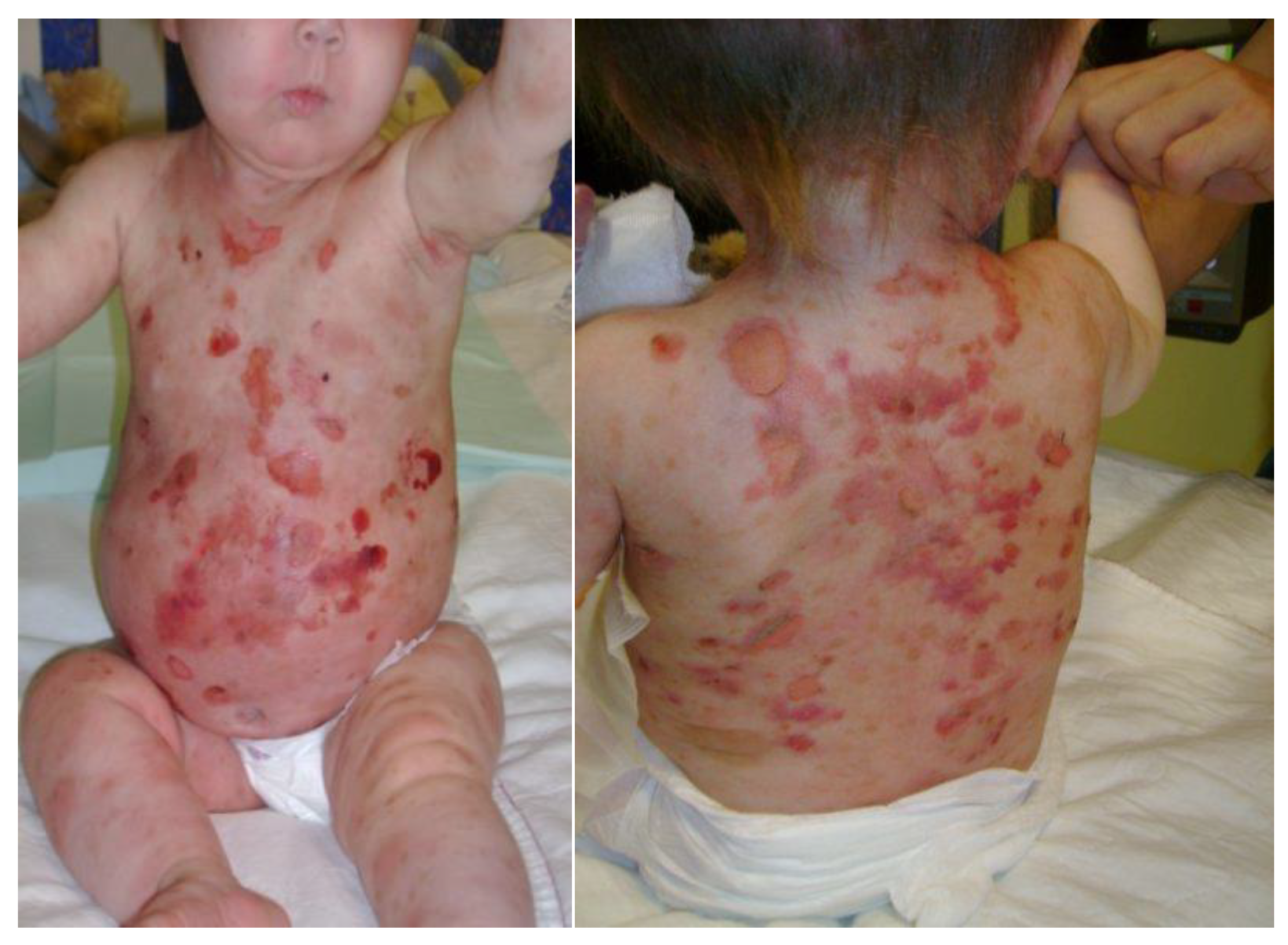
5. Clinical Evolution
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALT | Alanine Aminotransferase |
| BHCG | Beta-human chorionic gonadotropin |
| BM | Bone marrow |
| CGH | Comparative Genomic Hybridization |
| CSF | Cerebrospinal fluid |
| CT | Computed tomography scan |
| FISH | Fluorescence In Situ Hybridization |
| GGT | Gamma-Glutamyltransferase |
| MCs | Mast cells |
| MIM | Mendelian Inheritance in Man |
| MRI | Magnetic Resonance Imaging |
| SM | Systemic mastocytosis |
| WHO | World Health Organization |
References
- Ghiasi, M.; Ghanadan, A.; Jesri, S.B.; Sotudeh, S.; Ramyar, A. Diffuse cutaneous mastocytosis: Report of a severe case with fatal outcome. Dermatol. Online J. 2011, 17, 7. [Google Scholar] [CrossRef]
- Koga, H.; Kokubo, T.; Akaishi, M.; Iida, K.; Korematsu, S. Neonatal onset diffuse cutaneous mastocytosis: A case report and review of the literature. Pediatr. Dermatol. 2011, 28, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Angus, J.; Leach, I.H.; Grant, J.; Ravenscroft, J.C. Systemic mastocytosis with diffuse cutaneous involvement and haematological disease presenting in utero treated unsuccessfully with vincristine. Clin. Exp. Dermatol. 2008, 33, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.; von Komorowski, G.; Scheurlen, W.; Dorn-Beineke, A.; Back, W.; Bayerl, C. Neonatal mastocytosis with pachydermic bullous skin without c-Kit 816 mutation. Dermatology 2006, 212, 70–72. [Google Scholar] [CrossRef]
- Mann, C.; Sepp, N.; Simma, B. Congenital cutaneous mastocytosis. J. Pediatr. 2004, 145, 134. [Google Scholar] [CrossRef]
- Chemli, J.; Krid, S.; Tfefha, A.; Abroug, S.; Harbi, A. Systemic infantile mastocytosis: About a case with respiratory and digestive involvement. Arch. Pediatr. 2003, 10, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Kuint, J.; Bielorai, B.; Gilat, D.; Birenbaum, E.; Amariglio, N. Rechavi GC-kit activating mutation in a neonate with in-utero presentation of systemic mastocytosis associated with myeloproliferative disorder. Br. J. Haematol. 1999, 106, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef]
- Li, Z. New Insights into the Pathogenesis of Systemic Mastocytosis. Int. J. Mol. Sci. 2021, 22, 4900. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hatmann, K.; Alvarez-Twose, I.; Brockow, K.; Hermine, O.; Niedoszytko, M.; Schwaab, J.; Lyons, J.J.; Carter, M.C.; et al. Updated Diagnostic criteria and Classification of Mast Cell Disorders: A Consensus Proposal. HemaSphere 2021, 5, e646. [Google Scholar] [CrossRef]
- Price, C.J.; Green, J.; Kirsner, R.S. Mastocytosis in children is associated with mutations in c-KIT. J. Investig. Dermatol. 2010, 130, 639. [Google Scholar] [CrossRef] [PubMed]
