5-Methylcytosine (m5C) Modification Patterns and Tumor Immune Infiltration Characteristics in Clear Cell Renal Cell Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. ccRCC Datasets and Preprocessing
2.2. Unsupervised Clustering of m5C Regulators
2.3. Estimation of ccRCC Immune Cell Infiltration
2.4. Differentially Expressed Genes (DEGs) between m5C Clusters
2.5. Gene Set Variation Analysis (GSVA)
2.6. m5C Genetic Signature Generation
2.7. IPS Analysis
2.8. Statistical Analysis
3. Results
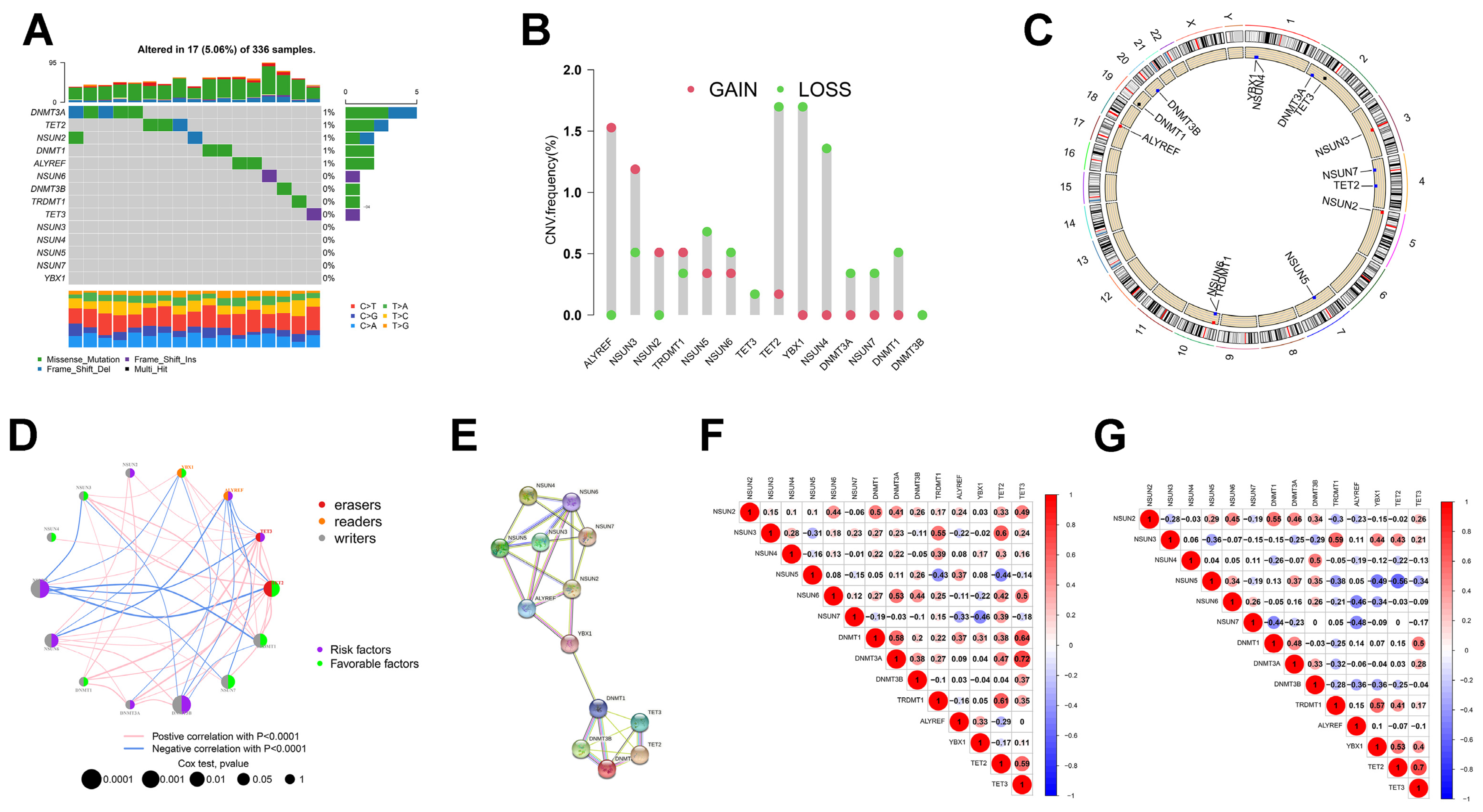
3.1. Somatic Gene Mutation and Expression Patterns of m5C Regulators in ccRCC
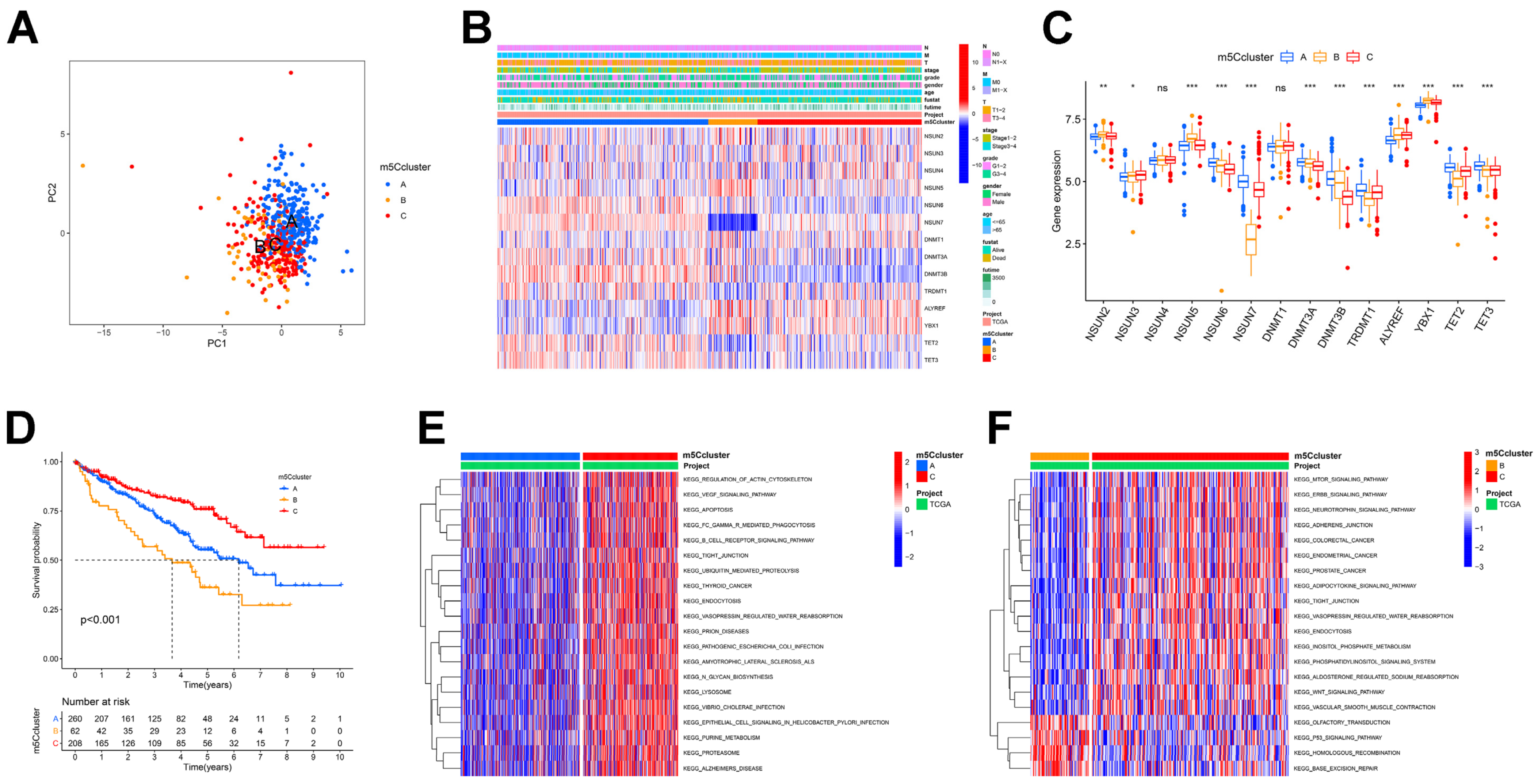
3.2. m5C Modification Patterns Mediated by 14 m5C Regulators
3.3. Analysis of Immune Infiltration in Different m5C Clusters
3.4. Establishment of m5C Gene Signature and Functional Annotation
3.5. Construction of m5Cscore and Its Clinical and Immunological Characteristics
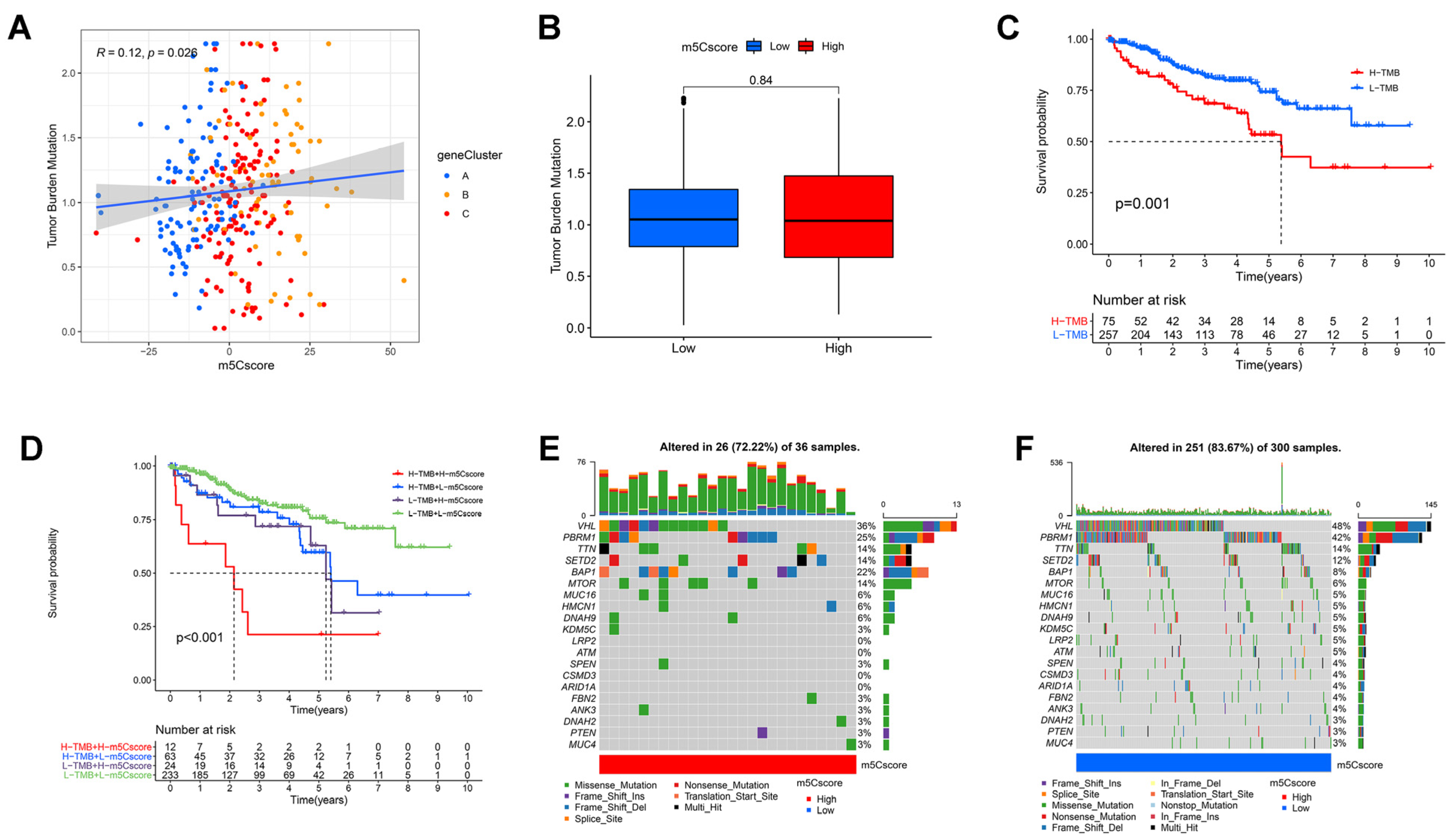
3.6. Correlation between m5C Modifications and Tumor Mutational Burden
3.7. Value of m5Cscore in Predicting Immunotherapy Efficacy
3.8. Validation of m5Cscore Performance in Predicting Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Trixl, L.; Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip. Rev. RNA 2019, 10, e1510. [Google Scholar] [CrossRef]
- Haag, S.; Warda, A.S.; Kretschmer, J.; Günnigmann, M.A.; Höbartner, C.; Bohnsack, M.T. NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. Rna 2015, 21, 1532–1543. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Spåhr, H.; Loguercio Polosa, P.; Meharg, C.; Becker, C.; Altmueller, J.; Habermann, B.; Larsson, N.G.; Ruzzenente, B. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet. 2014, 10, e1004110. [Google Scholar] [CrossRef]