- Bodemer, C.; Hermine, O.; Palmérini, F.; Yang, Y.; Grandpeix-Guyodo, C.; Leventhal, P.S.; Hadj-Rabia, S.; Nasca, L.; Georgin-Lavialle, S.; Cohen-Akenine, A.; et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J. Investig. Dermatol. 2010, 130, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, T.; Vestergaard, H.; Møller, M.B. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. J. Mol. Diagn. 2011, 13, 180–188. [Google Scholar] [CrossRef]
- Wasag, B.; Niedoszytko, M.; Piskorz, A.; Lange, M.; Renke, J.; Jassem, E.; Biernat, W.; Debiec-Rychter, M.; Limon, J. Novel, activating KIT-N822I mutation in familial cutaneous mastocytosis. Exp. Hematol. 2011, 39, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Smith, M.L.; Schultheis, B.; Fitzgibbon, J.; Lister, T.A.; Melo, J.V.; Cross, N.C.; Cavenagh, J.D. A novel K509I mutation of KIT identified in familial mastocytosis-in vitro and in vivo responsiveness to imatinib therapy. Leuk. Res. 2006, 30, 373-378. [Google Scholar] [CrossRef]
- Tang, X.; Boxer, M.; Drummond, A.; Ogston, P.; Hodgins, M.; Burden, A.D. A germline mutation in KIT in familial diffuse cutaneous mastocytosis. J. Med. Genet. 2004, 41, e88. [Google Scholar] [CrossRef]
- Chang, A.; Tung, R.C.; Schlesinger, T.; Bergfeld, W.F.; Dijkstra, J.; Kahn, T.A. Familial cutaneous mastocytosis. Pediatr. Dermatol. 2001, 18, 271–276. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Valent, P.; Akin, C. Mast Cells, Mastocytosis, and Related Disorders. N. Engl. J. Med. 2015, 373, 1885–1886. [Google Scholar] [CrossRef]
- Giona, F. Pediatric Mastocytosis: An Update: Children Mastocytosis. Mediterr. J. Hematol. Infect. Dis. 2021, 13, 69. [Google Scholar] [CrossRef]
- Kleinbaum, E.P.; Lazar, A.J.; Tamborini, E.; Mcauliffe, J.C.; Sylvestre, P.B.; Sunnenberg, T.D.; Strong, L.; Chen, L.L.; Choi, H.; Benjamin, R.S.; et al. Clinical, histopathologic, molecular and therapeutic findings in a large kindred with gastrointestinal stromal tumor. Int. J. Cancer 2008, 122, 711–718. [Google Scholar] [CrossRef]
- Carballo, M.; Roig, I.; Aguilar, F.; Pol, M.A.; Gamundi, M.J.; Hernan, I.; Martinez-Gimeno, M. Novel c-KIT germline mutation in a family with gastrointestinal stromal tumors and cutaneous hyperpigmentation. Am. J. Med. Genet. A 2005, 132, 361–364. [Google Scholar] [CrossRef]
- Hartmann, K.; Wardelmann, E.; Ma, Y.; Merkelbach-Bruse, S.; Preussner, L.M.; Woolery, C.; Baldus, S.E.; Heinicke, T.; Thiele, J.; Buettner, R.; et al. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology 2005, 129, 1042–1046. [Google Scholar] [CrossRef]
- Li, F.P.; Fletcher, J.A.; Heinrich, M.C.; Garber, J.E.; Sallan, S.E.; Curiel-Lewandrowski, C.; Duensing, A.; van de Rijn, M.; Schnipper, L.E.; Demetri, G.D. Familial gastrointestinal stromal tumor syndrome: Phenotypic and molecular features in a kindred. J. Clin. Oncol. 2005, 23, 2735–2743. [Google Scholar] [CrossRef]
- Beghini, A.; Tibiletti, M.G.; Roversi, G.; Chiaravalli, A.M.; Serio, G.; Capella, C.; Larizza, L. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001, 92, 657-662. [Google Scholar] [CrossRef]
- Torrelo, A.; Alvarez-Twose, I.; Escribano, L. Childhood mastocytosis. Curr. Opin. Pediatr. 2012, 24, 480–486. [Google Scholar] [CrossRef]
- Cheng, L.; Roth, L.M.; Zhang, S.; Wang, M.; Morton, M.J.; Zheng, W.; Abdul Karim, F.W.; Montironi, R.; Lopez-Beltran, A. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer 2011, 117, 2096–2103. [Google Scholar] [CrossRef]
- Terada, T. Mediastinal seminoma with multiple KIT gene mutations. Pathology 2009, 41, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Kemmer, K.; Corless, C.L.; Fletcher, J.A.; McGreevey, L.; Haley, A.; Griffith, D.; Cummings, O.W.; Wait, C.; Town, A.; Heinrich, M.C. KIT mutations are common in testicular seminomas. Am. J. Pathol. 2004, 164, 305–313. [Google Scholar] [CrossRef]
- Pauls, K.; Wardelmann, E.; Merkelbach-Bruse, S.; Büttner, R.; Zhou, H. c-KIT codon 816 mutation in a recurrent and metastatic dysgerminoma of a 14-year-old girl: Case study. Virchows Arch. 2004, 445, 651–654. [Google Scholar] [CrossRef]
- Tian, Q.; Frierson, H.F., Jr.; Krystal, G.W.; Moskaluk, C.A. Activating c-kit mutations in human germ cell tumors. Am. J. Pathol. 1999, 154, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.O.; Kang, H.K.; Na, S.Y.; Lee, J.R.; Roh, J.Y.; Lee, J.H.; Kim, H.J. Park SN822K c-kit mutation in CD30-positive cutaneous pleomorphic mastocytosis after germ cell tumour of the ovary. Br. J. Dermatol. 2012, 166, 1370–1373. [Google Scholar] [CrossRef]
- Lee, J.W.; Yang, W.S.; Chung, S.Y.; Kang, J.H.; Cho, B.; Kim, H.K.; Kim, K.M.; Jeong, D.C. Aggressive systemic mastocytosis after germ cell tumor of the ovary: C-KIT mutation documentation in both disease states. J. Pediatr. Hematol. Oncol. 2007, 29, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, S.; Hirota, S.; Park, Y.D.; Yamasaki, M.; Daikoku, N.; Morikawa, H.; Yoshioka, A.; Kitamura, Y.; Ichijima, K. Cutaneous mastocytosis associated with a mixed germ cell tumour of the ovary: Report of a case and review of the literature. Br. J. Dermatol. 2001, 145, 309–312. [Google Scholar] [CrossRef]
- Teitell, M.; Rowland, J.M. Systemic mast cell disease associated with primary ovarian mixed malignant germ cell tumor. Hum. Pathol. 1998, 29, 1546–1547. [Google Scholar] [CrossRef] [PubMed]