- Dor, Y.; Cedar, H. Principles of DNA methylation and their implications for biology and medicine. Lancet 2018, 392, 777–786. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Höbartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m⁵C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef]
- Chen, X.; Li, A.; Sun, B.F.; Yang, Y.; Han, Y.N.; Yuan, X.; Chen, R.X.; Wei, W.S.; Liu, Y.; Gao, C.C.; et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 2019, 21, 978–990. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011, 25, 2436–2452. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bossé, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e177. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, K.; Aufschnaiter, A.; Messner, S.; Derudder, E.; Herzog, S.; Villunger, A.; Rajewsky, K.; Labi, V. TET enzymes control antibody production and shape the mutational landscape in germinal centre B cells. Febs J. 2019, 286, 3566–3581. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Zhao, Y.; Han, L.; Ma, X.; Deng, J.; Yang, J.; Lü, S.; Shao, F.; Kong, W.; Wang, W.; et al. NSun2 regulates aneurysm formation by promoting autotaxin expression and T cell recruitment. Cell Mol. Life Sci. 2021, 78, 1709–1727. [Google Scholar] [CrossRef]
- Xu, Y.P.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.C.; Tan, X.M.; Cheng, M.; Li, Z.; Bovino, M.; Aubé, J.; et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019, 129, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013, 14, 7. [Google Scholar] [CrossRef]