- Delacrétaz, F.; Stalder, M.; Meugé-Moraw, C.; Schmidt, P.M.; Joris, F.; Kurt, A.M.; De Werra, P. Systemic mastocytosis following a malignant ovarian germ cell tumour. Histopathology 1997, 30, 582–584. [Google Scholar] [CrossRef]
- Valent, P.; Horny, H.P.; Escribano, L.; Longley, B.J.; Li, C.Y.; Schwartz, L.B.; Marone, G.; Nuñez, R.; Akin, C.; Sotlar, K.; et al. Diagnostic criteria and classification of mastocytosis: A consensus proposal. Leuk. Res. 2001, 25, 603–625. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, D.C.; Briuglia, S.; Concetta Cutrupi, M.; Gallizzi, R.; Rigoli, L.; Dallapiccola, B. Apparent third patient with cutaneous mastocytosis, microcephaly, conductive hearing loss and microtia. Am. J. Med. Genet. 2009, 149, 2270-2273. [Google Scholar] [CrossRef] [PubMed]
- Wolach, B.; Raas-Rothschild, A.; Metzker, A.; Choc, L.; Straussberg, R.; Lew, S.; Goodman, R.M. Skin mastocytosis with short stature, conductive hearing loss and microtia: A new syndrome. Clin. Genet. 1990, 37, 64–68. [Google Scholar] [CrossRef]
- Hennekam, R.C.; Breemer, F.A. Skin mastocytosis, hearing loss and mental retardation. Clin. Dysmorphol. 1992, 1, 85–88. [Google Scholar] [CrossRef]
- Trevisan, G.; Pauluzzi, P.; Gatti, A.; Semeraro, A. Familial mastocytosis associated with neurosensory deafness. J. Eur. Acad. Dermatol. Venereol. 2000, 14, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Ina, A.; Altintas, D.U.; Yilmaz, M.; Uguz, A.; Tuncer, U.; Kiroglu, M.; Herguner, O.; Bicakci, K. Congenital mastocytosis associated with neurosensory deafness. Pediatr. Dermatol. 2007, 24, 460–462. [Google Scholar]
- Murphy, M.; Walsh, D.; Drumm, B.; Watson, R. Bullous Mastocytosis: A fatal outcome. Pediatr. Dermatol. 1999, 16, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Wand, W. C-kit, a double-edged sword in liver regeneration ads diseases. Front. Genet. 2021, 2, 598855. [Google Scholar]
- Spritz, R.A.; Giebel, L.B.; Holmes, S.A. Dominant negative and loss of function mutations of the c-kit (mast/stem cell growth factor receptor) proto-oncogene in human piebaldism. Am. J. Hum. Genet. 1992, 50, 261–269. [Google Scholar]
- Spritz, R.A.; Holmes, S.A.; Berg, S.Z.; Nordlund, J.J.; Fukai, K. A recurrent deletion in the KIT (mast/stem cell growth factor receptor) proto-oncogene is a frequent cause of human piebaldism. Hum. Mol. Genet. 1993, 9, 1499–1500. [Google Scholar] [CrossRef] [PubMed]
- Pagano, L.; Valentini, C.G.; Caira, M.; Rondoni, M.; Van Lint, M.T.; Candoni, A.; Allione, B.; Cattaneo, C.; Marbello, L.; Caramatti, C.; et al. Advanced mast cell disease: An Italian hematological multicenter experience. Int. J. Hematol. 2008, 88, 483–488. [Google Scholar] [CrossRef]