- Zeng, D.; Li, M.; Zhou, R.; Zhang, J.; Sun, H.; Shi, M.; Bin, J.; Liao, Y.; Rao, J.; Liao, W. Tumor Microenvironment Characterization in Gastric Cancer Identifies Prognostic and Immunotherapeutically Relevant Gene Signatures. Cancer Immunol. Res. 2019, 7, 737–750. [Google Scholar] [CrossRef]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Z.; Qi, F.; Hu, X.; Luo, J. Exploration of the relationships between tumor mutation burden with immune infiltrates in clear cell renal cell carcinoma. Ann. Transl. Med. 2019, 7, 648. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Courtney, D.G.; Tsai, K.; Bogerd, H.P.; Kennedy, E.M.; Law, B.A.; Emery, A.; Swanstrom, R.; Holley, C.L.; Cullen, B.R. Epitranscriptomic Addition of m(5)C to HIV-1 Transcripts Regulates Viral Gene Expression. Cell Host Microbe 2019, 26, 217–227.e216. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, H.; Zhu, X.; Yadav, T.; Ouyang, J.; Truesdell, S.S.; Tan, J.; Wang, Y.; Duan, M.; Wei, L.; et al. m(5)C modification of mRNA serves a DNA damage code to promote homologous recombination. Nat. Commun. 2020, 11, 2834. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, X.; Tang, H.; Jiang, B.; Dou, Y.; Gorospe, M.; Wang, W. NSUN2-Mediated m5C Methylation and METTL3/METTL14-Mediated m6A Methylation Cooperatively Enhance p21 Translation. J. Cell Biochem. 2017, 118, 2587–2598. [Google Scholar] [CrossRef]
- Chellamuthu, A.; Gray, S.G. The RNA Methyltransferase NSUN2 and Its Potential Roles in Cancer. Cells 2020, 9, 1758. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Liu, Z.; Cai, C.; Duan, X.; Deng, T.; Zeng, G. m(6)A modification patterns and tumor immune landscape in clear cell renal carcinoma. J. Immunother. Cancer 2021, 9, e001646. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, M.; Calvo, E. Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell 2020, 38, 326–333. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Xu, C.; Chen, Y.P.; Du, X.J.; Liu, J.Q.; Huang, C.L.; Chen, L.; Zhou, G.Q.; Li, W.F.; Mao, Y.P.; Hsu, C.; et al. Comparative safety of immune checkpoint inhibitors in cancer: Systematic review and network meta-analysis. Bmj 2018, 363, k4226. [Google Scholar] [CrossRef]
- Abou Alaiwi, S.; Xie, W.; Nassar, A.H.; Dudani, S.; Martini, D.; Bakouny, Z.; Steinharter, J.A.; Nuzzo, P.V.; Flippot, R.; Martinez-Chanza, N.; et al. Safety and efficacy of restarting immune checkpoint inhibitors after clinically significant immune-related adverse events in metastatic renal cell carcinoma. J. Immunother. Cancer 2020, 8, e000144. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Bakouny, Z.; Hirsch, L.; Flippot, R.; Van Allen, E.M.; Wu, C.J.; Choueiri, T.K. Beyond conventional immune-checkpoint inhibition—Novel immunotherapies for renal cell carcinoma. Nat. Rev. Clin. Oncol. 2021, 18, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Pan, J.; Huang, Z.; Xu, Y. m5C RNA Methylation Regulators Predict Prognosis and Regulate the Immune Microenvironment in Lung Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 657466. [Google Scholar] [CrossRef]