- Droogendijk, H.J.; Kluin-Nelemans, H.J.C.; van Doormaal, J.J.; Oranje, A.P.; van de Loosdrecht, A.A.; van Daele, P.L.A. Imatinib Mesylate in the treatment of systemic mastocytosis. Cancer 2006, 107, 345–351. [Google Scholar] [CrossRef]
- Vega-Ruiz, A.; Cortes, J.E.; Sever, M.; Manshouri, T.; Quintás-Cardama, A.; Luthra, R.; Kantarjian, H.M.; Verstovsek, S. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk. Res. 2009, 33, 1481–1484. [Google Scholar] [CrossRef]
- Purtill, D.; Cooney, J.; Sinniah, R.; Carnley, B.; Cull, G.; Augustson, B.; Cannell, P. Dasatinib therapy for systemic mastocytosis: Four cases. Eur. J. Haematol. 2008, 80, 456-458. [Google Scholar] [CrossRef]
- Verstovsek, S.; Tefferi, A.; Cortes, J.; O’Brien, S.; Garcia-Manero, G.; Pardanani, A.; Akin, C.; Faderl, S.; Manshouri, T.; Thomas, D.; et al. Phase II study of Dasatinib in Philadelphia chromosome negative acute and chronic myeloid diseases, including systemic mastocytosis. Clin. Cancer Res. 2008, 14, 3906–3915. [Google Scholar] [CrossRef]
- Schittenhelm, M.M.; Shiraga, S.; Schroeder, A.; Corbin, A.S.; Griffith, D.; Lee, F.Y.; Bokemeyer, C.; Deininger, M.W.; Druker, B.J.; Heinrich, M.C. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006, 66, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Huang, Z.; Lin, T.; Liu, S.; Chang, H.; Yan, Z.; Zhang, H.; Liu, C. New insight into the anti-liver fibrosis effects of multitargeed tyrosine kinase inhibitors. Front. Pharmacol. 2016, 6, 300. [Google Scholar] [CrossRef] [PubMed]
- Mejias, M.; Garcia--Pras, E.; Tiani, C.; Miquel, R.; Bosch, J.; Fernandez, M. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology 2009, 49, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.-J.; Radia, D.-H.; George, T.-I.; Robinson, W.-A.; Quiery, A.-T.; Drummond, M.-W.; Bose, P.; Hexner, E.-O.; Winton, E.-F.; Horny, H.-P.; et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: The phase 1 EXPLORER trial. Nat. Med. 2021, 27, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Reiter, A.; Radia, D.-H.; Deininger, M.-W.; George, T.-I.; Panse, J.; Vannucchi, A.-M.; Platzbecker, U.; Alvarez-Twose, I.; Mital, A.; et al. Efficacy and safety of avapritinib in advanced systemic mastocytosis: Interim analysis of the phase 2 PATHFINDER trial. Nat. Med. 2021, 27, 2192–2199. [Google Scholar] [CrossRef]
- Pardanani, A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am. J. Hematol. 2021, 96, 508–525. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Criteria | Minor Criteria |
|---|---|
| Multifocal, dense infiltrates of MCs (≥15 mast cells in aggregates) detected in sections of bone marrow biopsies and/or sections of other extra-cutaneous organ(s). | 1. ≥25% of all mast cells are atypical cells on bone marrow smears or other extra cutaneous organs. |
| 2. Detection of KIT point mutation at codon 816 or in other critical regions of KIT in bone marrow, or in another extra-cutaneous organ. | |
| 3. Mast cells in bone marrow, blood, or another extra-cutaneous organ express one or more of the following: CD2, and/or CD25, and/or CD30. | |
| 4. Serum total tryptase > 20 ng/mL (no other associated myeloid neoplasm). |
| Clinical Manifestations | Wolach et al [38]. | Hennekam and Beemer [39]. | Salpetrio et al [37]. | Ina et al [41]. | Trevisan et al. [40]. | Present Case | |
|---|---|---|---|---|---|---|---|
| Case 1 | Case 2 | ||||||
| Cutaneous mastocytosis | + | + | + | + | + | + | + |
| Neonatal mastocytosis | + | + | − | + | − | − | + |
| Systemic mastocytosis | − | − | − | − | − | − | + |
| Hearing loss | + | + | − | + | + | + | + |
| Feeding problems | + | + | − | − | − | − | + |
| Hypotonia | + | + | + | − | − | − | + |
| Microcephaly | + | + | + | − | − | − | − |
| Mental retardation | − | + | + | − | − | − | + |
| Convulsion | − | + | − | − | − | − | + |
| Short stature | + | − | − | − | − | − | + |
| Microtia | + | − | + | − | − | − | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larouche, V.; Paré, M.-F.; Grenier, P.-O.; Wieckowska, A.; Gagné, E.; Laframboise, R.; Jabado, N.; De Bie, I. A Review of the Clinical Features and Management of Systemic Congenital Mastocytosis through the Presentation of An Unusual Prenatal-Onset Case. Curr. Oncol. 2023, 30, 8992-9003. https://doi.org/10.3390/curroncol30100649
Larouche V, Paré M-F, Grenier P-O, Wieckowska A, Gagné E, Laframboise R, Jabado N, De Bie I. A Review of the Clinical Features and Management of Systemic Congenital Mastocytosis through the Presentation of An Unusual Prenatal-Onset Case. Current Oncology. 2023; 30(10):8992-9003. https://doi.org/10.3390/curroncol30100649
Chicago/Turabian StyleLarouche, Valérie, Marie-Frédérique Paré, Pierre-Olivier Grenier, Anna Wieckowska, Eric Gagné, Rachel Laframboise, Nada Jabado, and Isabelle De Bie. 2023. "A Review of the Clinical Features and Management of Systemic Congenital Mastocytosis through the Presentation of An Unusual Prenatal-Onset Case" Current Oncology 30, no. 10: 8992-9003. https://doi.org/10.3390/curroncol30100649
APA StyleLarouche, V., Paré, M.-F., Grenier, P.-O., Wieckowska, A., Gagné, E., Laframboise, R., Jabado, N., & De Bie, I. (2023). A Review of the Clinical Features and Management of Systemic Congenital Mastocytosis through the Presentation of An Unusual Prenatal-Onset Case. Current Oncology, 30(10), 8992-9003. https://doi.org/10.3390/curroncol30100649