- Xue, M.; Shi, Q.; Zheng, L.; Li, Q.; Yang, L.; Zhang, Y. Gene signatures of m5C regulators may predict prognoses of patients with head and neck squamous cell carcinoma. Am. J. Transl. Res. 2020, 12, 6841–6852. [Google Scholar]
- Sun, Z.; Xue, S.; Zhang, M.; Xu, H.; Hu, X.; Chen, S.; Liu, Y.; Guo, M.; Cui, H. Aberrant NSUN2-mediated m(5)C modification of H19 lncRNA is associated with poor differentiation of hepatocellular carcinoma. Oncogene 2020, 39, 6906–6919. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Chen, L.-Y.; Zhang, J.-X.; Xu, H.-G. 5-Methylcytosine (m5C) Modification Patterns and Tumor Immune Infiltration Characteristics in Clear Cell Renal Cell Carcinoma. Curr. Oncol. 2023, 30, 559-574. https://doi.org/10.3390/curroncol30010044
Chen C, Chen L-Y, Zhang J-X, Xu H-G. 5-Methylcytosine (m5C) Modification Patterns and Tumor Immune Infiltration Characteristics in Clear Cell Renal Cell Carcinoma. Current Oncology. 2023; 30(1):559-574. https://doi.org/10.3390/curroncol30010044
Chicago/Turabian StyleChen, Can, Lin-Yuan Chen, Jie-Xin Zhang, and Hua-Guo Xu. 2023. "5-Methylcytosine (m5C) Modification Patterns and Tumor Immune Infiltration Characteristics in Clear Cell Renal Cell Carcinoma" Current Oncology 30, no. 1: 559-574. https://doi.org/10.3390/curroncol30010044
APA StyleChen, C., Chen, L.-Y., Zhang, J.-X., & Xu, H.-G. (2023). 5-Methylcytosine (m5C) Modification Patterns and Tumor Immune Infiltration Characteristics in Clear Cell Renal Cell Carcinoma. Current Oncology, 30(1), 559-574. https://doi.org/10.3390/curroncol30010044